

Abstract
Down's syndrome (DS) in humans is caused by trisomy of chromosome 21 (HSA 21). DS patients have a variety of pathologies, including mental retardation and an unusually high incidence of leukemia or lymphoma such as megakaryocytic leukemia. Individuals with DS develop the characteristic neuropathological hallmarks of Alzheimer's disease (AD) in early adulthood, generally by the fourth decade of life. There are several mouse models of DS that have a segmental trisomy of mouse chromosome 16 (MMU 16) with triplicated genes orthologous to HSA 21. These mice display neurodegeneration similar to DS. Although brain pathology in DS models is known, little information is available about other organs. We studied the extraneural pathology in aged DS mice (Ts65Dn, Ts2 and Ts1Cje aged 8 to 24 months) as well as other mouse models of neurodegeneration, including presenilin (PS), amyloid-β precursor protein (APP), and tau (hTau and JNPL) transgenic mice. An increased incidence of peripheral amyloidosis, positive for amyloid A (AA) but not amyloid-β peptide (Aβ), was found in APP over-expressing and tauopathic mice as compared to nontransgenic (ntg) littermates or to DS mouse models. A higher incidence of lymphoma was found in the DS models, including Ts1Cje that is trisomic for a small segment of MMU 16 not including the App gene, but not in the APP over-expressing mice, suggesting that high APP expression is not the cause of lymphoma in DS. The occurrence of lymphomas in mouse DS models is of interest in relation to the increased incidence of malignant conditions in human DS.
Keywords: Ts65DN, Amyloid-β Precursor protein, Aβ, Presenilin, Tau, Transgenic, Lymphoma, Down's syndrome, Amyloidosis, Splenomegaly
Introduction
Down's syndrome (DS) is the most common form of mental retardation with a known genetic etiology (Hook, 1989). DS occurs in one in every 800 to 1000 births, and afflicts approximately 350,000 individuals in the USA (Jacobs and Hassold, 1995). In most cases, DS results from the non-disjunction of human chromosome 21 (HSA 21) during meiosis, resulting in three copies of HSA 21 instead of the normal two copies. By the fourth decade of life, individuals with DS display many of the neuropathological features found in Alzheimer's disease (AD) and the majority of these individuals develop dementia (Mann et al., 1986; Visser et al., 1997; and Wisniewski et al., 1985). For these reasons, it has been suggested that DS may serve as a model for the study of pathophysiological factors observed in AD. The precise mechanism(s) by which trisomy 21 leads to either mental retardation or the early-onset of AD-like neuropathology and cognitive decline remains to be elucidated. It has been suggested that the neurodegenerative processes, at least partially, overlap in these disorders (Sendera et al., 2000). However, the phenotypic expression of the genetic defect extends beyond the nervous system. For example, patients with trisomy 21 may develop various forms of leukemia, solid tumors, and other non-neural diseases (Hasle, 2001; Hill et al., 2003; Satge et al., 1998; and Zipursky et al., 1992).
A segmental trisomy mouse model of DS, Ts65Dn, survives to adulthood and exhibits a number of the morphological, biochemical, and transcriptional changes seen in the human disease (Capone, 2001; Davisson et al., 1990; and Holtzman et al., 1996). Ts65Dn mice were produced as 15% of the distal end of mouse chromosome 16 (MMU 16) was translocated to <10% of the centromeric end of MMU 17 to form the small translocation chromosome. Ts65Dn mice possess a third copy of the distal region of MMU 16 spanning from the APP gene (App) to Mx1 orthologous to the DS critical region of HSA 21. Similar to DS individuals, these mice exhibit behavioral and cognitive abnormalities during early postnatal development that persist throughout adulthood (Galdzicki et al., 2001; and Reeves et al., 1995). In later life, they develop additional abnormalities seen in early stages of AD including memory and learning deficits. A particularly striking feature in Ts65Dn mice is the age-related loss of the low-affinity nerve growth factor receptor (p75NTR)-immunoreactive phenotype within the soma and axons of basal forebrain cholinergic neurons (Holtzman et al., 1996; and Salehi et al., 2006). Male Ts65Dn mice are subfertile, and these mice are difficult and expensive to breed. A second trisomic mouse model, Ts[Rb(12.1716)]2Cje (Ts2), is phenotypically similar to the Ts65Dn mouse except that a chromosomal rearrangement of the Ts65Dn genome has been translocated to mouse chromosome 12 (MMU 12) forming a Robertsonian chromosome (Villar et al., 2005). This genetic configuration enables fertility in males and transmits the DS segmental trisomy through the female germline. A third trisomic mouse, the Ts1Cje mouse, is trisomic for a smaller segment of MMU 16 (Sago et al., 1998). These mice were produced by superoxide dismutase 1 (Sod1) gene targeting by homologous recombination (Sago et al., 1998). They are trisomic for MMU 16 from Sod1 to Mx1 with Sod1 interrupted by the neomycin resistance gene. Ts1Cje mice survive to adulthood, exhibit no apparent dysmorphic features or basal forebrain cholinergic neuron degeneration, and do not display the severe cognitive deficits observed in adult Ts65Dn mice. The portion of the trisomic chromosomal region missing in Ts1Cje mice but present in Ts65Dn contains 28 genes, including App.
Although knowledge of brain abnormalities in DS models is evolving, the phenotypic expression beyond the nervous system of these mouse models has hardly been studied. A similar situation is occurring with related animal models of neurodegeneration, including mouse models of APP, PS1, and tau overexpression, whereby significant information is being gleaned concerning the central nervous system, yet little is being evaluated in other organs. In this study we used DS mice (Ts65Dn, Ts2, and Ts1Cje) and mouse models of APP and PS1 overexpression (PS1 mice) (Duff et al., 1996), APP mice (Tg2576) expressing the double Swedish APP mutation K670N (Hsiao et al., 1996; Kurt et al., 2001; and Sadowski et al., 2004), APP/PS1 double transgenic mice that are a cross between Tg2576 mice (Hsiao et al., 1996) and H8.9 mice expressing the M146L mutant PS1 (McGowan et al., 1999), and APP knockout mice (APP KO) (Zheng et al., 1995). Tau overexpressing mice that deposit neurofibrillary-tangle like inclusions were also evaluated including the hTau transgenic line expressing the normal human tau transgene (all 6 isoforms represented) in the absence of the native mouse tau (hTau/8c) (Andorfer et al., 2003; and Duff et al., 2000) and the JNPL line which expresses four-repeat human tau with a P301L missense mutation (Lewis et al., 2000 and Noble et al., 2003).
We present findings on certain viscera from a large cohort of transgenic mice of several genotypes and non-transgenic (ntg) littermates that lived long enough to allow for the development of some neoplasms and systemic amyloidosis.
Materials and methods
All mice used in this study were bred and maintained at the animal facility within the Nathan S. Kline Institute. The procedures and protocols were approved by the Institutional Animal Care and Use Committee (IACUC). Ts65Dn female carriers of translocations involving MMU 16 are mated to male C57BL/6J × C3H/HeJ F1 hybrid mice, as Ts65Dn males are subfertile (Davisson et al., 1990). Ts2 mice were generated by mating female carriers of the T(1716)65Dn marker chromosome and Rb(12.1716)2Cje, respectively, with (C57BL/6JEi · C3H/HeSnJ)F1 males (Villar et al., 2005). Disomic (2N) littermates for Ts65Dn, Ts2, and Ts1Cje mice are (B6EiC3SnJ)F1 (Chen et al., 2008). APP mice are from the Tg2576 line, expressing the double Swedish APP mutant APP K670N/M671L gene (Hsiao et al., 1996). The double transgenic APP/PS1 mice (Kurt et al., 2001; and Sadowski et al., 2004) are the progeny of a cross between Tg2576 mice (Hsiao et al., 1996) and line H8.9 mice expressing the M146L mutant PS1 gene (McGowan et al., 1999). The background strain for the APP, PS1, and APP/PS1 mice is (B6×Dba/2F1)×SW. The background strain for APP KO is C57BL6. hTau mice (Andorfer et al., 2005; and Andorfer et al., 2003) are generated by crossing 8c mice (Duff et al., 2000) that express a tau transgene derived from a human PAC H1 haplotype of the six human tau isoforms with mouse tau KO mice that have a targeted disruption of exon 1 of the mouse tau gene (Tucker et al., 2001). JNPL3 mice are generated by overexpressing a construct of the most common frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) mutation (P301L), which results in motor dysfunction and behavioral deficits by approximately 7 months in hemizygous and 5 months in homozygous JNPL3 mice (Lewis et al., 2000).
Mice were anesthetized with ketamine and xylazine and perfused through the left ventricle with 4% paraformaldehyde in phosphate buffer (0.12 M, pH 7.4) (Ginsberg, 2005; and Ginsberg et al., 2006). The brain and organs were removed and embedded in paraffin, and sectioned at 6 μm for immunocytochemical studies of Aβ and AA. For immunocytochemistry, deparaffinized tissue sections were subjected to microwave antigen retrieval in citrate buffer (Shi et al., 1991) prior to blocking tissue sections in a 0.1 M Tris (pH 7.6) solution containing 2% donor horse serum (DHS; Sigma, St. Louis, MO) and 0.01% Triton X-100 for 1 h and then incubated with primary antibodies in a 0.1 M Tris/2% DHS solution overnight at 4 °C in a humidified chamber. Primary antibodies were directed against Aβ (4G8; monoclonal, Covance, Princeton, NJ; 1:100 dilution) and AA (GTX27505; monoclonal, Gene Tex; San Antonio, TX; 1:10 dilution). Sections were processed with the ABC kit (Vector Labs, Burlingame, CA) and developed with 0.05% diaminobenzidine (Sigma), 0.03% hydrogen peroxide, and 0.01 M imidazole in Tris buffer for 10 min (Ginsberg, 2005). Selected dissected pieces of organs other than brain were post-fixed in Bouin's fluid, embedded in paraffin, sectioned at 5 μm and stained with hematoxylin and eosin. For histochemical amyloid assessments, sections were stained with 1.65% Congo red, as well as periodic acid-Schiff-hematoxylin (PAS). Tissues examined in all aged mice were brain, spleen, liver, kidney, heart, gonad, and any tumors or cysts. Thymus, adrenal, lung, pancreas, and bone were studied in some mice where tumors and/or amyloidosis were suspected. Statistical analysis was performed using a Fisher's exact test.
Results and discussion
Malignant lymphoma was observed in the spleen and lymph nodes of 11 male and 14 female mice. Some of these mice may have had leukemia but blood and bone marrow were not available for analysis. Mice with lymphoma included DS mice (Ts65Dn, Ts2, and Ts1Cje) as well as hTau and PS1 mice (Table 1). In each case with malignant lymphoma, the spleens were enlarged. The splenic architecture was replaced by proliferated immature mononuclear cells arranged in follicular aggregates (follicular lymphoma) (Fig. 1). These follicles were much larger and more irregular in shape than follicles in normal splenic white pulp and often fused with adjacent follicles. The splenic red pulp was largely obliterated. In some affected mice, the proliferated immature cells were diffusely distributed throughout the spleen, nearly obliterating all traces of splenic architecture (Fig. 1). Megakaryocytes were numerous in a few of these spleens, suggesting a relation to megakaryocytic leukemia. Liver and kidney infiltrates were present in a few mice. Statistical analysis of the prevalence of tumors/lymphoma indicated a significant increase in the Ts65Dn (p < 0.04), Ts1Cje (p < 0.002), and hTau (p < 0.04) mice as compared to age matched disomic and ntg controls. No significant differences were observed for the other transgenic lines as compared to ntg littermates.
Table 1.
Incidence of tumors, lymphoma, and amyloidosis in aged DS and AD mouse models
| Mouse lines | Number and gender of micea | Systemic amyloidosisb | Malignant tumorc | Lymphoma in spleen |
|---|---|---|---|---|
| Trisomic DS models | ||||
| Ts65Dn | 65M, 23F | 1M, 1F | 2M, 1F | 5M, 2F |
| Ts2 | 3M, 7F | 1M, 3F | ||
| Ts1Cje | 9M, 17F | 1M | 1M, 3F | |
| Disomic controlsd | 19M, 14F | 2M, 1F | ||
| AD mouse models | ||||
| APP | 15M | 3M | ||
| APP/PS1 | 12M | 3M | ||
| APP/KO | 5M, 3F | |||
| PS1 | 2M, 4F | 2F | 1M, 3F | |
| hTau | 20M, 12F | 2M, 1F | 1M | 3M, 3F |
| JNPL | 5M, 3F | |||
| Controlse | 62M, 18F | 2M, 1F |
All mice were older than 8 months, and most mice were between 12 and 24 months old.
Amyloid deposits in spleen, kidneys, liver, ovary and/or heart.
Malignant tumors of liver, colon, lung (see next column for malignant lesions in spleen).
Disomic littermates of Ts65Dn, Ts2, and Ts1Cje mice.
Non-transgenic littermates of APP, PS1, APP/PS1, APP KO, JNPL, and hTau mice.
Fig. 1.
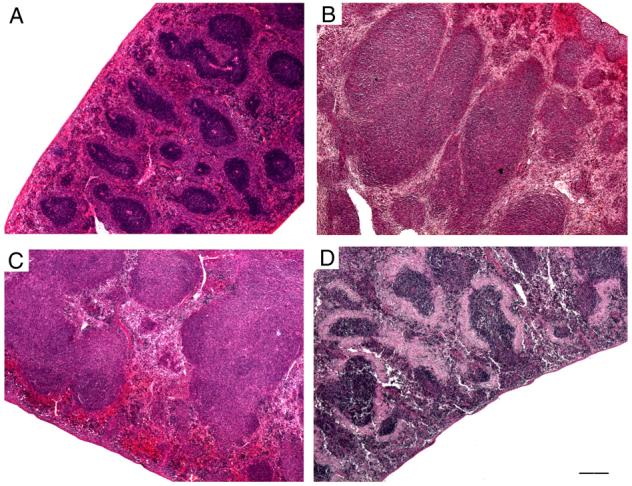
Photomicrographs of mice spleens stained with hematoxylin and eosin, all at the same magnification. (A) In a normal spleen from a disomic mouse, the follicles are small and darkly stained, with abundant red pulp surrounding each follicle. (B and C) The follicles in follicular lymphoma spleen from Ts65Dn mice vary greatly in size and shape, and many of the follicles are more than 10-fold larger than normal. Fusion of adjacent follicles and obliteration of red pulp are seen. (D) In this micrograph of a spleen from an APP mouse, the follicles are only slightly larger than normal, but each one is surrounded by a perifollicular sheath of lightly stained amyloid. The amyloid has replaced most of the red pulp. Scale bar: A-D, 200 μm.
None of the aged disomic and ntg controls had lymphoma. We attribute this negligible incidence rate to the clean facility and caretaking practices of the NKI vivarium. However, 7 control mice and 21 mice of most strains had slightly enlarged spleens with large follicles in the white pulp (Supplementary Table 1). The follicles were relatively uniform in size and shape and the red pulp was not obliterated. The preservation of splenic architecture indicated hyperplasia, not neoplasia. This benign lesion occurred in several DS, APP, and tauopathic genotypes in addition to ntg mice. It is possible that follicular hyperplasia can progress to lymphoma, perhaps only in certain genotypes. Extramedullary hematopoiesis was noted very frequently in splenic red pulp and often included many megakaryocytes. This benign pathology also occurred in control mice and in various genotypes.
An additional 8 mice of various strains and controls had other malignant tumors: hepatoma (n = 4), adenocarcinoma of colon (n = 1), and adenocarcinoma in lung (n = 3; probably metastatic, source unknown) (Table 1). Minor lesions encountered occasionally in all groups of mice included moderate fatty infiltration of liver, small foci of chronic inflammation in kidney, vacuoles in renal tubules, hydronephrosis, cysts of ovary, focal atrophy of testicular tubules, hemosiderosis or hemangioma of spleen, and herniated intervertebral disc. Thymus glands appeared normal and did not show the enlarged Hassall's corpuscles reported in human DS (Levin et al., 1979). Despite the large number of aged mice studied, the incidence of visceral disease was low, indicating the overall good health of these aged cohorts of mice, enabling comparisons across the entire cohort of control and genetically altered mice. It was striking that lymphomatous disorders were restricted to the Ts65, Ts2, Ts1Cje, and hTau genotypes (and a few PS1 mice that did not meet statistical significance). This restricted distribution suggests a relation to the occurrence of leukemias and lymphomas in HSA 21 (Patja et al., 2006; Satge et al., 1996; Smrcek et al., 2001; and Zipursky et al., 1992). However, no increase in the incidence of lymphoma was found in APP overexpressing mice compared to ntg mice. Moreover, a high incidence was observed in Ts1Cje mice which are trisomic for a small segment of MMU 16 not including the App gene. These data suggest that increased APP expression is not the cause of lymphoma in DS.
Systemic amyloid, or an amyloid-like, PAS-positive material, was encountered in 11 male and 5 female mice, all but one of which were > 18 months or older. Specifically, amyloidosis was observed in a significantly greater proportion of APP (p< 0.0001), APP/PS1 (p< 0.002), and hTau transgenic mice (p< 0.02) as compared to ntg littermates (Table 1). APP KO and JNPL mice did not display systemic amyloidosis. Only two Ts65Dn mice displayed amyloidosis and this frequency did not differ significantly from normal 2N mice. Ts2 and Ts1Cje mice did not display systemic amyloidosis. The amyloid was deposited in spleen, kidney and occasionally in liver, ovary, heart, or adrenal. In spleen, the amyloid was deposited concentrically in the perifollicular sheath, partly or completely encircling the follicles (Fig. 1D). In some instances the red pulp was severely reduced. In liver, the amyloid deposit was in the walls of sinusoids or central veins. In the kidney, amyloid material was in the glomeruli. Frequently some or most of the glomeruli were dilated. The dilation was probably caused by obstruction from the amyloid in the lumen or walls of medullary tubules especially in the papilla. Only minor accumulations of amyloid occurred in liver and heart. Deposit of amyloid in the zona reticularis of the adrenal cortex was observed in one mouse (adrenal not studied systematically). The broad distribution of amyloidosis among mouse strains is in accord with the extensive morphologic and histologic experience in the older literature (Andervont and Dunn, 1970) and contrasts with the restricted distribution of lymphomas.
Aβ amyloid deposits have been identified in brain and rarely in certain extraneural tissues (e.g., skin, intestine) in AD patients (Joachim et al., 1989). Moreover, curly fibers that are identified by several antibodies directed against ubiquitin and tau have been identified systemically in AD subjects (Miklossy et al., 1999). Staining of amyloid deposits in the heart with anti-Aβ antibodies was negative (Fig. 2D), indicating that the systemic amyloid deposits seen in the aged mice in the present cohort were not composed of Aβ. This finding was expected, given that the only mice with Aβ amyloid in their brains (APP and APP/PS1) were generated using neuronal-specific promoters to drive human gene expression only in the brain (Hsiao et al., 1996). However, the peripheral amyloid was positive for AA immunoreactivity (Fig. 2B). This is in accordance with the high age-related incidence of this spontaneous amyloidosis (Andervont and Dunn, 1970). It is notable that there is a higher prevalence of AA-immunoreactive deposits in APP and PS1 overexpressing mice, which may have relevance towards understanding the pathobiology of AD. In human pathology, AA amyloidosis in spleen, liver and kidney is typically associated with inflammation, for example secondary to rheumatoid arthritis, tuberculosis, and chronic suppuration as in osteomyelitis, among others (Urieli-Shoval et al., 2000). Inflammation in the brain of AD patients has been well documented (Heneka and O'Banion, 2007; and McGeer et al., 2006) and may be a target of pharmacotherapeutic interventions, but a relationship of AD neuropathology to systemic amyloidosis remains to be determined.
Fig. 2.

(A) Mouse heart from an aged APP mouse stained by hematoxylin and eosin demonstrating lightly stained amyloid (asterisk) in adjacent portions of several muscle fibers. (B) Immunostaining of an adjacent section to (A) for AA. Note darkly stained AA immunoreactivity within scattered muscle fibers (arrow). (C) Adjacent negative control tissue section of heart, prepared as in (B) but without primary antiserum. (D) Lack of positive immunostaining of mouse heart from the same APP transgenic mouse in A-C containing AA amyloid with an antibody directed against Aβ. Note that Aβ is not present in the amyloid fibrils within the heart. (E) Immunostaining of the brain of the same mouse in (D) with the anti-Aβ antibody, indicating the presence of profuse amyloid plaques throughout the cortex and brain parenchyma. Scale bar: A-E, 100 μm.
In summary, lymphoma was over represented in a cohort of aged DS mice as compared to disomic controls, which is relevant to the human DS condition, where lymphomas and leukemias are common negative sequelae. In contrast, aged APP and PS1 transgenic mice displayed a higher propensity for peripheral amyloidosis, which was AA positive, and may point towards a general inflammatory response that occurs in AD and related cerebral amyloidoses.
Supplementary Material
Acknowledgments
Supported by NIH grants AG17617 (EL, SDG), and NS48447 (SDG). We thank Monika Pawlik for expert technical assistance.
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.yexmp.2008.10.006.
References
- Andervont HB, Dunn TB. Amyloidosis in wild house mice during inbreeding and in hybrids derived from inbred strains and wild mice. J. Natl. Cancer Inst. 1970;44:719–727. [PubMed] [Google Scholar]
- Andorfer C, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Andorfer C, et al. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone GT. Down syndrome: advances in molecular biology and the neurosciences. J. Dev. Behav. Pediatr. 2001;22:40–59. doi: 10.1097/00004703-200102000-00007. [DOI] [PubMed] [Google Scholar]
- Chen Y, et al. In vivo MRI identifies cholinergic circuitry deficits in a Down syndrome model. Neurobiol. Aging. 2008;2:2. doi: 10.1016/j.neurobiolaging.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson MT, et al. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog. Clin. Biol. Res. 1990;360:263–280. [PubMed] [Google Scholar]
- Duff K, et al. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Duff K, et al. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- Galdzicki Z, et al. On the cause of mental retardation in Down syndrome: extrapolation from full and segmental trisomy 16 mouse models. Brain Res. Brain Res. Rev. 2001;35:115–145. doi: 10.1016/s0926-6410(00)00074-4. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD. Glutamatergic neurotransmission expression profiling in the mouse hippocampus after perforant-path transection. Am. J. Geriatr. Psychiatry. 2005;13:1052–1061. doi: 10.1176/appi.ajgp.13.12.1052. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, et al. Cell and tissue microdissection in combination with genomic and proteomic applications. In: Zaborszky L, et al., editors. Neuroanatomical Tract Tracing 3: Molecules, Neurons, and Systems. Springer; New York: 2006. pp. 109–141. [Google Scholar]
- Hasle H. Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol. 2001;2:429–436. doi: 10.1016/S1470-2045(00)00435-6. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J. Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Hill DA, et al. Mortality and cancer incidence among individuals with Down syndrome. Arch. Intern. Med. 2003;163:705–711. doi: 10.1001/archinte.163.6.705. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook EB. Issues pertaining to the impact and etiology of trisomy 21 and other aneuploidy in humans; a consideration of evolutionary implications, maternal age mechanisms, and other matters. Prog. Clin. Biol. Res. 1989;311:1–27. [PubMed] [Google Scholar]
- Hsiao K, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Jacobs PA, Hassold TJ. The origin of numerical chromosome abnormalities. Adv. Genet. 1995;33:101–133. doi: 10.1016/s0065-2660(08)60332-6. [DOI] [PubMed] [Google Scholar]
- Joachim CL, et al. Amyloid beta-protein deposition in tissues other than brain in Alzheimer's disease. Nature. 1989;341:226–230. doi: 10.1038/341226a0. [DOI] [PubMed] [Google Scholar]
- Kurt MA, et al. Neurodegenerative changes associated with beta-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp. Neurol. 2001;171:59–71. doi: 10.1006/exnr.2001.7717. [DOI] [PubMed] [Google Scholar]
- Levin S, et al. Thymic deficiency in Down's syndrome. Pediatrics. 1979;63:80–87. [PubMed] [Google Scholar]
- Lewis J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Mann DM, et al. The topography of plaques and tangles in Down's syndrome patients of different ages. Neuropathol. Appl. Neurobiol. 1986;12:447–457. doi: 10.1111/j.1365-2990.1986.tb00053.x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, et al. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J. Alzheimer's Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- McGowan E, et al. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol Dis. 1999;6:231–244. doi: 10.1006/nbdi.1999.0243. [DOI] [PubMed] [Google Scholar]
- Miklossy J, et al. Alzheimer disease: curly fibers and tangles in organs other than brain. J. Neuropathol. Exp. Neurol. 1999;58:803–814. doi: 10.1097/00005072-199908000-00003. [DOI] [PubMed] [Google Scholar]
- Noble W, et al. Cdk5 is a key factor in tau aggregation and tangle formation. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Patja K, et al. Cancer incidence of persons with Down syndrome in Finland: a population-based study. Int. J. Cancer. 2006;118:1769–1772. doi: 10.1002/ijc.21518. [DOI] [PubMed] [Google Scholar]
- Reeves RH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Sadowski M, et al. Amyloid-beta deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2004;63:418–428. doi: 10.1093/jnen/63.5.418. [DOI] [PubMed] [Google Scholar]
- Sago H, et al. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. 1998;95:6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Satge D, et al. A case report of Down syndrome and centroblastic lymphoma. Pathol. Res. Pract. 1996;192:1266–1269. doi: 10.1016/S0344-0338(96)80165-8. [DOI] [PubMed] [Google Scholar]
- Satge D, et al. A tumor profile in Down syndrome. Am. J. Med. Genet. 1998;78:207–216. [PubMed] [Google Scholar]
- Sendera TJ, et al. Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down's syndrome. J. Neurochem. 2000;74:1185–1196. doi: 10.1046/j.1471-4159.2000.741185.x. [DOI] [PubMed] [Google Scholar]
- Shi SR, et al. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J. Histochem. Cytochem. 1991;39:741–748. doi: 10.1177/39.6.1709656. [DOI] [PubMed] [Google Scholar]
- Smrcek JM, et al. Fetal hydrops and hepatosplenomegaly in the second half of pregnancy: a sign of myeloproliferative disorder in fetuses with trisomy 21. Ultrasound Obstet. Gynecol. 2001;17:403–409. doi: 10.1046/j.1469-0705.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- Tucker KL, et al. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Urieli-Shoval S, et al. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr. Opin. Hematol. 2000;7:64–69. doi: 10.1097/00062752-200001000-00012. [DOI] [PubMed] [Google Scholar]
- Villar AJ, et al. Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(171)65Dn and mouse chromosome 12. Mamm. Genome. 2005;16:79–90. doi: 10.1007/s00335-004-2428-7. [DOI] [PubMed] [Google Scholar]
- Visser FE, et al. Prospective study of the prevalence of Alzheimer-type dementia in institutionalized individuals with Down syndrome. Am. J. Ment. Retard. 1997;101:400–412. [PubMed] [Google Scholar]
- Wisniewski KE, et al. Alzheimer's disease in Down's syndrome: clinicopathologic studies. Neurology. 1985;35:957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- Zheng H, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- Zipursky A, et al. Leukemia in Down syndrome: a review. Pediatr. Hematol. Oncol. 1992;9:139–149. doi: 10.3109/08880019209018329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.