

Abstract
A dual-chip microfluidic platform that coupled perfusion of cultured adipocytes with on-line fluorescence-based enzyme assay was developed to monitor glycerol secretions in real-time from cultured adipocytes. The perfusion cell chip, which could be reversibly sealed to allow reloading of cells and reuse of the chip, was shown by modeling to generate low shear stress on the cells under study. Effluent from the perfusion chip was pumped into an enzyme assay chip for monitoring of secretion from the cells. The on-line enzyme assay had a LOD of 4 μM glycerol. The temporal resolution of the combined system for detecting changes in glycerol concentration was 90 s. The microfluidic device was used to continuously monitor glycerol secretion from murine 3T3-L1 adipocytes, grown and differentiated on glass cover slips, for at least 2 h. An average basal glycerol concentration of 28 μM was detected in the effluent. Pharmacological treatment with a β-adrenergic agonist to stimulate lipolysis evoked a 3-fold increase in glycerol secretion followed by sustained release that was 40% higher than basal concentration. The ability to monitor changes in cellular secretion over time may provide insight into adipocyte metabolism and the dysregulation that occurs with obesity-related disorders.
Keywords: Enzyme assay, microfluidics, adipocytes
INTRODUCTION
Physiological studies frequently require maintaining cells or tissues in a controlled environment while detecting their physical, electrical, or chemical properties. Microfluidics may greatly facilitate such research by allowing creation of highly controlled cell-compatible environments integrated with sophisticated measurement and cell manipulation methods. Examples of using microfluidics for cell physiology include screening for ligand binding and reporter gene expression from immobilized cells using fluorescence microscopy,1 determining metabolic flux from electrically stimulated cells using amperometry2, and characterizing drug dose-response using patch clamp methods.3 Another common physiological method that may benefit from microfluidics is monitoring secretion of chemicals from perfused cells. Without microfluidics, such studies are typically laborious as they require collection and subsequent chemical analysis of large numbers of fractions. Microfluidics may improve perfusion and secretion measurements by reducing costs through reduction of reagent usage, allowing automated analysis, reducing cells required, improving throughput, allowing novel perfusion patterns, and improving temporal resolution.4–6
Several examples of using chips to monitor cellular secretions have been reported.7–13 These include using electrophoretic immunoassay to measure insulin secretion from islets of Langerhans,7, 8 amperometry to monitor catecholamine release from PC12 cells,9 and chemiluminescence to detect glucose and ethanol secretions from S. cerevisiae.10 Enzyme assays have also proven to be a viable approach to monitoring cellular secretions on microfluidic devices. Enzyme assays have been utilized on microfluidic devices to study metabolic secretions from preimplantation embryos,11 activity of expressed enzymes from E. coli cells,12 and immune response from macrophages.13
In this study, we extend such measurements to near real-time monitoring of glycerol secretion from an adipocyte cell line (differentiated murine 3T3-L1 cells)14, 15 using a continuous-flow enzyme assay. The prevalence of obesity and obesity-related disorders underscores the necessity to study and to understand adipocyte physiology. A primary function of adipocytes is to store and release energy. Adipocytes store energy as triacylglycerol and release fatty acids and glycerol by lipolysis to supply energy for other tissues and organs. Measurement of glycerol secretion is often used to ascertain the function and physiological state of adipocytes.16–19 Studying adipocytes in microfluidic devices is hampered by several fat cell properties including high buoyancy, lengthy culture times (~2 weeks) to differentiate adipocyte cell lines, fragility of lipid-laden adipocytes, and secretion of hydrophobic moieties which are difficult to transport through PDMS-based devices. Despite these difficulties, adipocytes have been differentiated,20, 21 studied for incorporation of toxins,20, 22 and examined for size23 on chips; however, integration of cultured adipocytes with chemical measurement of cellular secretions has yet to be performed.
We have developed a dual-chip microfluidic system for culturing adipocytes, perfusing them, and then monitoring glycerol release using a continuous-flow fluorescent enzyme assay. One chip was a cell perfusion chamber. Cells grown on conventional cover slips could be loaded into the reversibly-sealed chip for perfusion experiments. These features allowed the chip to be reused and allowed cells to be cultured for long periods in conventional incubators prior to use. The second chip was used for continuous-flow enzyme assay. To achieve sufficient sensitivity, a commercially available absorbance-based enzyme assay for glycerol was converted to a fluorescence-based enzyme assay by the inclusion of the hydrogen peroxide-sensitive dye Amplex UltraRed. For actual measurements, the chips were coupled so that effluent from the perfusion chip was transferred to the enzyme assay chip allowing adipocyte secretions to be monitored on-line with 90 s temporal resolution. The system was used to demonstrate transient increases in glycerol secretion during exposure of the cells to isoproterenol, a β-adrenergic agonist. The ability to modify enzyme assays for fluorescence-based detection and analyze cells grown in culture on cover slips demonstrates the potential to monitor metabolite secretions in real-time from various cell types utilizing the microfluidic system presented.
EXPERIMENTAL SECTION
Chemicals and Reagents
Free glycerol reagent, glycerol standard solution, isoproterenol, dimethyl sulfoxide (DMSO), cell culture reagents, and 1x Hanks balanced salt solution (HBSS) were purchased from Sigma (St. Louis, MO). Amplex UltraRed was purchased from Invitrogen (Carlsbad, CA). All other chemicals were purchased from Fisher (Pittsburgh, PA). Glycerol standards and the glucose solution perfused over cells were made using HBSS as solvent. All other solutions were made using Milli-Q (Millipore, Bedford, MA) 18-MΩ deionized water. Solutions perfused through microfluidic devices were filtered using 0.2-μm nylon syringe filters (Fisher).
Fabrication of Microfluidic Chips
Microfluidic chips were fabricated using a method previously described.7 The perfusion chip was comprised of two etched, reversibly-sealed glass wafers. The enzyme assay chip was comprised of two hermetically sealed glass wafers, one of which was etched. In brief, Borofloat glass wafers (2.54 × 7.62 × 0.1 cm) coated with 530 nm of AZ1518 positive photoresist on a 120 nm chrome layer were purchased from Telic Co. (Santa Monica, CA). A 50,800 dpi photomask (Photoplot Store, Colorado Springs, CO) was printed from an AutoCAD file of the chip design. To transfer the fluidic network design onto the glass wafer, a collimated UV light source (Optical Associates, Inc., Milpitas, CA) was exposed for 9 s at 26 mW cm−2 through the photomask. The exposed photoresist-coated wafers were developed in AZ 726 MIF developer (AZ Electronic Materials USA Corp., Somerville, NJ) followed by treatment with CEP-200 chrome etchant (HTA Enterprises., San Jose, CA). Channels were formed by etching exposed glass in a 48:17:35 (v/v/v) HF:HNO3:H2O solution. Dicing tape (Semiconductor Equipment Corp., Moorpark, CA) was placed on the uncoated backside of the photoresist-coated wafers and over shallower features to protect from back-etching and to create differential etching depths. Fluidic access holes were created by drilling holes into the etched enzyme assay chip photoresist-coated wafer and the top photoresist-coated wafer of the perfusion chip with 360 μm diameter diamond-tipped drill bits (Kyocera, Irvine, CA). The remaining photoresist and chrome were removed. For bonding of the enzyme assay chip, the enzyme assay etched wafer and a blank coverplate were cleaned in Piranha solution (3:1, v/v, H2SO4:H2O2) for 20 min followed by RCA solution (5:1:1, v/v/v, H2O:NH4:H2O2) for 40 min at 60 C. The cleaned wafers were placed together when wet and then bonded under 400 g weight at 610 C for 8 h in a Neytech Centurian Qex furnace (Pacific Combustion, Los Angeles, CA). Microfluidic reservoirs purchased from Upchurch (Oak Harbor, WA) were bonded on top of the fluidic access holes of both the bonded enzyme assay chip and the top wafer of the perfusion chip. High-pressure perfusion inlets (Upchurch N-124H) were used for all reservoirs except for the waste collection reservoir of the mixing chip (Upchurch N-131).
Adipocyte Culture
Murine 3T3-L1 preadipocytes were cultured in 10 cm Petri dishes at 37 ºC and 10% CO2 until confluent. Preadipocyte media consisted of Dulbecco’s modified Eagle’s medium containing 10% calf serum. Two days post-confluence, differentiation was induced with the addition of fetal bovine serum, insulin, dexamethasone, and isobutylmethylxanthine.24 Media was replaced every two days. Cell experiments were performed using adipocytes 8–10 days after differentiation was induced. The proliferation and differentiation of 3T3-L1 preadipocytes into lipid-laden adipocytes was monitored for cells plated on glass cover slips. Neutral lipid was stained with Oil Red-O as described previously.25
To facilitate ease of use for coupling with the microfluidic device, cells were grown on glass cover slips (24 × 40 × 0.15 mm, Sigma) precut with a diamond-tipped scorer to fit the cell chamber. The cover slips were sterilized with 70% ethanol and placed in the Petri dishes prior to plating cells. The cover slip with differentiated adipocytes was transferred from the Petri dish to the perfusion chip with sterilized tweezers for cellular secretion experiments.
Cell Perfusion Chip and Reversible Sealing
For secretion measurements, glass cover slips supporting ~50 000 differentiated adipocytes were transferred into the cell chamber of the perfusion chip (Figure 1A). The etched depths of the 4 cm long × 0.5 cm wide cell chamber area were 450 μm for the bottom wafer and 100 μm for the top wafer for a total volume of 80 μL. The overlapping cell chamber area was used for alignment as the cell chamber design was etched into both the top and the bottom wafer. A side view of the two perfusion chip wafers together depicts the cells located in the bottom cell chamber area (Fig. 1B). For reversible sealing of the perfusion chip, a pre-cut PTFE sheet (125 μm thick, Small Parts, Inc., Miramar, FL) and high vacuum grease (Fisher) were applied to the 100 μm deep moat prior to pressing the top and bottom wafers together. An in-house built compression frame consisting of two sheets of clear, acrylic plastic (7 × 12 × 0.3 cm per sheet, Small Parts) and screws was used to ensure sufficient contact between the two glass wafers comprising the perfusion chip. A thin-film resistive heater (Minco Products, Inc., Fridley, MN) was placed under the compression frame to maintain the cell chamber at 37 ºC as measured by digital thermometer (Fisher). Additionally, the perfusate solution was pre-warmed prior to flowing over the cells. During cellular secretion experiments, cells were perfused with HBSS containing 5 mM glucose in the absence or presence of 20 μM isoproterenol. Pressure-driven flow was generated through the perfusion chip using a stainless steel reservoir pressurized with He that was connected by fused silica capillary (60 cm long by 150 μm i.d. from Polymicro, Phoenix, AZ) to the chip inlet as reported previously8. The gas pressure system generated 80 μL min−1 volumetric flow through the perfusion chip with 18 psi applied. The flow rate resulted in the chamber being washed out once per minute. The pressure limit for the reversibly-sealed perfusion chip was ~40 psi as determined by observing when fluid leaked out from the seal of the perfusion chip.
Figure 1.

Schematics of the two microfluidic devices used in this work. (a) A diagram of the perfusion cell chip depicts the two separate wafers employed in this work. The wafers were reversibly sealed with the aid of an in-house built compression frame. (b) A side view of the perfusion chip displays the cell chamber, which contained 50 000 differentiated adipocytes. Perfusion solution washed over the cells to sample secretions released from the cells. (c) The enzyme assay chip was capable of performing on-line mixing of three solutions and on-line detection of the enzymatic product. The layout shows the initial mixing channel connected to the incubation channel.
Computational Modeling of Perfusion Chip
The fluid flow dynamics of the perfusion cell chip were modeled using COMSOL Multiphysics 3.3 (Comsol, Inc., Burlington, MA). The flow was defined to be laminar and with a volumetric flow rate of 80 μL min−1. Constants used in the model were 1 × 10−9 m2 s−1 as diffusion coefficient for glycerol, 6.85 × 10−10 m2 s−1 as diffusion coefficient for isoproterenol, 993 kg m−3 for density of water, and 6.90 × 10−4 Pa s for viscosity of water (37 C). The cell chamber volume used was 4 cm long, 0.5 cm wide, and 0.04 cm deep (0.01 cm from the top wafer and 0.03 cm from the bottom wafer after accounting for the presence of the 0.015 cm thick cover slip).
Enzymatic Assay and On-line Mixing
A commercially available absorbance-based, enzyme assay (Free Glycerol Reagent, Sigma) for glycerol was converted to fluorescence-based detection by the inclusion of the hydrogen peroxide sensitive dye Amplex UltraRed resulting in the assay scheme shown in Figure 2A. Amplex UltraRed was diluted with DMSO to result in a 100 μM solution upon on-chip mixing. The glycerol enzyme reagent was reconstituted according to manufacturer’s instructors. The enzyme reagent, which contained all necessary enzymes and co-factors, was mixed with dye and sample and allowed to react for 5 min prior to detection.
Figure 2.
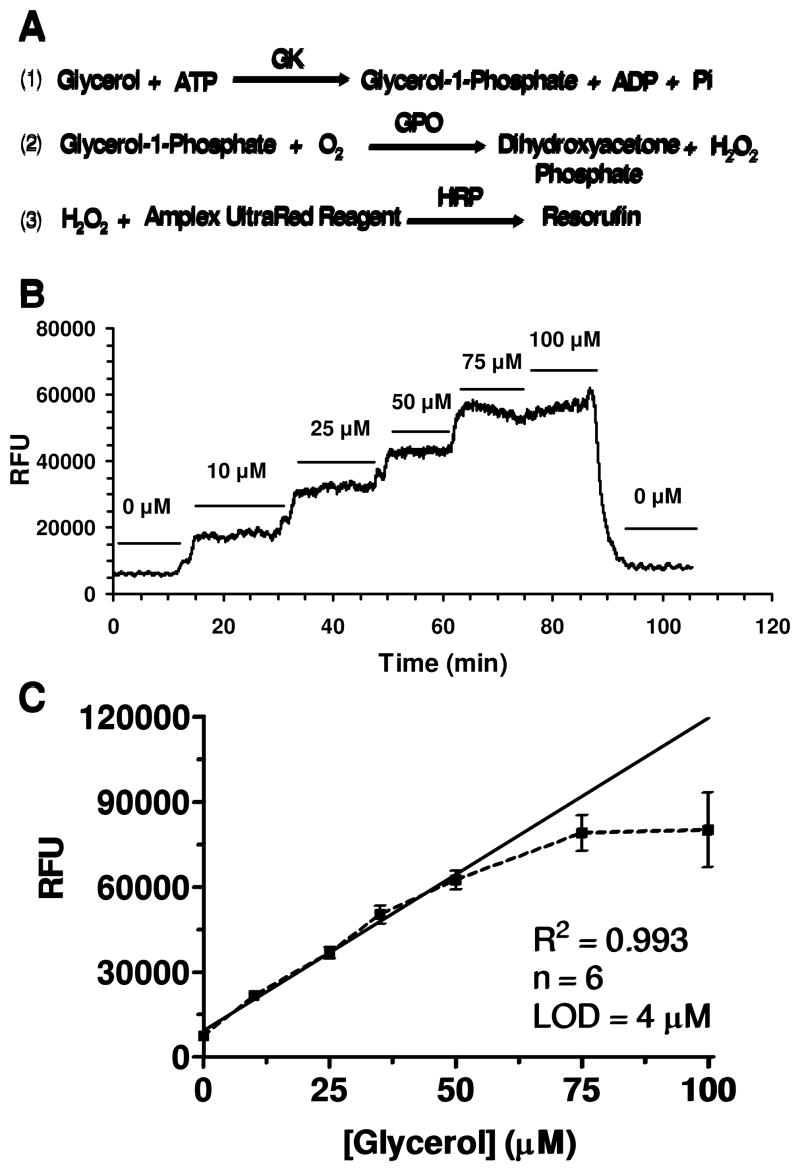
Characterization of on-line fluorescence-based enzyme assay. (a) The scheme of the fluorescence-based enzyme assay for glycerol employed for on-chip mixing and detection is depicted. Glycerol kinase (GK), glycerol phosphate oxidase (GPO), peroxidase (HRP), and Amplex UltraRed were mixed on-chip with glycerol (either from standards or cell effluent) to form the fluorescent product resorufin. (b) An example step-change calibration utilizing the dual-chip system is shown. Glycerol concentrations ranging between 0–100 μM were perfused through the system to determine the corresponding changes in fluorescence. (c) The overall calibration generated a LOD of 4 μM and was linear from 0–50 μM glycerol.
To perform this assay on the microfluidic enzyme chip (Fig. 1C), sample or perfusate from the perfusion chip was transferred via capillary to the enzyme assay chip as a continuous flow. The flow was split so that only 0.31% of the flow from the perfusion chip entered the enzyme chip with the rest going to waste. The flow split was achieved using a Valco tee between the chips with the following capillary lengths and inner diameters for connection: 20 cm, 150 μm from perfusion chip to flow split; 10 cm, 50 μm from flow split to enzyme chip; 7 cm, 150 μm from flow split to waste. All capillary outer diameters were 360 μm. The enzyme reagent and fluorogenic dye were each delivered to the enzyme assay chip via capillary (40 cm, 50 μm i.d.) by a syringe pump (model 402, CMA Microdialysis, North Chelmsford, MA) fitted with 100 μL Hamilton syringes (Reno, NV) resulting in a 1:1:1 ratio of solutions. The inlet flows from each of the 3 channels were 250 nL min−1 each resulting in a total volumetric flow rate through the device of 750 nL min−1. The merged streams flowed into a 7.1 cm long mixing channel (90 μm width at half-height), which was kept relatively narrow to facilitate mixing, and then a 3.75 cm long incubation channel (648 μm width at half-height). The latter channel was wider to reduce the pressure required for pumping. All enzyme assay chip channels were etched to a depth of 60 μm. The combined lengths allowed a 5 min mixing and incubation time.
LIF Detection
For fluorescence detection, a Zeiss Axiovert 100 inverted microscope with epi-fluorescence optics was used to monitor the on-line enzyme assay. The 543.4-nm line of a He-Ne laser (Melles Griot) was directed through a 40x, 0.6 numerical aperture, long working distance objective (Carl Zeiss, Inc. Thornwood, NY) for detection of resorufin. Fluorescence was collected through an appropriate filter set (Omega Optical, Inc., Brattleboro, VT) and a 1 mm pinhole spatial filter and was detected by a photon counting detector (Thermo Oriel, Stratford, CT) at 2 Hz using a LabView program written in-house. Microsoft Excel 2000 (Microsoft, Redmond, WA), Prism 3.03 (GraphPad Software, Inc., La Jolla, CA) and Cutter 5.026 were used for data analysis and graphing. Visual images were collected using a Canon EOS Digital Rebel XT camera (Canon U.S.A., Inc., Lake Success, NY).
RESULTS
On-line Enzyme Assay Characterization
To characterize the response of the system, glycerol standards were pumped through the cell perfusion chip and into the enzyme assay chip where they were mixed with reagents for the glycerol assay, while monitoring fluorescence (Fig. 2B). For this experiment, glycerol standards were pumped through the perfusion chip at 80 μL min−1. The LOD of the on-line fluorescence-based enzyme assay, determined by the concentration that gave a signal 3 times the standard deviation of the blank, was 4 μM for glycerol. The assay gave a linear response from the detection limit to 50 μM glycerol (Fig. 2C). RSDs were from 1–5% indicative of a stable reaction and efficient mixing of the reagents. The signal generated by the fluorescence-based glycerol assay yielded similar RSDs over 3 h for both the blank and a 25 μM glycerol standard, typical basal concentration of glycerol detected from mature adipocytes, indicating good stability and relatively long term operation. Day-to-day reproducibility of the calibration was good with slope equal to 1105 ± 56 RFU μM−1 and intercept 9329 ± 1658 RFU (n = 6).
The rise time of the system, as determined by a 10–90% change in signal for a step-change in glycerol concentration, was 90 s. If the cell perfusion chip was by-passed, the rise time improved to 80 s. Little change in rise time was observed at various points along the incubation channel of the enzyme chip. These observations combined indicate that the greatest source of dispersion was due to the fluidic connections such as the connection volumes and transfer capillaries, not the incubation time or the cell chip.
Glycerol Secretion from Adipocytes
To test the system for monitoring dynamic changes in glycerol release from adipocytes, we monitored glycerol secretion during treatment of adipocytes with the β-adrenergic agonist isoproterenol, which is known to elevate cellular cAMP, activate protein kinase A and stimulate lipolysis.27 For these experiments, cells were loaded into the cell chamber and perfused with glucose-HBSS while monitoring the enzyme reaction as described above for the standards. The cells remained adherent during transfer and perfusion. Basal measurements were collected for at least 60 min and yielded an average concentration of 28 ± 5 μM (SEM, n = 5). Upon switching to a perfusion fluid containing 20 μM isoproterenol, a transient burst of glycerol secretion of approximately 3-fold over basal secretion was detected followed by a sustained release that was ~40% over basal levels (see Fig. 3 for sample individual traces and averaged responses). The burst in glycerol release rose within the first 2 min of exposure to isoproterenol and decreased after ~6 min. The cell system gave stable responses allowing glycerol secretion to be monitored for at least 3.5 h, although most experiments were completed in 2 h (see Supporting Information Figure S1).
Figure 3.

Glycerol secretion data from differentiated adipocytes and response to isoproterenol treatment. (a, b) Representative traces of glycerol release from differentiated adipocytes and response upon 20 μM isoproterenol treatment are shown. (c) Five glycerol secretion traces were averaged and shown with ± SEM. The SEM above and below the average was plotted to enable visualization of the error between measurements. The bars above traces represent exposure to 20 μM isoproterenol. The traces were shortened to depict only the time surrounding the initial exposure to isoproterenol.
In several experiments, fractions were collected from the waste capillary and analyzed for glycerol using the standard assay yielded glycerol concentrations. These experiments yielded glycerol concentrations that on average were with 6% of the on-line measurement (n = 10 fractions from 4 experiments). Moreover, Getty-Kaushik and colleagues reported an increase in overall glycerol secretion upon perfusion of primary rat adipocytes with isoproterenol.17 These results indicate the reliability of the online measurements.
Control Experiments for Secretion Data
Although the measurements were in good agreement with previous results and off-line measurements, we performed several control experiments to ensure that the glycerol recordings were not due to artifacts. Secretion from preadipocytes was used as a negative control as these cells were expected to release little glycerol relative to differentiated adipocytes due to minimal triacylglycerol stores. Prior to differentiation of 3T3-L1 preadipocytes into adipocytes, little triacylglycerol was accumulated within the cells as was confirmed by Oil Red-O staining. Perfusion of 95% confluent preadipocytes resulted in glycerol release below the LOD of the assay (Fig. 4A). Thus, the released levels were at least 9 fold less than from differentiated adipocytes. Moreover, isoproterenol treatment did not result in a detectable increase in glycerol release from treated preadipocytes.
Figure 4.
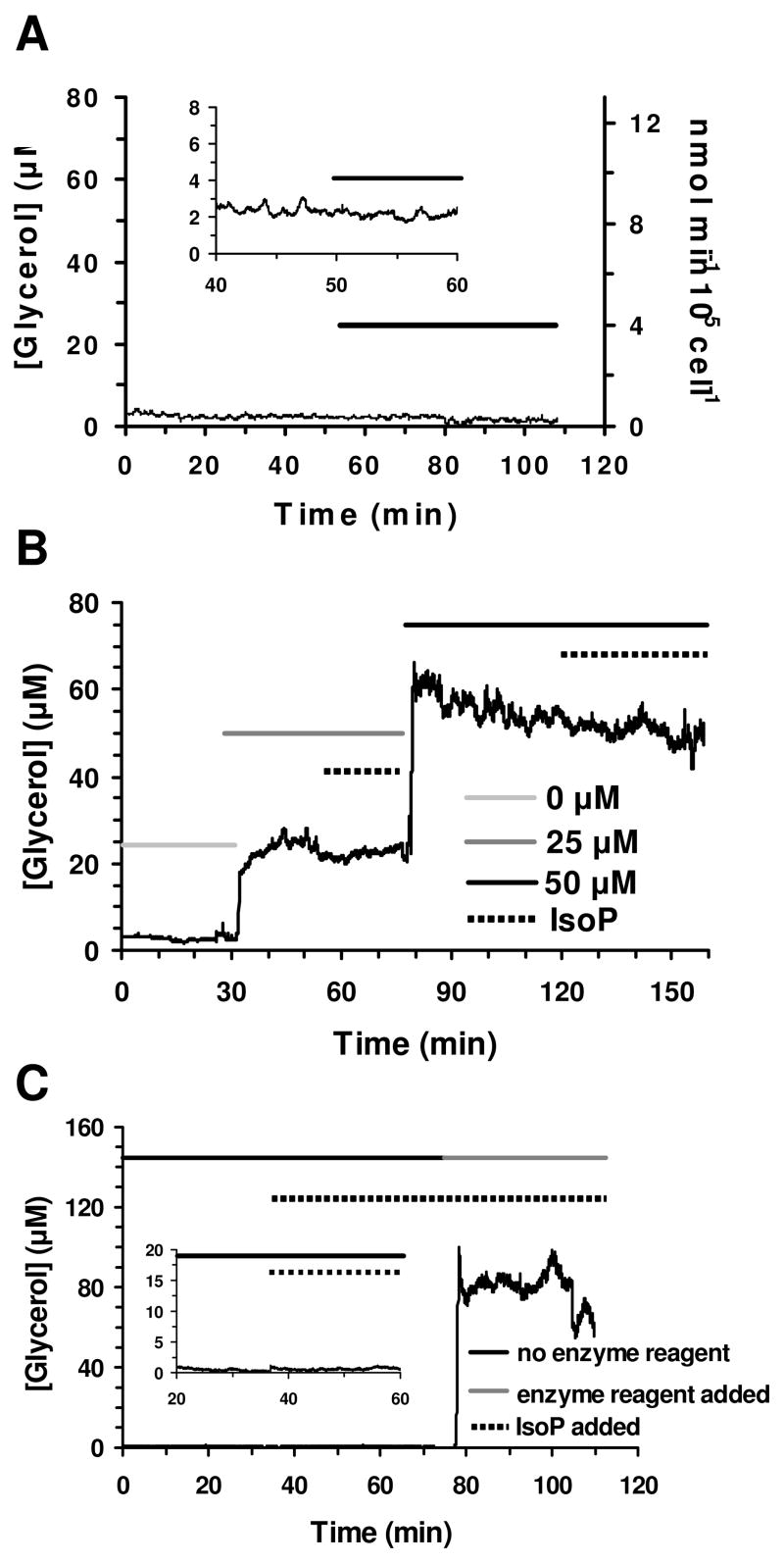
Control secretion data. (a) Glycerol secretion data was obtained from 95% confluent preadipocytes. The bar indicates perfusion of 20 μM isoproterenol. The y-axis of the insert is 10x greater to enable visualization of the glycerol release measured. (b) The effect of isoproterenol introduction on the on-line enzyme assay was determined to be minimal at both 25 μM and 50 μM glycerol. The light grey line indicates perfusion of 0 μM glycerol standard, medium grey 25 μM, and black 50 μM, respectively. The dashed line represents exposure to 20 μM isoproterenol. (c) The response of the system without the enzyme reagent present and upon inclusion of the enzyme reagent during a cellular secretion experiment was tested. The black and grey lines represent the absence and presence of the enzyme reagent, respectively. The dashed line represents treatment with 20 μM isoproterenol. The y-axis scale of the insert is 8x greater for visualization of the release monitored when isoproterenol was introduced in the absence of the enzyme reagent.
To test that the introduction of isoproterenol did not cause a change in fluorescence signal or affect the on-line enzyme assay in the presence of glycerol, the assay was performed in the absence and presence of 20 μM isoproterenol and 25 μM or 50 μM glycerol standards. As shown in Figure 4B, the addition of drug had no effect on signal proving that perfusion of isoproterenol alone did not affect the fluorescence signal and that activation of the cells via isoproterenol treatment was necessary to enhance glycerol release.
An additional control experiment was performed by removing the enzyme reagent during a cellular secretion experiment to ensure that the signal detected was due to enzymatic conversion of glycerol and not direct chemical effects on the dye. In the place of enzyme reagent, buffer was flowed into the enzyme reagent channel to enable correct mixing and incubation times. Without the enzyme reagent present, the fluorescence signal detected was lower than the LOD of the on-line enzyme assay and was approximately the same value as obtained with perfused unreacted dye alone (Fig. 4C). The lack of fluorescence signal shows that Amplex UltraRed was not oxidized by some other moiety released by the cells or intrinsic to the system. Instead, the data show that hydrogen peroxide generated from the series of enzymatic reactions was necessary for generation of the fluorescence signal. Upon introduction of the enzyme reagent, fluorescence signal was detected that corresponded to typical stimulated glycerol levels. Adding the enzyme reagent back to the system ensured that the cells were viable and secreting glycerol.
Characterization of Cell Perfusion Chip
We also characterized the perfusion chip with simulations and experiments to determine the shear stress on cells and uniformity of drug application to the cells. Due to the fragility of adipocytes, the perfusion chip was designed to have a recessed cell chamber area relative to the inlet and outlet flow stream to reduce shear stress and permit laminar flow of fluid over the cells. This design was inspired by previous work that showed that shear force is diminished by placing the fluid inlet above the cells and not inline with them.28 The COMSOL model of our chip estimated the shear stress upon the cells at 0.006 N m−2. This value is in the same range as reported in other microfluidic systems employing cell perfusion5 despite the high volumetric flow rate employed. This low value, plus the observation that the flow did not cause cells to detach or deform, suggests that the chip design prevents excessive shear on the cells.
A potential concern with this design is that drugs are not uniformly applied to the cells. To test for uniformity of flow across the width of the cell chamber, food dye was flowed into the perfusion chip to visualize the flow profile. Pictures of the flow (Supporting Information, Figure S2A) showed that fluid flow was dispersed evenly across the width of the chip, an observation confirmed by COMSOL modeling (not shown). This study also confirmed that: 1) the solution stays confined to the cell chamber, i.e., it does not leak past the moat, 2) the chip washes out rapidly, and 3) flow exhibits the characteristic parabolic profile of laminar flow. To determine if drugs applied would uniformly reach the bottom of the chip, we used a COMSOL model because of the difficulty of directly imaging the depth of penetration of dyes (See Supporting Information, Figure S2B). The model predicted that fluid flowed throughout the entire depth of the cell chamber region and that substances in the perfusion flow, such as isoproterenol, would rapidly reach a uniform concentration throughout the depth of the chip due to a combination of flow and diffusion. Washout of the cell chamber using the model was predicted to occur in 55 s, which closely matched the observed washout time of 60 s and further validated the efficacy of the model.
DISCUSSION
The results show that the dual-chip system is a useful approach to adipocyte culture and monitoring. Although an integrated chip would likely result in a better temporal response by reducing the connection volume between the cells and assay, the dual chip platform allowed both components of the system, cell perfusion and enzyme assay, to be optimized separately. The two-chip approach also had other practical advantages including: 1) ability to operate the perfusion chip while inverted if necessary to prevent cells floating in the direct flow stream without affecting detection in the assay chip (floating cells being a more likely problem with primary adipocytes), and 2) compatibility of a reversibly sealed chip, useful for cell loading, and a bonded chip, useful for enzyme assay.
The perfusion chip was designed to overcome challenges associated with coupling adipocytes with microfluidic devices. Glass was chosen as the material for the microfluidic chips as adipocytes secrete hydrophobic moieties that may interact with or adhere to polymeric materials. The inline weir at the inlet and exit of the chip (Fig. 1B), created through multi-step etching, confined the cells to the cell chamber area and prevented cells from creating clogs in downstream channels as adipocytes may become non-adherent and float. The inlet and outlet locations also minimized shear stress on the cells while allowing drugs to be uniformly and quantitatively applied. To circumvent the long culture time, murine 3T3-L1 adipocytes were grown on glass cover slips that could be placed into the perfusion chip for experiments. Growing cells on cover slips had numerous advantages including: 1) enabling growth of the cells in Petri dishes according to previously optimized culture conditions, 2) increased throughput for secretion experiments relative to growing cells directly in the cell chamber, 3) decreased variability of measurements as multiple cover slips could be tested from the same Petri dish of cells, and 4) potential to return cells to culture following an experiment for re-use. Furthermore, testing cultured cells in a resealable chip enabled the same device to be used numerous times. Indeed, the same chips were used for the entirety of the experiments presented in this paper (data collected over the course of 6 months). The robustness of the chip increases the convenience of use and negates the cost disadvantage of working with glass as the chip material.
Coupling the perfusion chip with the on-line enzyme assay proved efficacious for monitoring cellular activity. Transient secretion dynamics from adipocytes were able to be observed upon treatment with isoproterenol. The sharp rise in glycerol release within the first two minutes of exposure to isoproterenol likely reflects the fast signaling associated with phosphorylation and activation of hormone sensitive lipase and other proteins involved in lipolysis. The decrease in secreted levels after ~6 min may be due to internalization of the β-adrenergic receptor and pathway desensitization upon continuous perfusion of ligand. Previous work with primary rat adipocytes also has shown a burst in glycerol secretion in response to isoproterenol treatment as well as sustained, elevated release.17 The duration of the pulse was ~3 times longer with primary rat adipocytes compared to the pulse detected in this work using immortalized 3T3-L1 adipocytes. However, a similar increase in overall glycerol levels upon treatment with isoproterenol was reported. In this work, the secretion data also consistently showed two peaks during this initial burst of glycerol release. This effect needs further study but may reflect internal dynamics such as mobilization of triacylglycerol stores.
While adipocyte secretions have previously been monitored off-line fraction collection,16–19 this microfluidic-based platform facilitates automated mixing and detection to greatly save on labor and reagent costs. Compared to the same experiments performed using off-line assays, i.e. following the manufacturer’s instructions and using an equivalent concentration of fluorogenic dye with readout on a plate reader, the chip system employing the on-line enzyme assay consumed less than 1% Amplex UltraRed and less than 0.05% enzyme reagent (i.e., 800 μL per aliquot of enzyme reagent is consumed off-line with effluent fractions collected every min compared to 250 nL min−1 on-line). Additionally, the on-line format diminished labor by removing numerous pipetting steps resulting in shorter analysis time. This work also complements other work, such as use of calorimetry for monitoring adipocyte function on chips.29 The fluorescence assay offers specific chemical detection while the calorimetry method offers an overview of cell metabolism.
CONCLUSIONS
We developed a microfluidic platform consisting of two separate chips to integrate cell perfusion, sample handling, and reagent mixing as well as to enable near real-time monitoring of glycerol release from cultured adipocytes. The microfluidic platform was able to detect changes in glycerol levels from ~50 000 adipocytes under basal conditions and upon pharmacological treatment with isoproterenol. The use of a microfluidic device for on-line mixing and detection reduced the consumption of costly reagents to less than 1% of off-line volumes and decreased labor by automating the mixing and detection on-chip. Culturing adipocytes on cover slips increased the throughput of cells available for study as well as decreased biological variability as multiple experiments could be tested from cells cultured in the original Petri dish. This device may be applied to studying the oscillatory nature of glycerol release and long-term changes in metabolism. While the device has been employed to monitor glycerol secretion from 3T3-L1 adipocytes, it is possible that it can be configured to detect other moieties secreted by adipocytes such as non-esterified fatty acids, to test primary adipocytes that have been genetically modified and/or exposed to pharmacological treatment, or to monitor secretions from other adherent cell types such as osteoblasts.
Supplementary Material
Acknowledgments
This work was supported by NIH R37 DK046960 (R.T.K.), NIH NIBIB T32 EB005582 (A.M.C.), DE007057-32 (K.M.S), DK51563 and DK62876 (O.A.M.). The authors thank Dr. Zechariah D. Sandlin and Ammar Salhadar for technical assistance and useful discussions.
References
- 1.Davidsson R, Boketoft A, Bristulf J, Kotarsky K, Olde B, Owman C, Bengtsson M, Laurell T, Emneus J. Anal Chem. 2004;76:4715–4720. doi: 10.1021/ac035249o. [DOI] [PubMed] [Google Scholar]
- 2.Cheng W, Klauke N, Sedgwick H, Smith GL, Cooper JM. Lab Chip. 2006;6:1424– 1431. doi: 10.1039/b608202e. [DOI] [PubMed] [Google Scholar]
- 3.Lau AY, Hung PJ, Wu AR, Lee LP. Lab Chip. 2006;6:1510–1515. doi: 10.1039/b608439g. [DOI] [PubMed] [Google Scholar]
- 4.El-Ali J, Sorger PK, Jensen KF. Nature. 2006;442:403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 5.Kim L, Toh YC, Voldman J, Yu H. Lab Chip. 2007;7:681–694. doi: 10.1039/b704602b. [DOI] [PubMed] [Google Scholar]
- 6.Meyvantsson I, Beebe DJ. Annu Rev Anal Chem. 2008;1:423–449. doi: 10.1146/annurev.anchem.1.031207.113042. [DOI] [PubMed] [Google Scholar]
- 7.Roper MG, Shackman JG, Dahlgren GM, Kennedy RT. Anal Chem. 2003;75:4711–4717. doi: 10.1021/ac0346813. [DOI] [PubMed] [Google Scholar]
- 8.Shackman JG, Dahlgren GM, Peters JL, Kennedy RT. Lab Chip. 2005;5:56–63. doi: 10.1039/b404974h. [DOI] [PubMed] [Google Scholar]
- 9.Li MW, Dana M, Spence DM, Martin RS. Electroanal. 2005;17:1171–1180. [Google Scholar]
- 10.Davidsson R, Johansson B, Passoth V, Bengtsson M, Laurell T, Emnéus J. Lab Chip. 2004;4:488–494. doi: 10.1039/b400900b. [DOI] [PubMed] [Google Scholar]
- 11.Urbanski JP, Johnson MT, Craig DD, Potter DL, Gardner DK, Thorsen T. Anal Chem. 2008;80:6500–6507. doi: 10.1021/ac8010473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huebner A, Olguin LF, Bratton D, Whyte G, Huck WTS, de Mello AJ, Edel JB, Abell C, Hollfelder F. Anal Chem. 2008;80:3890–3896. doi: 10.1021/ac800338z. [DOI] [PubMed] [Google Scholar]
- 13.Goto M, Sato K, Murakami A, Tokeshi M, Kitamori T. Anal Chem. 2005;77:2125–2131. doi: 10.1021/ac040165g. [DOI] [PubMed] [Google Scholar]
- 14.Kawamura M, Jensen DF, Wancewicz EV, Joy LL, Khoo JC, Steinberg DP. Natl Acad Sci-Biol. 1981;78:732–736. doi: 10.1073/pnas.78.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosen ED, Spiegelman BM. Annu Rev Cell Biol. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 16.Getty L, Corkey B. Diabetes. 2003;52:A82–A82. [Google Scholar]
- 17.Getty-Kaushik L, Richard AMT, Corkey BE. Diabetes. 2005;54:629–637. doi: 10.2337/diabetes.54.3.629. [DOI] [PubMed] [Google Scholar]
- 18.Getty-Kaushik L, Richard AMT, Corkey BE. Obes Res. 2005;13:2058–2065. doi: 10.1038/oby.2005.255. [DOI] [PubMed] [Google Scholar]
- 19.Getty-Kaushik L, Song DH, Boylan MO, Corkey BE, Wolfe MM. Obesity. 2006;14:1124–1131. doi: 10.1038/oby.2006.129. [DOI] [PubMed] [Google Scholar]
- 20.Viravaidya K, Shuler ML. Biotechnol Prog. 2004;20:590–597. doi: 10.1021/bp034238d. [DOI] [PubMed] [Google Scholar]
- 21.Ni XF, Crozatier C, Sensebé L, Langonne A, Wang L, Fan Y, He PG, Chen Y. Microelectron Eng. 2008;85:1330–1333. [Google Scholar]
- 22.Nakayama H, Kimura H, Komori K, Fujiii T, Sakai Y. AATEX. 2007;14:619–622. [Google Scholar]
- 23.Wang HW, Bao N, Le TT, Lu C, Cheng JX. Opt Express. 2008;16:5782–5789. doi: 10.1364/oe.16.005782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El-Jack AK, Hamm JK, Pilch PF, Farmer SR. J Biol Chem. 1999;274:7946–7951. doi: 10.1074/jbc.274.12.7946. [DOI] [PubMed] [Google Scholar]
- 25.Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Science. 2000;289:950–953. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 26.Shackman JG, Watson CJ, Kennedy RT. J Chrom A. 2004;1040:273–282. doi: 10.1016/j.chroma.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Arner P. Best Pract Res Cl En. 2005;19:471–482. doi: 10.1016/j.beem.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Figallo E, Cannizzaro C, Gerecht S, Burdick JA, Langer R, Elvassore N, Vunjak-Novakovic G. Lab Chip. 2007;7:710–719. doi: 10.1039/b700063d. [DOI] [PubMed] [Google Scholar]
- 29.Johannessen EA, Weaver JMR, Bourova L, Svoboda P, Cobbold PH, Cooper JM. Anal Chem. 2002;74:2190–2197. doi: 10.1021/ac011028b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.