

Abstract
The development of small molecules that disrupt protein-protein interactions is a key goal in addressing a number of disease states. The α-helix is commonly found at protein interaction interfaces and has been the focus of substantial small molecule mimetic efforts. One of the primary drawbacks of many small molecule α-helix mimetics is their hydrophobic core structures. To address this problem we have developed a novel scaffold based on a more water soluble 5-6-5 imidazole-phenyl-thiazole core. An inhibitor of this class has been shown to disrupt the Cdc42/Dbs protein-protein interaction at micromolar concentrations and may be useful in overcoming Cdc42 induced tumor resistance to anti-cancer therapies.
Protein-protein interactions have been implicated in numerous disease states including HIV and cancer, making them important targets for disruption by small molecules.1 α-Helices are common protein secondary structure elements and are often responsible for recognition between proteins via one helical face where interacting residues occupy predominantly the i, i + 3 or i + 4, and i + 7 positions. Accordingly, these surfaces are critical targets for small molecule mimicry. In an effort to disrupt α-helix mediated protein-protein interactions, we have reported several small molecules that act as structural and functional mimetics of α-helices.2 Often the binding hot spots targeted by α-helix mimetics lie in hydrophobic clefts that are solvent exposed when the two interacting proteins are not associated.3 We have previously focused on scaffolds that rely on six-membered rings (either aromatic or hydrogen bonded) to maintain their proper orientation.1 Earlier α-helix mimetics, such as those based on terpyridine2a and terphenyl2b scaffolds, are inherently disadvantaged because of poor solubility while others, based on terephthalamide2c and trispyridylamide2d scaffolds, rely on hydrogen-bonding networks to maintain their correct orientation. Recent work by Rebek and König has begun to address the demand for more water soluble α-helix mimetics, by making one side of the scaffold hydrophilic4a-b or via a 1,4-dipiperazino benzene scaffold.4c Our design focuses on two variations of a novel 5-6-5 imidazole-phenyl-thiazole scaffold that replaces sixmembered aromatic end-units with water-soluble fivemembered heterocyclic groups.
Using this novel class of α-helix mimetics we have chosen to target a protein-protein interaction governed largely by hydrophilic contacts, that between Cdc42 (cell division cycle 42) and Dbs (Dbl’s big sister).5 Cdc42 is a GTPase (Guanine nucleotide Triphosphatase) shown to mediate cancer cell resistance to both cytotoxic therapies and cytotoxic T-lymphocyte induced tumor suppression.6 Cdc42 has also been linked to diabetes, cardiovascular, and neurodegenerative diseases.7 However, Cdc42 is only capable of these aberrant activites in its activated, GTP bound, form.7 Cdc42 is activated by interaction with the GEF (Guanine nucleotide Exchange Factor), Dbs. We chose to target the Cdc42/Dbs interaction in an effort to block the most upstream event involved in Cdc42 related disease. In addition, to the best of our knowledge, no rationally designed small molecule inhibitors of this protein-protein interaction currently exist. The interaction between these proteins is mediated by the Q770, K774, and L777 residues of Dbs, which correspond to the i, i + 4 and i + 7 of a key Dbs α-helix, making it an ideal target for our mimetics (Figure 1c).5 Herein we detail the design and synthesis of a 5-6-5 imidazole-phenyl-thiazole based, water soluble α-helix mimetic of these key residues.
Figure 1.
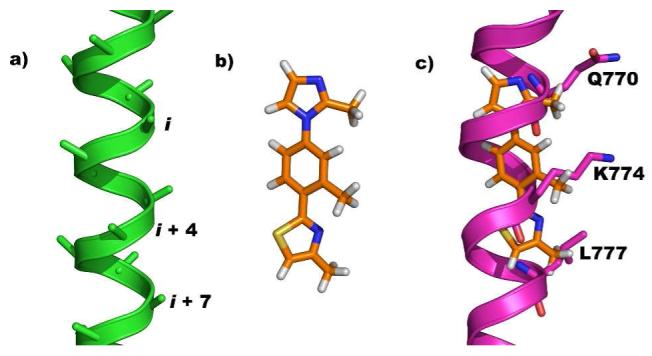
a) Energy minimized poly-alanine α-helix displaying i, i + 4, and i + 7 positions. b) Energy minimized structure of 4. c) Overlay of energy minimized 4 onto targeted region of Dbs, residues Q770, K774, and L777.
Scheme 1 depicts the synthetic route to a model derivative, 4-methyl-2-(2-methyl-4-(2-methyl-1H-imidazol-1-yl)phenyl) thiazole (4), with methyl groups in the key substituent positions. Treatment of commercially available 1, with phosphorous pentasulfide gave thioamide 2 which after heating with chloroacetone furnished thiazole 3.8 Ullman coupling with 2-methylimidazole gave 4, in excellent yield.9 A crystal structure of 4 (Figure 2) confirmed the extended shape of the molecule and the staggered projections in a non planar orientation (5-6-5 dihedral angles, 43.0° and 38.2°). The log P (o/w) of the corresponding trimethyl substituted terphenyl,10 terpyridine2a and 4 were calculated as 7.3, 3.4 and 2.3, respectively, suggesting a significant improvement in aqueous solubility of the 5-6-5 heterocyclic system and this was seen with 12 and 19 which were soluble to ≥500 μM in 2% DMSO/buffer.11
Scheme 1.
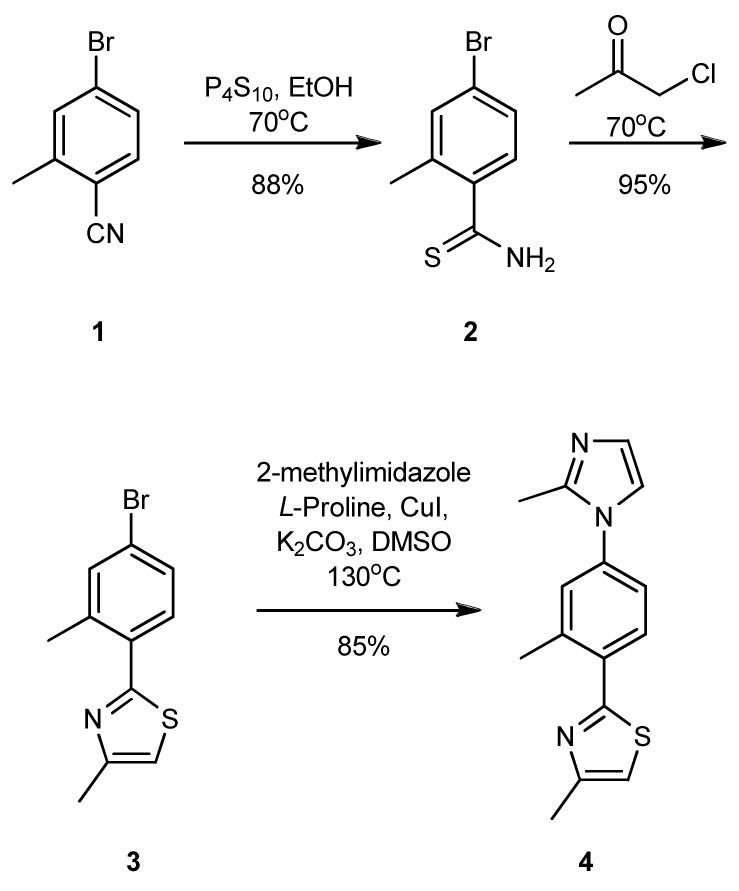
Synthesis of Model 5-6-5 scaffold 4.
Figure 2.
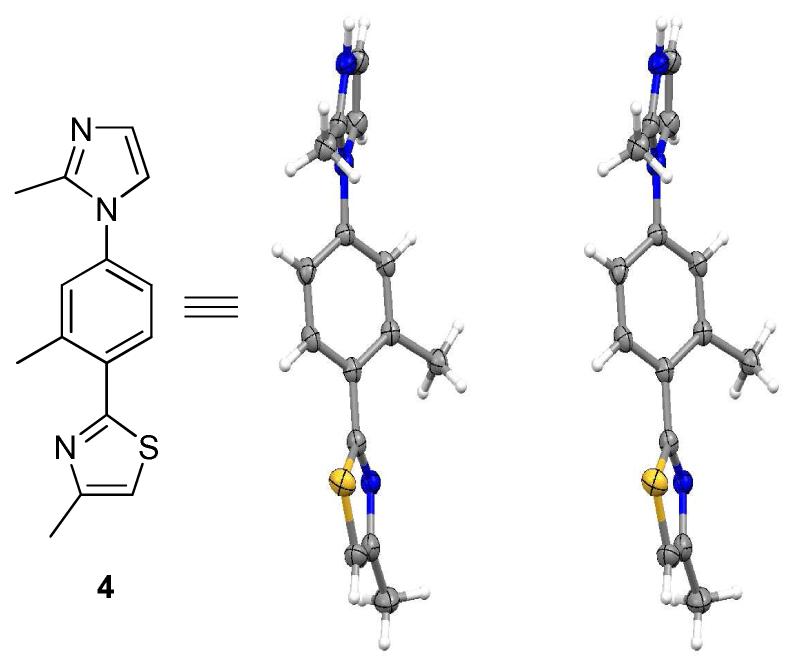
Crystal Structure of 4 in stereoview.
Functionalization of the core scaffold with amino acidlike side chains similar to those of Dbs involved modification of the synthetic route. Nucleophilic aromatic substitution of 5 with t-butyl 3-hydroxypropylcarbamate (KHMDS in THF) gave 6, which was coupled under Ullman conditions with methyl 3-(1H-imidazol-2-yl)propanoate to form 7 in excellent yield.8 Oxidation with (NH4)2S(aq) gave thioamide 8, which on treatment with isopropylchloroactetate, formed 9.9 Saponification of 9 followed by oxidation and deprotection gave 12 in high overall yield. The synthetic approach to 12 is flexible allowing for the ready introduction of diverse side chains.
Compound 12 was assayed for its ability to disrupt the Cdc42/Dbs interaction using a gel pull-down assay that has identified inhibitors of other GTPase/GEF pairs.12 However, 12 showed no detectable inhibitory activity, possibly due to the highly flexible side groups.
In an attempt to reduce the flexibility of the side chains in 12, we introduced into a second generation inhibitor design a series of substituents based on constrained analogs of the key functional groups. A benzimidazole diacid replaced the flexibly substituted imidazole and a 2-substituted tolyl group was used in place of the isopropyl ether. Although it has a lower pKa, 3-substituted aniline was used to rigidify the alkyl amine group in 12 with the expectation that the arylamine would still participate in hydrogen bonding to Cdc42 active site residues. The synthetic route (Scheme 3) involves nucleophilic aromatic substitution of 2-bromo-4-fluorobenzonitrile with 14 to give N-arylbenzimidazole 15. Hantzsch conditions with 2-bromo-1-o-tolylethanone and thioamide 16 produced thiazole 17 which was subjected to Suzuki coupling with 3- aminophenyl boronic acid to give, after deprotection, target 19.9,13 Diacid 19, in a nonplanar conformation, could match the positions of the i, i + 4, and i + 7 side chains in the key Dbs α-helix.
Scheme 3.
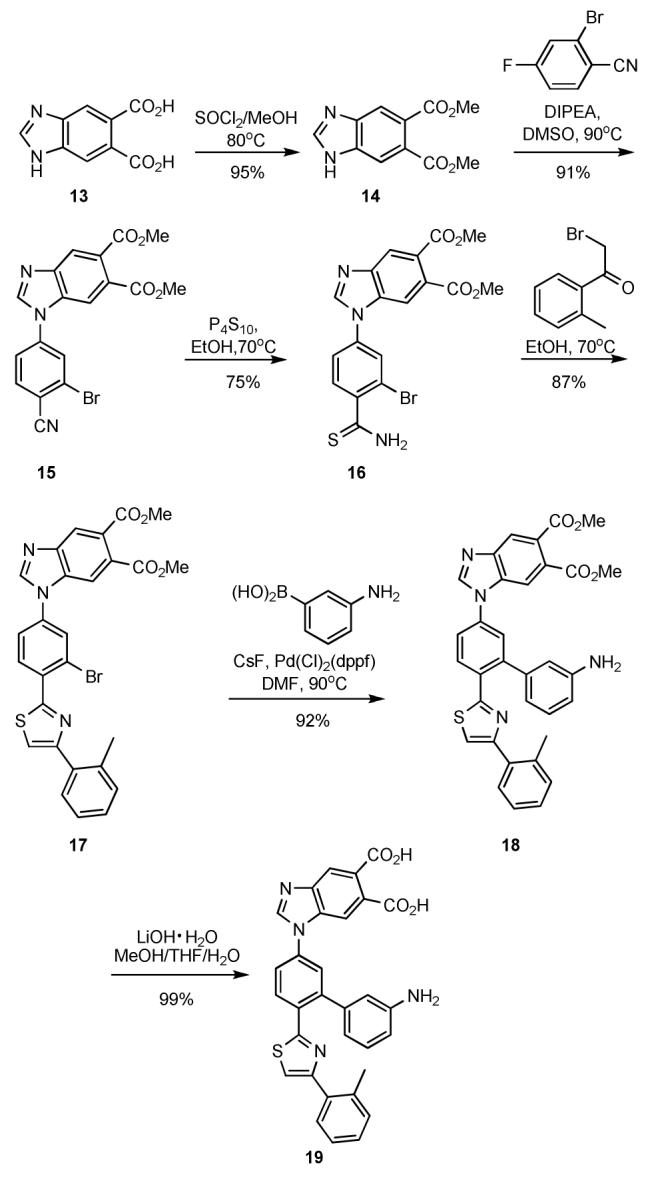
Synthesis of 19
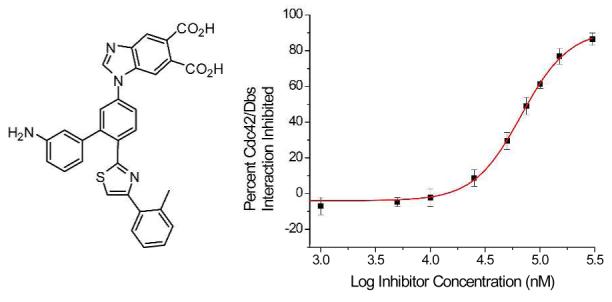
Mimetic 19, was assayed for its ability to disrupt the Cdc42/Dbs interaction using a mant-GDP fluorescence based activity assay, in which the uptake of this GDP analog into Cdc42 is monitored.14 Compound 19 was screened at varying concentrations with 100% inhibition being defined as the extent of mant-GDP uptake when no Dbs or inhibitor was present.
Dbs accelerated uptake of mant-GDP by Cdc42 was inhibited in a dose-response fashion by 19 (Figure 3). Fitting this data to a one-site binding model yielded an IC50 of 67 μM. To further validate that the binding site of 19 was in fact the Cdc42/Dbs interface, the ability of 19 to disrupt non-Dbs assisted uptake of mant-GDP was monitored. At 50 μM 19 less than 10% of mant-GDP uptake was blocked, indicating that the mechanism of action of this compound is not simply directly interfering with mant-GDP uptake by Cdc42.
Figure 3.
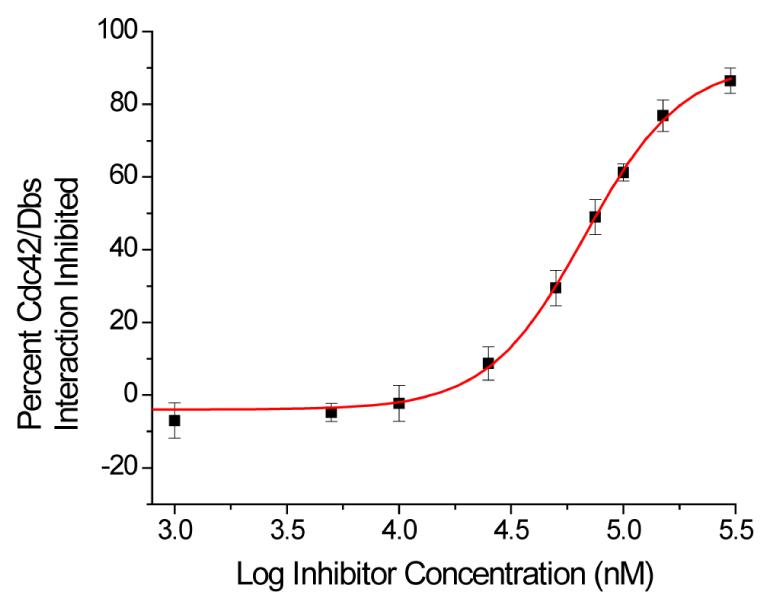
IC50 of 19 against the Cdc42/Dbs interaction
In summary, we have generated a novel class of α-helix mimetics based on a 5-6-5 imidazole-phenyl-thiazole scaffold. X-ray crystallographic studies confirm the core structure, which has an α-carbon RMSD of 1.014 Å between 4 and the side chain positions on a poly-alanine α-helix. In addition, to demonstrate the potential of our 5-6-5 imidazole-phenyl-thiazole scaffold to disrupt a biologically relevant protein-protein interaction, we have generated a mimic with rigidified side chains that is a 67 μM inhibitor of the Cdc42/Dbs interaction. We are currently preparing a diverse set of rigidified mimetics and testing their affinity and specificity for Cdc42/Dbs as well as other members of the Rho family of GTPases.
Supplementary Material
Scheme 2.
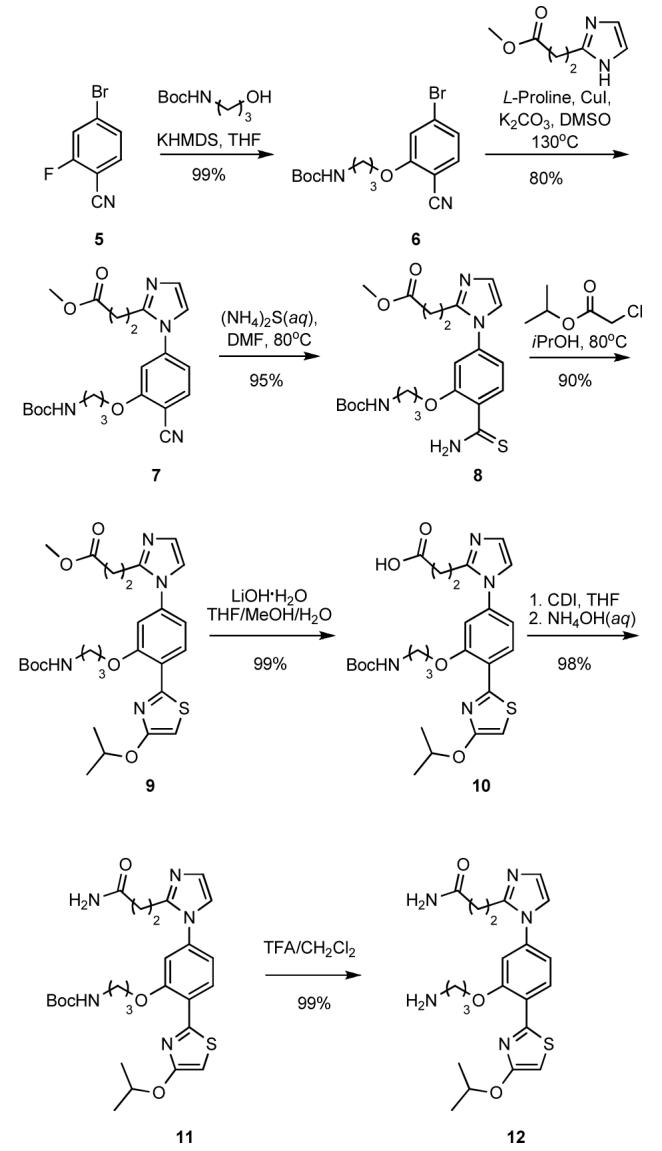
Synthesis of 12
Acknowledgement
The authors would like to thank the National Institutes of Health for financial support of this work (CA67771) and our collaborators Channing Der, Adrienne Cox, Jon Sondek (UNC) and Said Sebti (H. Lee Moffitt Cancer Center and Research Institute) for invaluable discussions. We would also like to thank Dr. Steven Fletcher and Dr. Patrick Gunning for initial discussions and Dr. Christopher Incarvito for X-ray crystallographic analysis.
Footnotes
Supporting Information Available: X-ray crystallographic data, experimental procedures, characterizations of all compounds and assay protocols are available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For a review, see: Davis JM, Tsou LK, Hamilton AD. Chem. Soc. Rev. 2007;36:326–334. doi: 10.1039/b608043j.
- (2) (a).Davis JM, Truong A, Hamilton AD. Org. Lett. 2005;7:5405–5408. doi: 10.1021/ol0521228. [DOI] [PubMed] [Google Scholar]; (b) Yin H, Lee GI, Sedey KA, Kutzki O, Park HS, Omer BP, Ernst JY, Wang HG, Sebti SM, Hamilton AD. J. Am. Chem. Soc. 2005;127:10191–10196. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]; (c) Yin H, Lee GI, Sedey KA, Rodriguez JM, Wang HG, Sebti SM, Hamilton AD. J. Am. Chem. Soc. 2005;127:5463–5468. doi: 10.1021/ja0446404. [DOI] [PubMed] [Google Scholar]; (d) Ernst JT, Becerril J, Park HS, Yin H, Hamilton AD. Angew. Chem. Int. Ed. 2003;42:535–539. doi: 10.1002/anie.200390154. [DOI] [PubMed] [Google Scholar]
- (3).Fletcher S, Hamilton AD. J.R. Soc. Interface. 2006;3:215–233. doi: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4) (a).Volonterio A, Moisan L, Rebek J., Jr. Org. Lett. 2007;9:3733–3019. doi: 10.1021/ol701487g. [DOI] [PubMed] [Google Scholar]; (b) Biros SM, Moisan L, Mann E, Carella A, Zhai D, Reed JC, Rebek J., Jr. Bioorg. Med. Chem. Let. 2007;17:4641–4645. doi: 10.1016/j.bmcl.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maity P, König B. Org. Lett. 2008;10:1473–1476. doi: 10.1021/ol8002749. [DOI] [PubMed] [Google Scholar]
- (5).Rossman KL, Worthylake DK, Snyder JT, Siderovski DP, Campbell SL, Sondek J. EMBO J. 2002;21:1315–1326. doi: 10.1093/emboj/21.6.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Marques CA, Hahnel PS, Wolfel C, Thaler S, Huber C, Theobald M, Schuler M. Blood. 2008;111:1413–1419. doi: 10.1182/blood-2007-05-089458. [DOI] [PubMed] [Google Scholar]
- (7).Sinha S, Yang W. Cell. Sig. 2008 doi: 10.1016/j.cellsig.2008.05.002. doi:10.1016. [DOI] [PubMed] [Google Scholar]
- (8).Kouwer PHJ, Swager TM. J. Am. Chem. Soc. 2007;129:14042–14052. doi: 10.1021/ja075651a. [DOI] [PubMed] [Google Scholar]
- (9).Rudolph J, Chen L, Majumdar D, Bullock WH, Burns M, Claus T, Dela Cruz FE, Daly M, Ehrgott FJ, Johnson JS, Livingston JN, Schoenleber RW, Shapiro J, Yang L, Tsutsumi M, Ma X. J. Med. Chem. 2007;50:984–1000. doi: 10.1021/jm061299k. [DOI] [PubMed] [Google Scholar]
- (10).Orner BP, Ernst JT, Hamilton AD. J. Am. Chem. Soc. 2001;123:5382–5383. doi: 10.1021/ja0025548. [DOI] [PubMed] [Google Scholar]
- (11).Chemical Computing Group MOE: Molecular Operating Environment. 2007.
- (12).Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Proc. Nat. Acad. Sci. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457–2483. [Google Scholar]
- (14).Hemsath L, Ahmadian MR. Methods. 2005;37:173–182. doi: 10.1016/j.ymeth.2005.05.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.