

Due to its potential importance in drug delivery and value in understanding ion transport across biomembranes, facilitated ion transfer (FIT) at liquid/liquid (L/L) interfaces has been widely investigated since the pioneering work of Koryta in 1979.1 To date, most reports in this area have focused on facilitated cation transfer (FCT).2 In fact, in spite of great advances in the area of anion recognition chemistry,3 few studies involving facilitated anion transfer (FAT) at L/L interfaces have been published,4 and even fewer reports regarding a kinetic analysis of the underlying phenomenon have appeared.4j Given this lack of available dynamic information, we have used electrochemical methods to study the thermodynamic and kinetic transfer behavior of several monovalent anions at a micropipet-supported micro-water/1,2-dichloroethane (μ-W/DCE) interface. As detailed below, little effective transfer is observed in the absence of an additive or when calix[4]pyrrole 1 is used as a potential transfer agent (cf. Fig. 1A for structures). On the other hand, in the presence of β-octafluoro-meso-octamethylcalix[4]pyrrole 2 effective transfer of Cl-, Br-, NO2- and CH3CO2- (Ac-) at the μ-W/DCE interface is observed. However, the kinetic rate constants are 1-2 orders of magnitude smaller than those for analogous FCT processes involving crown ethers as the ion receptor.2
Figure 1.

A) Structures of calix[4]pyrroles, 1 and 2. B) Cyclic voltammograms for the transfer of Cl-, Br-, NO2- and Ac- at a μ-W/DCE interface facilitated by 2 using Cell 1 (x = 2, y = 100) and a pipet radius, r, of 20, 20, 22, and 10 μm, respectively. The scan rate is 50 mV/s.
In simplified terms, FAT at a μ-W/DCE interface involves the following process:
| (1) |
where A- and R represent a monovalent anion and the putative facilitating anion transfer agent (e.g., receptors 1 or 2, respectively). Key goals of the present study were therefore to i) find a receptor that would allow for facilitated anion transfer and ii) determine the transfer kinetics of anion transport in the event that such a transfer agent is identified.
By their nature, anions are relatively challenging substrates for FIT studies. They are usually larger than isoelectronic cations.3,5,6 They thus have lower charge densities and are less prone to bind strongly to potential transfer agents through electrostatic interactions. Compared to cations, inorganic anions also have larger Gibbs energies of transfer.4 Therefore, transfer of most free inorganic anions occurs at the negative limit or outside of the potential window accessible using typical μ-W/DCE setups. This means that it is inherently harder to find a receptor for FAT than it is for FCT.2 In fact, to date, only a limited number of systems have been used to effect FAT and only when relatively large L/L interfaces (on the order of cm2) were employed.4 We thus sought to explore whether FAT could be achieved in a μ-W/DCE setup using a calix[4]pyrrole as the receptor.7
Calix[4]pyrroles, such as 1, have attracted much attention as anion receptors over the past decade.5 In organic solution and in solid phase, calix[4]pyrroles can bind anions such as F-, Cl-, Br-, I-, and H2PO4- effectively.5 β-Octafluorocalix[4]pyrrole 2 was shown to have an increased anion binding affinity relative to the parent system 1 due to the presence of electron withdrawing fluorine substitutents.8 To date, calix[4]pyrroles have been studied in a number of applications, including as response elements in ion-selective electrodes.9 In this work, the FAT of small anions at the μ-W/DCE interface was studied electrochemically using both 1 and 2 as the possible receptors.
The μ-W/DCE electrochemical cell (Cell 1) employed for the cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements (see SI for experimental detail) is as follows:
| Cell 1 |
Of the anions tested, four (Cl-, Br-, NO2-, and Ac-) displayed reasonable steady-state voltammograms within the potential window when 2 was used as the receptor (Fig. 1B). The negative current reflects the transfer of anions from the aqueous to the DCE phase.10 In the case of F-, which has the strongest interaction with 2 in organic media,5 no wave corresponding to its facilitated transfer can be observed within the potential window. This finding likely reflects the fact that F- is very hydrophilic and retained in the aqueous phase. From the CV and DPV curves, the thermodynamic parameters (such as the diffusion coefficient DR, the stoichiometric ratio m:n, and the association constant can be easily obtained (see the SI for experimental details); these values are listed in Table 1.
Table 1.
Thermodynamic Data for Anion Transfer Facilitated by 2
| Anion | m: n | DR (cm2/s) | ||
|---|---|---|---|---|
| Cl- | -51411a | 1:1 | 8.26 | 4.2×10-6 |
| Br- | -40511a | 1:1 | 6.52 | 3.6×10-6 |
| NO2- | -33211b | 1:1 | 3.28 | 3.2×10-6 |
| CH3CO2- | -56011c | 1:1 | 5.77 | 3.0×10-6 |
| Mean | (3.5±0.5)×10-6 |
Bard et al. have developed a three-point method to determine the kinetic parameters of a heterogeneous electron transfer reaction from a quasi-steady state voltammogram.12 Three parameters, namely the half-wave potential, E1/2, and the quartile potentials E1/4 and E3/4, which can be obtained experimentally, are used to determine the transfer coefficient (α) and the standard rate constant (kο).13 However, it is subject to several caveats, including the need for well-defined steady state voltammograms. In the particular case of the reaction shown in Eq. 1, if the three potentials obtained at a W/DCE interface cannot satisfy the conditions that i) |ΔE1/4=E1/2-E1/4| ≥ 30.5 mV, ii) |ΔE3/4=E3/4-E1/2| ≥ 31.0 mV, and iii) |ΔE3/4| ≥ |ΔE1/4|, then the reaction at the μ-W/DCE interface is reversible and no kinetic data can be obtained using this method. Reversibility depends on kο and the mass transport rate (kd); therefore, it is useful to increase the kd, e.g., by using a smaller nano-L/L interface, to determine even larger kο values. This is what has been done for the previously reported FCT studies.13 In accord with what would be expected based on Eq. S1 (see SI), the E1/2 was found to shift to a more positive potential as the anion concentration increased. Using this approach, well-defined steady state voltammograms for Cl- and Ac- could be observed (cf. Fig. 2 for Cl-). This allowed kο values of 2.11±0.90 and 0.75±0.50 (× 10-2 cm/s), as well as α values of 0.57±0.07 and 0.62±0.04 for Cl- and Ac-, respectively, to be determined (see Table T1 in the SI). Efforts to extend this analysis to Br- and NO2- failed since their waves proved too close to the negative end of potential window.
Figure 2.
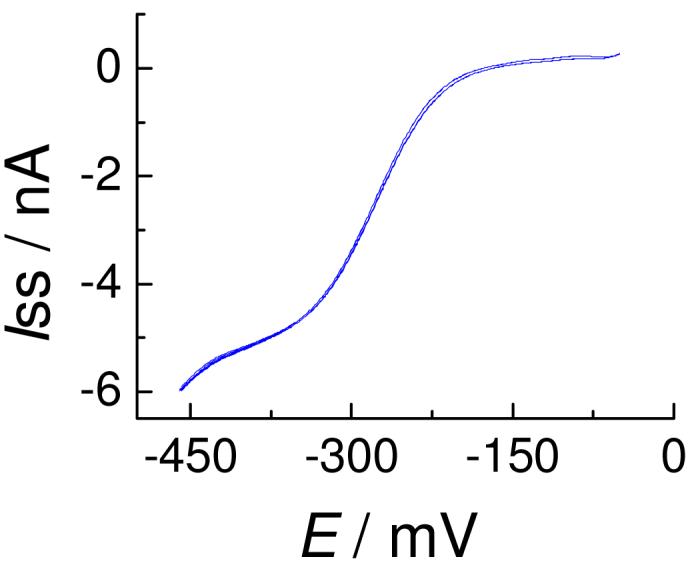
Steady-state voltammogram of Cl- transfer at a μ-W/DCE interface facilitated by 2 using Cell 1, where x = 2, y = 5000. The scan rate is 50 mV/s and r = 7 μm.
From the above results it is clear that the FAT at the W/DCE interface mediated by 2 is much slower than analogous FCT processes observed for alkali metal cations using crown ether as receptors.13 While too many differences exist to allow for direct comparisons, there are a number of likely explanations for this apparent dichotomy. First, as noted above, most anion binding agents, including 2, generally display lower affinities for their targeted anions than do receptors for similarly sized cations. While an obvious oversimplification, such a thermodynamic disadvantage is likely to translate into slower ion transfer kinetics.14 Second, anions of similar charge and size are usually characterized by higher hydration energies (ΔGhyd); cf. e.g., F- -465/Na+ -365 kJ mol-1, Cl- -340/K+ -295 kJ mol-1.15 According to the Marcus theory,16 an ion transfer reaction at a L/L interface involves initial desolvation of an ion from the first phase and then concerted solvation by the second phase. The higher the hydration energy, the harder it is to overcome this barrier. As above, this thermodynamic “penalty” (in the case of anions) is likely to be reflected in slower facilitated ion transfer kinetics. Anions also usually display shorter solvation times than cations (on the order of picoseconds and nanoseconds, respectively);17 however, the effect of this difference on the FIT kinetics is unclear. Nevertheless, this clear difference between cation and anion behavior is of inherent interest and could be useful in the design of ion sensors.
In summary, we have demonstrated the facilitated ion transfer of four anions, namely Cl-, Br-, NO2-, and Ac-, by receptor 2 at a μ-W/DCE interface. We have also shown for the first time that the dynamics of this process can be studied by micropipet-voltammetry. Studies such as these are expected to be useful in understanding the mechanism of anion transport at soft interfaces and for the design of yet-improved anion receptors and carriers.
Supplementary Material
Acknowledgment
This work was supported by the National Natural Science Foundation of China (20735001, 20628506, 20525518, 20775005), the Foundation of Doctoral Programs of the Ministry of Education of China, and the special 985 project of Peking University. The work in Austin was supported by the National Institutes of Health (Grant No. 58907 to J.L.S.) and the INEST Group of Philip Morris USA.
Footnotes
Supporting Information Available: Details of experiments and electrochemical measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Koryta J. Electrochim. Acta. 1979;24:193. [Google Scholar]
- (2).(a) Shao Y, Osborne MD, Girault HH. J. Electroanal. Chem. 1991;318:101. [Google Scholar]; (b) Shao Y, Mirkin MV. Anal. Chem. 1998;70:3155. doi: 10.1021/ac980244q. [DOI] [PubMed] [Google Scholar]; (c) Matsuda H, Yamada Y, Kanamori K, Kudo Y, Takeda Y. Bull. Chem. Soc.Jpn. 1991;64:1497. [Google Scholar]; (d) Zoski CG. In Handbook of Electrochemistry. Elsevier; Amsterdam: 2007. p. 785. [Google Scholar]; (e) Volkov AG. In Liquid Interfaces in Chemical, Biological, and Pharmaceutical Applications. Marcel Dekker, Inc.; New York. Basel: 2001. p. 38. [Google Scholar]
- (3).(a) Sessler JL, Gale PA, Cho WS. Royal Society of Chemistry. Anion Receptor Chemistry. 2006:413. [Google Scholar]; (b) Beer PD, Gale PA. Angew. Chem. Int. Ed. 2001;40:486. [PubMed] [Google Scholar]; (c) Schmidtchen FP, Berger M. Chem. Rev. 1997;97:1609. doi: 10.1021/cr9603845. [DOI] [PubMed] [Google Scholar]
- (4).(a) Shao Y, Linton B, Hamilton AD, Weber SG. J. Electroanal. Chem. 1998;441:33. [Google Scholar]; (b) Katano H, Murayama Y, Tatsumi H. Anal. Sci. 2004;20:553. doi: 10.2116/analsci.20.553. [DOI] [PubMed] [Google Scholar]; (c) Shioya T, Nishizawa S, Teramae N. J. Am. Chem. Soc. 1998;120:11534. [Google Scholar]; (d) Nishizawa S, Yokobori T, Shioya T, Teramae N. Chem. Lett. 2001:1058. [Google Scholar]; (e) Nishizawa S, Yokobori T, Kato R, Shioya T, Teramae N. Bull. Chem. Soc. Jpn. 2001;74:2343. [Google Scholar]; (f) Nishizawa S, Yokobori T, Kato R, Yoshimoto K, Kamaishi T, Teramae N. Analyst. 2003;128:663. doi: 10.1039/b301141k. [DOI] [PubMed] [Google Scholar]; (g) Qian QS, Wilson GS, James KB, Girault HH. Anal. Chem. 2001;73:497. doi: 10.1021/ac000806h. [DOI] [PubMed] [Google Scholar]; (h) Qian QS, Wilson GS, James KB. Electroanalysis. 2004;16:1343. [Google Scholar]; (i) Dryfe RAW, Hill SS, Davis AP, Joos JB, Roberts EPL. Org. Biomol. Chem. 2004;2:2716. doi: 10.1039/B408118H. [DOI] [PubMed] [Google Scholar]; (j) Rodgers PJ, Jing P, Kim Y, Amemiya S. J. Am. Chem. Soc. 2008;130:7436. doi: 10.1021/ja800568q. [DOI] [PubMed] [Google Scholar]
- (5).(a) Gale PA, Sessler JL, Král V, Lynch V. J. Am. Chem. Soc. 1996;118:5140. [Google Scholar]; (b) Gale PA, Sessler JL, Král V. Chem. Commun. 1998:1. [Google Scholar]; (c) Gale PA. Coord. Chem. Rev. 2000;199:181. [Google Scholar]; (d) Gale PA. Coord. Chem. Rev. 2001;213:79. [Google Scholar]
- (6).Shannon RD. Acta Crystallogr.Sect. A. 1976;32:751. [Google Scholar]
- (7).A number of reports describing anion transfer under interfacial conditions have appeared lately; see, for instance: Davis AP, Sheppard DN, Smith BD. Chem. Soc. Rev. 2007;36:348. doi: 10.1039/b512651g. and references therein.. Santacroce PV, Davis JT, Light ME, Gale PA, Iglesias-Sanchez JC, Prados P, Quesada R. J. Am. Chem. Soc. 2007;129:1886. doi: 10.1021/ja068067v.. Eller LR, Stepien M, Fowler CJ, Lee JT, Sessler JL, Moyer BA. J. Am. Chem. Soc. 2007;129:11020. doi: 10.1021/ja074568k.. Wintergerst MP, Levitskaia TG, Moyer BA, Sessler JL, Delmau LH. J. Am. Chem. Soc. 2008;130:4129. doi: 10.1021/ja7102179..
- (8).Anzenbacher P, Jr., Try AC, Miyaji H, Jurisíková K, Lynch VM, Marquez M, Sessler JL. J. Am. Chem. Soc. 2000;122:10268. [Google Scholar]
- (9).Král V, Sessler JL, Shishkanova TV, Gale PG, Volf R. J. Am. Chem. Soc. 1999;121:8771. [Google Scholar]
- (10).Under conditions where the concentration of the anion (CA-) in the aqueous phase is much higher than that of 2 (C2) in the DCE phase (i.e., CA-≫C2), the current is limited by the hemispherical diffusion of 2 to the interface. The mechanism can be verified as involving transfer by interfacial complexation (TIC) and interfacial dissociation (TID).2a
- (11).(a) From http://lepa.epfl.ch/cgi/DB/InterrDB.pl.; (b) Chen Y, Gao Z, Li F, Ge L, Zhang M, Zhan D, Shao Y. Anal. Chem. 2003;75:6593. doi: 10.1021/ac034674e. [DOI] [PubMed] [Google Scholar]; (c) Shao Y, Weber S. J. Phys. Chem. 1996;100:14714. [Google Scholar]
- (12).Mirkin MV, Bard AJ. Anal. Chem. 1992;64:2293. [Google Scholar]
- (13).This method has also been used to study charge transfer kinetics at a L/L interface for examples see: Shao Y, Mirkin MV. J. Am. Chem. Soc. 1997;119:8103.. Yuan Y, Shao Y. J. Phys. Chem. B. 2002;106:7809.
- (14).In simplified terms, the binding energy (ΔGbind) associated with the interaction between an ion and a receptor in two phases can be described as ΔGbind = Gint - Ghyd, where ΔGint is the complexing energy between the ion and receptor and ΔGhyd is the energy of hydration.18 Under interfacial conditions, the situation is more complex, making it difficult to compare directly the energetics of anion vs. cation binding, let alone the underlying kinetics of ion transfer
- (15).Marcus Y. J. Chem. Soc, Faraday Trans. 1991;87:2995. [Google Scholar]
- (16).Marcus RA. J. Chem. Phys. 2000;113:1618. [Google Scholar]
- (17).Girault HH. Analytical and Physical Electrochemistry. EPFL Press; 2004. p. 101. [Google Scholar]
- (18).Blas JR, Márquez M, Sessler JL, Luque FJ, Orozco M. J. Am. Chem. Soc. 2002;124:12796. doi: 10.1021/ja020318m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.