

Synopsis
Since the histological description of the hamartomatous polyp in 1957 by Horrilleno et al., several different syndromes have been described with the propensity to develop these polyps in the upper and lower GI tracts. These include Juvenile Polyposis, Peutz-Jeghers syndrome, hereditary mixed polyposis syndrome, and the PTEN hamartoma tumor syndromes (Cowden and Bannayan-Riley-Ruvalcaba syndromes), which are autosomal-dominantly inherited, and Cronkhite-Canada syndrome, which is acquired. The clinical aspects, the molecular pathogenesis, the organ systems affected, the risks of cancer, and the management of these hamartomatous polyposis syndromes will be reviewed in this paper. Although the incidence of these syndromes is low, it is important for clinicians to recognize these disorders in order to prevent morbidity and mortality in these patients, and to perform presymptomatic testing in patients at risk.
Keywords: Hamartoma, Peutz-Jeghers syndrome, Cowden and Bannaya-Riley-Ruvalcaba syndrome, Hereditary mixed polyposis syndrome, Juvenile-Polyposis syndrome, and Cronkite-Canada syndrome
INTRODUCTION
The hamartomatous polyposis syndromes are a fascinating group of disorders which have in common relatively benign appearing polyps of the GI tract, but also an increased risk of cancer. These conditions include Juvenile Polyposis Syndrome (JPS), Peutz-Jeghers Syndrome (PJS), Bannayan-Riley-Ruvalcaba Syndrome (BRRS), Cowden Syndrome (CS), Cronkhite-Canada Syndrome (CCS), and Hereditary Mixed Polyposis Syndrome (HMPS). The progression of these polyps to cancer is not well understood, and represents a different mechanism than that seen in adenomatous polyposis. This has been dubbed the landscaper effect, where changes predominantly affecting the lamina propria may lead to epithelial cancers80. In this review, we will discuss the clinical features of these hamartomatous polyposis syndromes, their histopathologic characteristics, risk of cancer, genetics, and screening recommendations for each.
JUVENILE POLYPOSIS SYNDROME (JPS)
HISTORY
In 1939, Diamond described a 30 month-old child with a prolapsed polyp that he felt was of congenital origin. The child primarily had constipation, bright red blood per rectum, with a pedunculated, and sessile polyp on proctoscopy28. Ravitch described a 10-month-old child that on autopsy was found to have multiple GI polyps, from the stomach to the anus. The child’s symptoms were bloody diarrhea, failure to gain weight, cachexia, recurrent rectal prolapse, intussuception, and severe anemia117. In 1957, Horrilleno et al. performed a review of the literature on children with rectal and colonic polyps, and concluded that most polyps contained mucus-filled glands, with retention cysts, abundant connective tissue, and a chronic cellular infiltration of eosinophils. They introduced the term “hamartomatous polyp” based upon these observations56. In 1964, McColl et al. coined the term Juvenile Polyposis (JP) after careful analysis of the syndrome, and concluded that JP was a different entity than adenomatous polyposis coli98. In 1970, Sachatello et al. described a JP family with affected members in three generations with disease involving the stomach, small bowel, large bowel, and rectum125. In 1975, Stemper et al. reported a family with 10 members affected with JP. Individuals had juvenile polyps of the stomach, small bowel, colon, and rectum, and several also developed colon cancer, gastric cancer, and one had pancreatic cancer139.
CLINICAL FEATURES
In 1974, Sachatello et al. defined the criteria for the diagnosis of JP, which was fulfilled by the findings of any one of the following: (1) ten juvenile polyps in the colorectum; (2) juvenile polyps throughout the GI tract; (3) any number of juvenile polyps with a family history of juvenile polyposis124. The criteria were later revised by Jass et al., who decreased the requisite number of polyps from 10 to 5 to make the diagnosis of JP71. Sachatello et al. also proposed a classification of JP into: (1) juvenile polyposis coli, where the only site of disease is the colon (Figure 1); (2) JP of infancy, a subtype that carries a poor prognosis due to a very early age of onset, severe hypoalbuminemia, and failure to thrive; (3) generalized JP, where individuals have juvenile polyps of both the upper and lower GI tract123. Veale et al. reviewed 145 cases of JP and found an average age onset of 6 years, with familial cases presenting at a later age (9.5 years), and sporadic cases younger (4.5 years). The most common symptom was rectal bleeding, prolapse, mucus per rectum, diarrhea, and abdominal pain. They found an equal incidence between males and females145.
Figure 1.
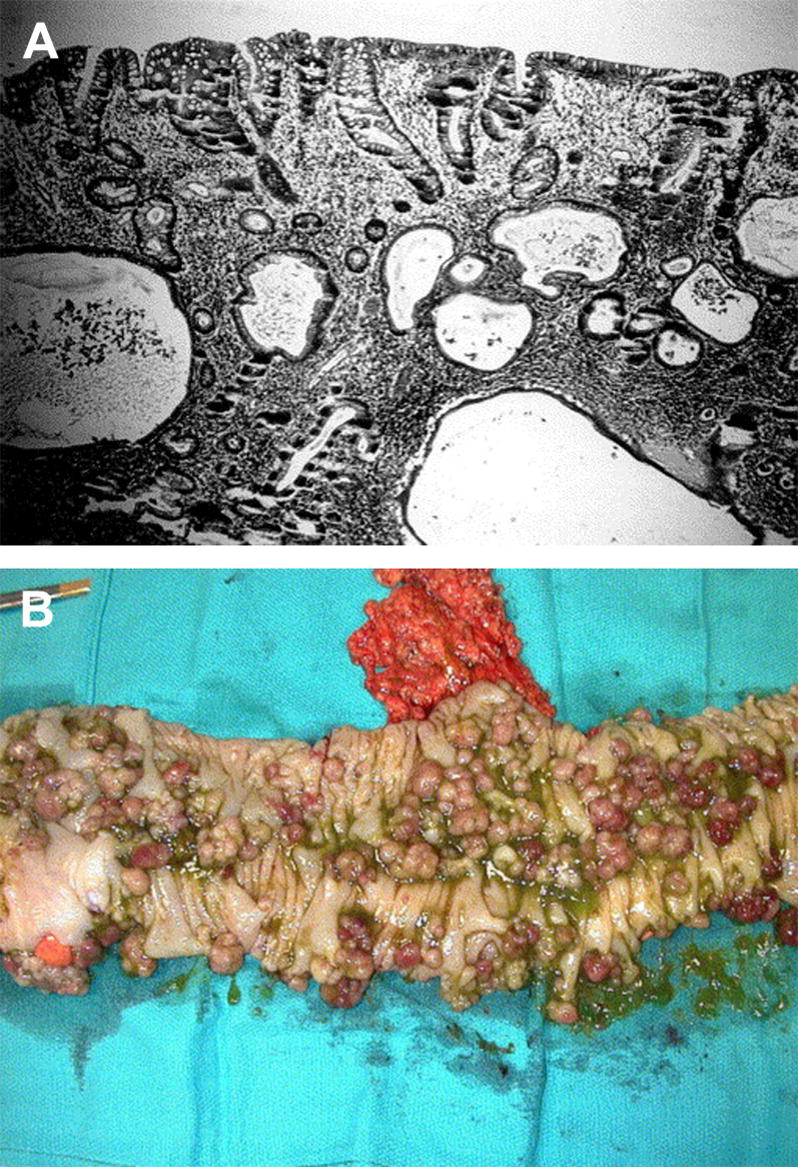
(A) A typical juvenile polyp with the three classic histological features that define a hamartomatous polyp, which are dilated cystic glands with retention of mucus and lined by tall columnar epithelium, a markedly expanded lamina propria, and diffuse chronic infiltration of inflammatory cells. (B) Gross picture of the colon in a JP patient that is carpeted with juvenile polyps (reprinted with permission from101).
In 1986, Grosfeld et al. described the differences between multiple versus solitary juvenile polyps in children. Patients with multiple polyps have considerable mucus secretion presenting with more severe hypokalemia, and protein losing enteropathy, leading to hypoalbuminemia and hypoprotenimia. In addition, they have more severe anemia from blood loss compared with those having solitary juvenile polyps47. Jass stated that solitary juvenile polyps (as opposed to JPS) usually present in childhood, with a peak incidence at age 4–5 years, and were mainly located in the colon or rectum, with the most common symptom being rectal bleeding. The polyps have a tendency to auto-amputate or prolapse. In JPS, he found that most cases were sporadic (66%), while there was family history of the disease less frequently (33%)70. In 1995, Desai reported that 85% of JP patients presented in the first or second decade of life, and 98% of patients had colorectal polyps that are relatively evenly distributed throughout the colon, and 14% had involvement of the stomach, 2% of the duodenum, and 7% of the jejunum or ileum27. Coburn et al. found that the mean age at diagnosis was 18.5 years, that 50% of patients had a family history of JP, and 15% had extracolonic anomalies. Anemia, rectal bleeding, prolapse, enteropathy, and intussusception were the most common symptoms23.
Associated Anomalies
The extracolonic anomalies described in JP patients include macrocephaly, hypertelorism, amyotonia congenita, extra toes on the foot, Meckel’s diverticulum with umbilical fistula, mild communicating hydrocephalus, malrotation of the small bowel, undescended testes, mesenteric lymphangioma, malrotation of the cecum, and acute porphyria27,98,145. Bussey et al. estimated that 20% of JP patients suffered from congenital anomalies16. Coburn et al. performed a large review of JP patients, and found multiple thoracic anomalies, which included atrial septal defects, arteriovenous malformations (AVMs) of the lung, pulmonary stenosis, tetralogy of Fallot, coarctation of the aorta, patent ductus arteriosus, and subvalvular aortic stenosis. CNS defects included macrocephaly, hydrocephalus, and spina bifida. In the GI system, Meckel’s diverticulum, gastric and duodenal diverticuli, and malrotation were observed. Urogenital anomalies included undescended testes, unilateral renal agenesis, bifid uterus and vagina, and abnormal ureteropelvic insertion. Other abnormalities included osteoma, lymphangioma, pectus excavatum, hereditary telangiectasia, familial congenital lymphedema, hypertelorism, thyroglossal duct cyst, and amyotonia congenita23. Jass stated that congenial defects were more common in sporadic cases, compared to familial JP70.
HISTOPATHOLOGY
Horrilleno et al. described juvenile polyps as having proliferation of mucus glands with formation of cystic structures in abundant connective tissue, with a background of chronic cellular infiltration of eosinophils56. Morson described the juvenile polyp as a hamartoma, with alteration of the layers above the muscularis mucosa. The stroma of the polyp has tubules lined by columnar epithelium and many goblet cells, with atrophy of the lining epithelium in the tubules, showing cystic dilation and retention of mucus. The epithelium, which covers the entire surface of the polyp, has no signs of hyperplasia, hyperchromatism, or increased mitotic activity. He also noted that ulceration, infection, and auto-infarction was evident is some of the polyps, which could be the reason for infiltration of inflammatory cells (Figure 1)102.
Jass reported that 20% of polyps are multilobated or papillary, however 80% had typical features, with the lamina propria thinned out, and the epithelial cells possibly showing some evidence of dysplasia. Solitary polyps generally have a smooth, spherical, red head, with a narrow stalk. The surface of solitary polyps has cysts filled with mucin, with no muscularis mucosa in the expanded layer, and the epithelium is normal and has no evidence of excess mitotic activity. There is also a strong component of infiltration of inflammatory cells in the lamina propria70. Subramony et al. described juvenile polyps greater than 3 cm as being mostly pedunculated, with epithelium showing mild to moderate dysplasia resembling adenomas. The largest polyp they described had adenocarcinoma in the stalk of the polyp. In addition, they found that gastric polyps in JP patients had histological features identical to hyperplastic polyps, however, the surrounding gastric mucosa showed a diffuse process consistent with chronic gastritis140.
CANCER PREDISPOSITION
Colorectal Cancer
In 1975, Stemper et al. described the Iowa kindred, which consisted of 56 members at the time. There were 15 affected members, and 11 had GI malignancies (5 colon, 2 stomach, 2 duodenum, 1 pancreatic, and 1 unknown GI cancer). At that time, the authors did not infer the malignant potential of juvenile polyps, although it was evident that cancer was common within the three successive generations of this family139. Goodman et al. described the spectrum of changes in JP polyps in a 23 year-old male with sporadic JP who had rectal cancer. They found typical juvenile polyps, juvenile polyps with focal adenomatous changes, adenomas, and adenocarcinoma. They conclude that there must be a sequence of events that leads to adenocarcinoma formation within the juvenile polyp43. Other authors supported the notion of a pathological sequence of events and that the polyps in JP patients had malignant potential46,69,116,120.
Jones et al. found an intramucosal carcinoma arising from a typical juvenile polyp in a 24-year-old JP patient that only had 4 juvenile polyps in the rectum. They concluded that individuals with solitary polyps are also at risk for malignancy76. However, after an extensive review of the clinical and pathological characteristics of juvenile polyps, Jass et al. concluded that patients with JP are at risk for developing malignancies but not patients with solitary juvenile polyps71. Bentley et al. found that the subgroup of patients with JP that had the greatest risk of malignancy were those with typical juvenile polyps and adenomatous features6. In 1990, Jass quantified the risk of developing cancer, and concluded that in patients with solitary polyps, the risks are minimal, but for patients with JP, the cumulative risks is greater than 50%70. Giardiello et al. reported that the mean age of diagnosis of colorectal neoplasia was 37 years for both sporadic and familial JP, and found that the risk for neoplasia was approximately the same for both familial and sporadic JP41. Howe et al. updated the records of the Iowa JP kindred in 1998, which now consisted of 117total individuals, with 29 affected members. Eleven had been diagnosed with colon cancer, 4 with gastric cancer, 1 with cancer of the duodenum/ampulla, and 1 with pancreatic cancer. They concluded that the overall risk for colorectal cancer in this family was 38%, and of upper GI cancer was 21%, with the overall risk for GI malignancies was 55%59.
Gastric Cancer
Watanabe et al. described a family that consisted of 2 siblings with JP with disease localized mainly to the upper GI tract. The mother had passed away from gastric cancer at 37 years of age. Both siblings underwent a total gastrectomy, and although their stomachs were found to be diffusely involved with typical juvenile polyps, there was no evidence of dysplasia or cancer150. Yoshida et al. reported a 31 year old JP patient that was diagnosed with a well-differentiated gastric adenocarcinoma, and concluded that JP patients are also at risk for gastric cancer156. Sassatelli et al. reported a 16 year old JP patient with diffuse polyposis of the stomach, who later developed an infiltrating adenocarcinoma from one of the JP polyps in the stomach at the age of 21129. Howe et al. reported that 6 of 29 affected patients in the Iowa kindred had upper GI malignancies (4 in the stomach), and the overall risk of developing upper GI cancer in affected members of the family was 21%59.
Pancreatic Cancer
Stemper et al. reported the first JP patient with pancreatic adenocarcinoma in 1975139, and in 1989, Walpole and Cullity described another JP patient presenting with epigastric mass at age 19. Exploration revealed a poorly differentiated adenocarcinoma that had replaced the majority of the pancreas, with and metastasis to the left lung148.
MANAGEMENT
The standard methods used to screen and survey JP patients are colonoscopy and EGD. Although capsule endoscopy has the potential for evaluation of the entire GI tract, its usefulness is primarily for evaluation of the small bowel. Howe et al. published general guidelines (Figure 2) which stated that in patients at risk for JP, screening should begin with a thorough history and physical to evaluate for JP (rectal bleeding, prolapse, anemia, constipation, obstruction, diarrhea, abdominal discomfort), and this should be done soon after birth. EGD and colonoscopy, however, should be done at the age of onset of symptoms, or in asymptomatic patients at risk, at 15 years of age. If screening reveals no polyps, it should be repeated every three years. If polyps are found, they should be removed endoscopically, if possible. Then, the procedure should be repeated yearly until no polyps are seen, at which point screening should be repeated every 3 years. Individuals that do not have the mutation found in the family, should undergo baseline screening at age 15, and if screening is negative, then this should be repeated every 10 years (instead of every 3 years) until the age of 45. If no polyps are ever found, screening should then be done as one would do for the normal population. However, if a genetic mutation is found in an individual at risk, they should continue to be screened every 3 years, or yearly if polyps are found. If the individual or the family does not have a genetic mutation found in one of the 2 known JP genes, then screening should follow the same guidelines as those for patients at risk for JP60.
Figure 2.

Algorithm for the surveillance and management of JP patients, incorporating genetic testing (reprinted with permission from60).
Grosfeld et al. felt that subtotal colectomy with ileorectal anastomosis was the procedure of choice in selected JP patients, with indications for the procedure being children with anemia from chronic bleeding, hypoproteinemia, failure to thrive, and non-reducible intussusception47. Jarvinen et al. recommended prophylactic colectomy with ileorectal anastomosis for (1) children with JP that have severe or repeated bleeding (leading to failure to thrive or death), and (2) adults with JP because of the greater than 50% life time risk for cancer. They felt that the optimal age for surgery was 20–25 years of age, since the risk for cancer is greater than the risk of surgery68. Subtotal colectomy was also favored by Howe et al., who stressed that the rectum needs to be screened with flexible sigmoidoscopy every 1–3 years due to the risk of recurrence in the rectal remnant. However, they felt that surgery should be reserved for those patients with large numbers of polyps, significant anemia, or other complications of JP. Most patients can have repeated colonoscopic removal of polyps rather than colectomy, especially if the polyp burden is low60. Many authors have published their experience of a high rate of recurrence of polyps after various surgical procedures48,114,120,124,131, and therefore Scott-Conner et al. recommended total colectomy with J pouch ileoanal anastomosis as their procedure of choice131. Some believe that the frequency and urgency of bowel movements, and reduced continence associated with this procedure may be less desirable than the required surveillance for recurrent polyps in the rectosigmoid60.
Patients with generalized JP should undergo close surveillance of the upper GI tract because of the high risk of malignancy43,55,60,68, Jarvinen, 1984 #570,131,156. Howe et al. recommended screening the upper GI tract by EGD starting at the age 15 for asymptomatic individuals at risk, or as soon as signs or symptoms develop. Screening should be repeated every 3 years, and if polyps are present, every year to make sure there are no dysplastic polyps present. Polyps of the stomach tend to be diffuse, and difficult to remove endoscopically. Therefore, total or subtotal gastrectomy should be performed for bleeding, gastric outlet obstruction, dysplasia, adenomatous changes, or adenocarcinoma60. Particularly close surveillance should be directed towards patients with SMAD4 mutations, since they are at greater risk for developing gastric cancer38,130.
GENETICS
Stemper et al. suggested that JP had an autosomal dominant mode of inheritance based upon observations in the Iowa kindred, with a high degree of penetrance139. In 1978, Bussey, Veale, and Morson speculated that in 25% of JP cases there must be an inherited defect in a gene, while in the other 75% of JP patients a de novo mutation or environmental factor promoted disease16.
In 1998, Howe et al. performed genetic linkage analysis on affected members of the Iowa kindred, and found linkage to chromosome 18q21. This region contained the tumor suppressor genes DCC and SMAD4/DPC461. They then searched for germline mutations in both genes by direct sequencing, and identified a 4 base pair (bp) deletion in exon 9 of SMAD4 in all affected members of the kindred, and mutations in 4 other JP kindreds62. Soon thereafter, these findings were confirmed in other JP families37,57,119. In 2001, Howe et al. performed genetic linkage analysis in 4 unrelated JP families without SMAD4 or PTEN mutations, and found linkage to markers on chromosome 10q22-23. Mutations were found in all affected members of each family in the bone morphogenetic protein receptor type IA gene (BMPR1A)58. These findings were later confirmed by other authors38,79,160. The overall prevalence of germline mutations is 20% for SMAD4 and 20% for BMPR1A in JP probands63, which has been validated in other large studies of JP patients3,115.
SMAD4 is a protein that functions as the common intracellular mediator of the TGF-β, bone morphogenetic protein (BMP), and activin signaling pathways. The BMP signaling pathway has a wide range of actions, from regulation of cell proliferation, differentiation, survival, and apoptosis. BMPR1A is a transmembrane protein, which is a type I receptor. TGF-β superfamily ligands (TGF-β, BMPs, activins, and inhibins) bind to serine/threonine kinsase type 2 receptors that then bind to and phosphorylate the serine and threonine domains of the type I receptors. These then phosphorylate intracellular SMAD proteins (SMAD2 and 3 in the TGF-β pathway, and SMAD1, 5 and 8 in the BMP pathway), which then form heteromers with SMAD4 and the complexes recruit DNA binding proteins as they migrate into the nucleus. Here, they bind directly to DNA sequences and regulate the transcription of various genes51,95.
Counseling
Since 2 genes have been identified for JP, individuals at risk can be tested for mutations prior to the onset of symptoms. In 1999, Howe et al. suggested that screening individuals at risk within a JP family for a known genetic mutation would allow clinicians to make the diagnosis at an earlier age, and would lead to closer endoscopic screening and follow up. Individuals without the mutation would need less frequent endoscopic screening and perhaps only endoscopy after age 50 as in the normal population. Therefore, genetic testing can help to define the recommended interval for screening and surveillance, and hopefully to prevent malignancy by early polypectomy and heightened surveillance60.
PEUTZ-JEGHERS SYNDROME (PJS)
HISTORY
In 1896, Hutchinson described twin sisters that were healthy, and at the age of 9 developed pigmentation around the mouth, lips, and oral mucous membranes. One of the sisters died at age 20 from intessuseption65. In 1921, Peutz described a family with 10 affected members within 3 generations. Seven of the affected members had pigmentation of the lips, mouth and oral mucosa, and also had polyps confined to the small intestine. Two of these family members also had nasal polyps, and 1 had bladder polyps. One of the patients underwent resection of a portion of the jejunum secondary to intussusception, and adenomatous polyps were described in the specimen112. In 1949, Jeghers et al. reviewed the literature on patients with pigmentation of the mouth, lips and oral cavity that were associated with intestinal polyposis, and found 10 cases. They came to the conclusion that the syndrome consists of two distinctive clinical features, the melanin deposition around the mouth, oral mucosa, and lips (Figure 3), and polyposis of the small intestine. They concluded that the syndrome had an autosomal dominant pattern of inheritance73. In 1954, the term “Peutz-Jeghers syndrome” was coined by Bruwer et al.12.
Figure 3.
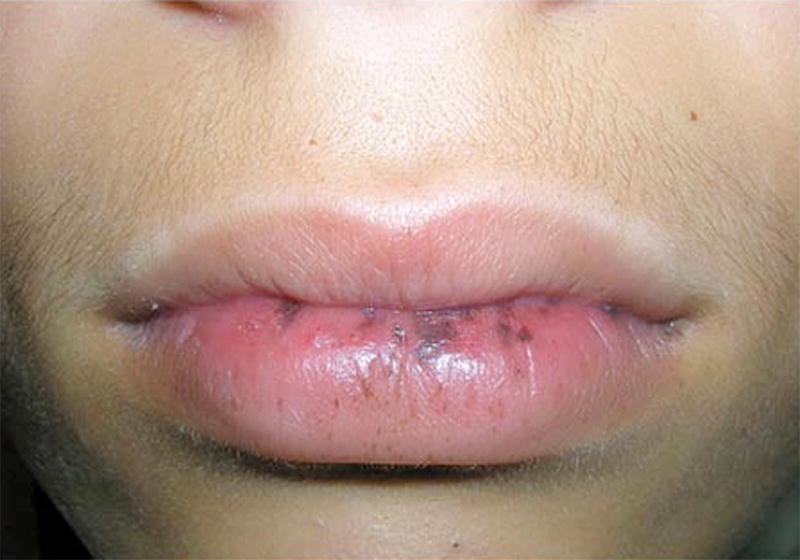
Oral melanosis in a patient with Peutz-Jeghers syndrome (reprinted with permission from25).
CLINICAL FEATURES
In 1962, Bartholomew et al. defined PJS as mucocutaneous melanosis and small bowel polyposis. The pigmentation may develop at any age, however, it most commonly manifests in infancy and early childhood. The melanosis occurs almost universally on the lips (>95%), with the buccal mucosa being the second most common site (83%) (Figure 3). Less frequent locations are the hands and feet, and areas around the mouth and nose. This pigmentation tends to fade with age. In terms of the polyposis, lesions vary in size from microadenomas within the intestinal wall to polyps several centimeters in size. Most of the polyps are found in the jejunum and ileum, and to a lesser extent the rectum, colon, stomach, and duodenum. The number of polyps varies from solitary polyps to hundreds of polyps where the intestine is virtually carpeted. There is no difference in prevalence between males and females, or in specific races or ethnic groups, and the average age of diagnosis is 24.3 years. The most common presenting symptoms are recurrent episodes of abdominal pain as a results of intussusception, anemia secondary to occult GI bleeding, melena, and hematochezia. Hematemesis was also reported in patients with gastric or duodenal polyposis, and prolapse of rectal polyps may also occur5.
In 1999, Westerman and Wilson performed a review of the literature on PJS, and stated that the polyps tend to be either several millimeters and are sessile, or several centimeters, in which case they tend to be pedunculated. They can be solitary, or can occur in clusters, at times carpeting the entire GI surface. These polyps can lead to intussusception most commonly in the small bowel, but 2 cases have been described of the colon. Another common presentation is bleeding from the polyps, which occurs in 81% of patients, and tends to present as hematochezia, or hematemesis in 10% of patients with polyps in the duodenum or stomach. It can also present as occult bleeding with anemia. Sixty per cent of patients present by their early twenties, and 33% present in the first decade of life151.
Associated Anomalies
In 1957, Dormandy et al. reviewed the PJS cases described up to that time, and concluded that bladder, renal pelvis, bronchial, and nasal polyps are associated with PJS. Skeletal anomalies include clubbed foot, scoliosis, and bony tumors, as well as ovarian lesions, such as cysts, cystadenomas, and malignant tumors of the ovaries may also be seen29. Polyps in the nasal cavity may present with as epixtasis, or symptoms of sinus obstruction. Ureteral polyps were described in 1 patient with hematuria and frequency137. Polyps in the gallbladder have also been reported, although histologically these were considered adenomatous polyps, while polyps removed from the ileum were hamartomatous36. Polyps of the biliary tract were described in two patients, both with signs and symptoms of obstructive jaundice39,109.
HISTOPATHOLOGY
Areas of pigmentation consist of melanin in the subepithelial regions both within the cells and around them, without any abnormal pathology at the microscopic level. Acanthosis with prominent melanin in the basal layers may also be seen. Polyps in the small intestine are made of columnar cells, goblet, and Paneth cells. Within the stroma there are arborizing strands of smooth muscle, which represents the muscularis mucosa branching in various directions. This is the major difference from juvenile polyps, where there is no muscularis mucosa in the lamina propria (Figure 4). There is no nuclear atypia or proliferation. Gastric polyps are also hamartomas, and contain all the cell types found in the gastric mucosa, and the duodenal polyps have Brunner’s glands. Colonic polyps, however, have a more adenomatous appearance, and therefore, they should be regarded as potentially malignant5. Farmer et al. described polyps as having minor variations depending upon the location in which they are found. Polyps in the small intestine have goblet cells that form the typical tubular glands within the stroma, and there are crypt-like formations resembling the crypts of Lieberkuhn. The epithelial component of gastric polyps resembles the pyloric or gastric mucosa with less prominent smooth muscle34.
Figure 4.
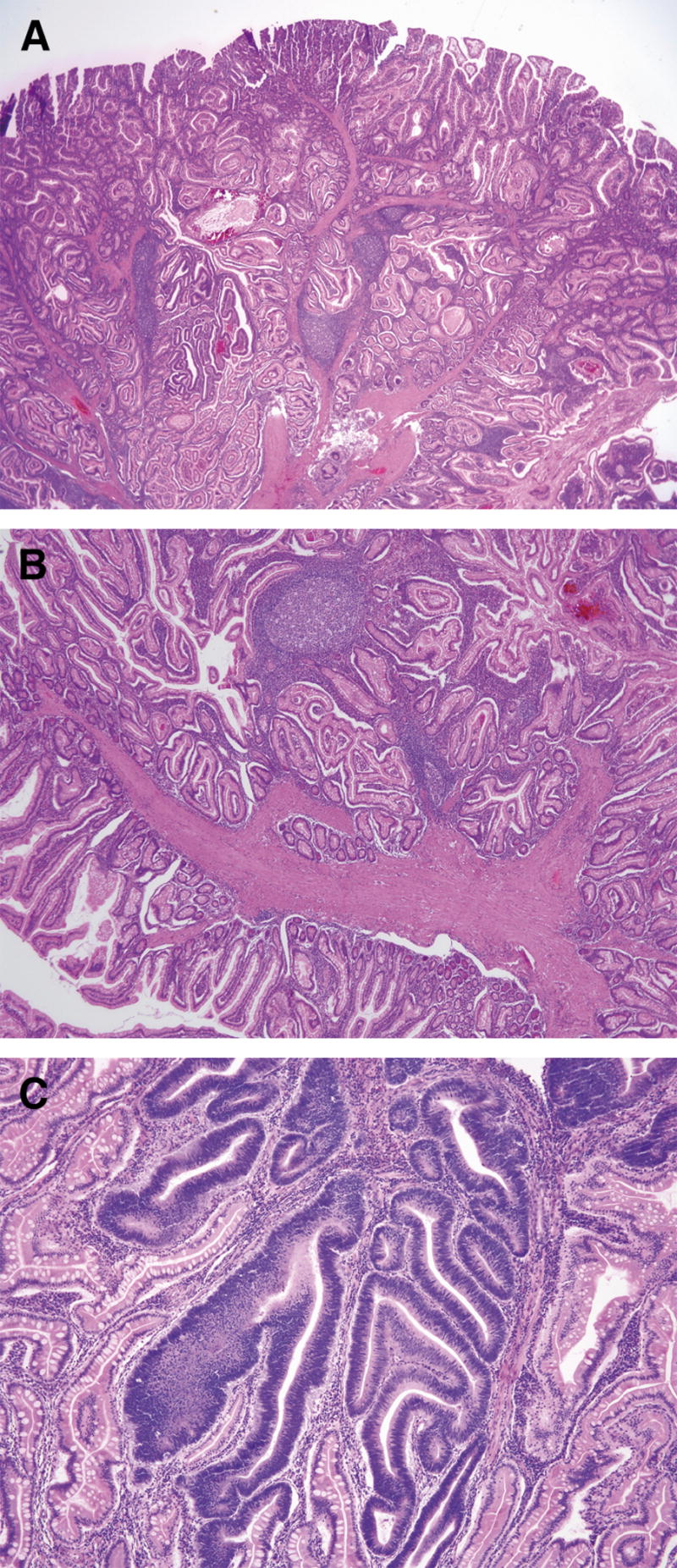
Polyp from a Peutz-Jegher syndrome patient. In (A), note the arborizing smooth muscle that branches into the stroma to where the absorptive epithelium, goblet, and Paneth cells are located. In (B), one can see the smooth muscle fibers that make up a large part of the stroma, originating from the muscularis mucosae. In (C), an area with low-grade dysplasia can be seen (reprinted with permission14).
CANCER PREDISPOSITION
Colorectal
Although many authors have described the incidence of colorectal cancer in patients with the PJS, most have been case reports and descriptions of kindreds that do not provide a full risk analysis64,107,144. Konishi et al. reviewed all cancer cases reported in PJS patients, and found 20 colorectal cancers in 103 patients (12 in Japan, and 8 in the western literature). The mean age of diagnosis was 48 years of age81. Giardiello et al. found similar results when they reviewed all PJS papers from 1966 to 1998 for reports of cancer. They found an 84 fold relative risk of developing colon cancer (Figure 5), and cumulative risk of 39% of developing colon cancer between ages of 15 to 64, with a mean age of diagnosis of 46 years. There was no statistically significant difference in the incidence of colon cancer between males and females40.
Figure 5.

Cumulative risk (percent) and relative risk (RR) of the various organs predisposed to the development of cancer in JPS, PJS, and PTEN hamartoma tumor syndromes (data derived from9,40,49,59,138).
Gastric and Small Bowel
The malignant potential of gastric and duodenal polyps was originally described in 1967 by Payson and Moumgis when they reported a 21 year old with a 2 year history of hematochezia, with weight loss, poor appetite, and fatigue. He developed hematemesis and on work up was found to have jejunal intussusception, multiple polyps throughout the colon, stomach, duodenum and jejunum. A large hard mass was found in the antrum and a subtotal gastrectomy with omentectomy was performed, in addition to small bowel resection for the intussusception. On pathology, the patient had gastric adenocarcinoma with multiple positive nodes and multiple polyps in the stomach. The rest of the small and large bowel polyps did not show any evidence of malignancy. A review of the literature at that time showed 6 other cases of upper GI cancers associated with PJS, and they concluded that polyps in the stomach and duodenum must be considered as having malignant potential, and should not be dismissed as being benign110. The malignant potential of PJS polyps in the small bowel and stomach has been well established and described by many authors13,82,86,96. Foley, McGarrity, and Abt did a clinicopathologic survey of the largest PJS kindred, the Harrisburg family, which was originally described by Jeghers in 1949. In their survey, they reported one member who died at the age of 40 from metastatic gastric carcinoma. Another member, at the age of 27 underwent an antrectomy, partial duodenectomy, and gastrojejunostomy for duodenal polyps, and was found to have a focus of adenomatous elements with dysplasia in the duodenum. He ended up having a pancreaticoduodenectomy, and 14 months later died from a malignant pleural effusion35. Giardiello et al. found a 93% overall cumulative risk for the development of cancer in PJS patients (15.2 fold relative risk), with the cumulative risk for gastric cancer being 29% (relative risk of 213), and of the small intestine 13% (relative risk of 520). It should be noted that this study only looked at familial cases of PJS, and therefore the risk in patients with sporadic PJS has not been as well studied40.
Reproductive Organs
The association of PJS and a functional ovarian tumor was made by Christian et al. in 1964, when they described a 4 year old girl with precocious puberty. At the age of 3, she developed vaginal spotting, and then heavier bleeding similar to menses. Her mother had pigmentation of her lips, but no evidence or symptoms of GI polyposis, and the rest of the family history was unremarkable. She underwent an exploratory laparotomy, and was found to have multiple polyps of the small intestine, stomach, duodenum, and a mass on the left ovary that turned out to be a granulosa-theca cell tumor22. Other authors have described ovarian tumors in patients with PJS64,136,157, and Scully et al. concluded that up to 5% of females with PJS develop an unusual tumor of the ovary, called ovarian sex cord tumor with annular tubules, which are microscopic and tend to be bilateral and multifocal132. Functional tumors of the testes (lead to feminizing charateristics) have also been described in boys with PJS, especially Sertoli cell tumors, which arise from the same embryonic cells17,154,158. Cases of well-differentiated adenocarcinoma, infiltrating ductal carcinoma, and papillary carcinoma from the breast have also been described in woman with PJS13,82. Giardiello et al. they found that PJS patients have an absolute risk for cancer of the breast of 54% (15.2 relative risk), the ovaries 21%(27 relative risk), the cervix 10% (1.5 relative risk), and the uterus and testes of 9% (16 fold and 4.5 fold relative risk, respectively)40.
Pancreatic and Gallbladder Cancer
In 1986, Bowlby et al. described a 14 year old boy with sporadic PJS found to have a retroperitoneal mass with a non-functioning left kidney. On autopsy, the mass replaced the body and tail of the pancreasand the mass was found to be a pancreatic adenocarcinoma7. Giardiello et al. found 6 cases of pancreatic cancer in the literature, and concluded that PJS patients have a 132 fold increased risk of developing pancreatic cancer (36% cumulative risk)40. Wada et al. described a 39-year-old woman with PJS that was found to have two polyps in the gallbladder, with cholelithiasis and choledocholithiasis on ERCP. Cholecystectomy and common bile duct exploration revealed a well-differentiated adenocarcionoma of the gallbadder arising from the mucosa close to the largest polyp147.
MANAGEMENT
In 2006, Giardiello and Trimbath published a review of PJS, in which they recommended screening from birth of all first degree relatives of PJS patients. Screening should be directed to identifying melanosis, and ruling out testicular or ovarian tumors that might cause precocious puberty. Asymptomatic first-degree relatives who do not show any clinical signs of the disease should be offered genetic testing for mutations in the STK11/LKB1 gene starting at the age of 8. Asymptomatic individuals at risk, without knowledge of the mutation status, should undergo careful screening with upper endoscopy, colonoscopy, and small bowel follow through at the ages of 12, 18, and 24. Another strategy is to have just the small bowel follow through every two years until the age of 25. In addition, the patient at risk should be advised to follow the same screening guidelines as affected individuals to make sure no other organ systems are involved. If a mutation is found in STK11/LKB1, then colonoscopies every 2–3 years should be done starting at the age of 18 or with symptoms. Upper endoscopy should start at the age of 8, and repeated every 2 years if any polyps are found, which should be removed endoscopically. Endoscopic ultrasound to look at the pancreas should be done every 1–2 years starting at the age of 25–30. Males should have careful testicular examination starting at birth, with ultrasounds every 2 years until the age of 12. Females should have regular self-breast exams starting at the age of 18, and annual mammograms or MRI as an alternative, with physician directed breast exams starting at the age of 25, or at the age when the first relative was diagnosed with the PJS if earlier than age 25. Pelvic exam with annual Pap smears should start at the age of 21 to rule out cervical cancer, with transvaginal ultrasound and serum CA-125 done annually at the age of 25 to rule out cancer of the uterus and ovaries42.
Soares et al. performed capsule endoscopy in two groups of PJS patients. Group A consisted of 14 patients known to have polyps in the small bowel, and had undergone small bowel surgical procedures. Group B was 6 first-degree relatives that had GI symptoms but no polyps found by prior endoscopic evaluations, nor had they undergone any surgical procedures. Capsule endoscopy identified multiple polyps in all patients from group A, but none of group B. Seven patients of group A had polyps >1 cm, and five of these patients underwent enteroscopy to remove the polyps. At the time of enteroscopy, 20 more polyps were identified in all 5 patients, which were missed by capsule endoscopy. There were no complications from the procedure, and all the capsules were expelled within 24 hours. The authors concluded that capsule endoscopy is a safe and well tolerated method for evaluating the small intestine in PJS patients, however, polyps were missed by this method134.
Plum et al. studied double balloon endoscopy (DBE) in patients with PJS. This technique features an endoscope, and a sliding tube with two balloons, one attached to the distal end of the scope and the other attached to a transparent tube, which slides over the endoscope. When the two balloons are inflated they can trap the small intestine and allow the scope to be advanced until the whole small bowel is visualized. Plum et al. performed the procedure on 16 patients with the diagnosis of PJS between the years 2003–2006. A total of 24 procedures were done, 39 were via the oral approach, and 8 by the anal approach. A total of 178 polyps were documented, mostly in the jejunum, and 47 endoscopic polypectomies were performed. Thirty-seven polyps were >1 cm with the largest being 5 cm, and 10 were removed due to an abnormal gross appearance. No evidence of malignancy was found in any of these polyps on pathologic evaluation. Four complications were reported, 1 perforation, 1 episode of hypoxia associated with a large dose of propofol, and 2 episodes of bleeding with a drop in hemoglobin. They concluded that DBE is not only a safe, effective, and well-tolerated method of screening, but it allows the clinician to evaluate and treat polyps which might cause intussusception, SBO, hemorrhage, or be suspicious for malignancy. Polyps can also be tattooed so their exact location in the small bowel can be determined at surgery113.
Westerman and Wilson stated that most cases of intussusception resolve on their own, but when paralytic ileus evolves, or the obstruction does not resolve within a few hours, laparotomy is required. Surgery should be conservative, since in the future resection of additional small bowel might be required. Surgery should focus on removing responsible polyps that caused the intussusception, as well as the diseased small bowel. The small bowel should be cleared of all large polyps by enterotomy at the same time151. Giardiello and Trimbath recommended polypectomy of >1 cm polyps in the stomach and colon if encountered during surveillance. In the small intestine, surgery was recommended for polyps that are rapidly enlarging (noted by serial exams with endoscopy or UGI series), or that are >1 cm in size. If the patient becomes symptomatic and requires a laparotomy, an attempt to clear the small intestine of polyps should be made at the same time, which can be done by enterotomy if the polyps are large, usually with the aid of intraoperative endoscopy42.
GENETICS
PJS can be either familial or sporadic, and it is transmitted in an autosomal dominant fashion5,97. In 1997, Hemminki et al. studied 12 families with PJS by comparative genomic hybridization (CGH), loss of heterozygosity (LOH) in polyps, and linkage analysis. They found that 6 out of 16 polyps had had a subtle loss of chromosome 19p on CGH, which was confirmed by LOH of markers on 19p. To verify that the region deleted from 19p was the area containing the PJS gene, linkage analysis was performed. They found a LOD score of 7.00 with the marker D19S886 at theta = 0.00. Their results showed that PJS is a genetically homogeneous syndrome with a very high penetrance. Based upon LOH in polyps they hypothezised that the gene responsible was a tumor-suppressor gene53. Mehenni et al. confirmed these findings in 6 different PJS families (with 39 affected, and 48 unaffected individuals), localizing the PJS locus to chromosome 19p13.3, with a multipoint LOD score of 7.51 at theta = 0.045100.
In 1998, Hemminki et al. performed a search for the predisposing gene in the PJS by constructing a cosmid contig across the putative PJS gene region from markers D19S886 to D19S883. Transcripts from this region were identified using various methods, and 27 transcripts were further screened for mutations by RT-PCR from lymphoblastoid cell lines from 12 affected individuals with PJS. The LKB1 gene, which encodes for a 433-aminoacid serine threonine kinase was chosen for further screening. Five PJS patients were found to have deletions of LKB1 (188 bp, 29 bp, 2 bp, and 1 bp in length leading to frameshifts, and another 174 bp in-frame with loss of 58 amino acids), and 5 had substitutions (4 nonsense, 1 missense). Another had a 1 bp insertion and frameshift, and one individual had no mutations found52. Jenne et al. independently found the PJS gene in a PJS kindred with 5 affected individuals within 3 generations, which they called STK1174. Volikos et al. reported that approximately 80% of PJS patients have mutations in the LKB1/STK11 gene. Sixty-three percent (48/76) of patients had mutations by direct sequencing, another 14% (11/28) had larger deletions (5 whole gene deletions, 2 with the promoter and exon 1 deleted, and 1 with exon 8 deleted) found by the multiple ligation probe-dependent amplification (MLPA) technique146. The LKB1/STK11 product localizes to both the nucleus and cytoplasm, has been found to interact with p53, some SMAD4 complexes, and is involved in cell polarity, chromatin remodeling, cell cycle arrest, and Wnt signaling90.
Counseling
Eighty per cent of PJS patients will have LKB1 mutations, of which 60% are identified by sequencing and another 20% by MLPA146. PJS patients need to be educated as to the resources available for genetic testing, which can be very helpful, although the diagnosis can be made clinically in most cases when there is a positive family history. A positive LKB1 mutation test will confirm that individuals need careful screening and follow up, while a negative test will allow individuals to be spared from unnecessary screening procedures 99. Patients need to be educated on the natural history of the disease, the high incidence of the various cancers, possible complications, and the signs/symptoms of different polyps and cancers associated with PJS. They need to learn about the recommended screening procedures and be set up with physicians who will carry out appropriate long-term follow up
HEREDITARY MIXED POLYPOSIS SYNDROME (HMPS)
HISTORY
In 1971, Kaschula described an 11-year girl with profuse diarrhea mixed with blood and mucus and no associated anomalies. She was found to have polyps throughout the colon and small bowel. A total colectomy with ileorectal anastomosis was done, and the polyps were found to have both adenomatous and juvenile features, but no link was made to a new syndrome78. In 1987, Sarles et al. proposed a new syndromehe called mixed familial polyposis, while describing a father and son having multiple polyps throughout the colon that on pathologic examination were adenomas and metaplastic polyps in the father, and adenomas, metaplastic polyps, and juvenile polyps in the son127.
CLINICAL FEATURES
In 1997, Whitelaw coined the term HMPS, although they stated that it is not entirely clear weather this was a new syndrome or a variant of JPS. They performed an extensive evaluation of St. Mark’s family 96 (SM96), which had been followed for over 40 years. The family had over 20 members in the second generation, 64 in the third generation, 102 in the fourth generation, and 42 members in the fifth generation. Family members are located throughout the world, and 42 (18 women and 24 men) had either colorectal cancer or polyps. The polyps in the family were tubular adenomas, villous adenomas, flat adenomas, hyperplastic polyps, and atypical juvenile polyps. All atypical juvenile polyps had mixed histological elements, with overlapping adenomatous and hyperplastic features. HMPS patients develop polyps only in the colon and rectum, and on initial colonoscopy they tend to usually have less than 15 polyps. Patients had a median age at presentation of 40 years. The most common symptoms were bright blood per rectum, altered bowel habits such as blood in the stool and/or diarrhea, abdominal pain, and on rare occasions bowel obstruction. On laboratory examination, signs of anemia were common. They found that one member had an epidermoid cyst removed from his face, and another had a lipoma overlying the left scapula, but otherwise no other extracolonic anomalies were found152.
In 2003, Rozen et al. published clinical data on members of a large family in Israel with HMPS. These patients are relatives of the SM96 family, which originated from Lithuania. There are 37 members in the family, and 17 are affected with either colorectal cancer or polyps, which ranged from juvenile, hyperplastic, or mixed juvenile or hyperplastic polyps with adenomatous features, and also serrated and tubular adenomas121. Cao et al. found the mean age of presentation to be 32.4 years. In their study they published data on two families from Singapore, and found that the patients had polyps that ranged from hyperplastic, adenomatous, juvenile, or mixed hyperplastic or juvenile with adenomatous elements. They also concluded that there are no extracolonic anomalies that are associated with HMPS18.
HISTOPATHOLOGY
According to Whitelaw et al., polyps show evidence of mixed elements, such as tubular, villous and sessile adenomas, and atypical juvenile polyps with adenomatous and/or hyperplastic features152. HMPS patients also tend to develop inflammatory and metaplastic polyps142. Rozen et al. described mixed hyperplastic-adenomatous polyps, as the adenomatous part of the polyp have crypts lined by dysplastic epithelium, and the hyperplastic part showing crypts with stellate lumens lined by non-dysplastic epithelium. The mixed juvenile-adenomatous polyp had adenomatous areas composed of serrated crypts, and the juvenile parts with dilated non-dysplastic crypts121.
CANCER PREDISPOSITION
Sarles et al. felt that in all cases with mixed polyposis, especially with adenomatous features, there was a risk of malignancy127. Jeevaratnam et al. described a 3 generation family with 6 members having colonic adenocarcionma and evidence of hyperplastic polyps. The grandmother in the family underwent surgical resection for colon cancer. She had 10 children, five of them with colon adenocarcinoma and evidence of mixed hyperplastic polyps with adenomatous changes. Three of these individuals had poorly differentiated adenocarcinoma, and passed away from metastatic disease. The other 2 were found to have moderately differentiated adenocarcionma. Two grandchildren from affected parents were also found to have hyperlastic polyps on colonoscopy. Jeevaratnam et al. suggested that cancer arises from within hyperplastic or mixed polyps that develop foci of dysplasia, or from adenomas that might also be present in the GI tract72. Whitelaw et al. also felt that HMPS carries a risk for the development of colorectal cancer (CRC). They found that in family SM96 13 individuals had CRC, with a median age at diagnosis of 47 years. In addition, they reported two female members with breast cancer, but no evidence of colonic polyps, and one member, who was a nonsmoker with bronchial carcinoma who at the time of the study had not been screened for polyps152. Rozen et al. also described a branch of this family in Israel that had five members with colorectal cancer121.
In 1999, Tomlinson et al. reported an Ashkenazi family, named St. Mark’s family 1311 (SM1311), where members had the predisposition to develop colorectal adenomas and carcinomas. The kindred was composed of 4 generations, in which 12 members developed colorectal cancer, one member had pancreatic cancer, and another had renal cancer. Affected members had tubulovillous, villous, and serrated adenomas throughout the colon and rectum143. Cao et al. reported six members with colorectal cancer in the two families from Singapore (3 from each family). One member of family 2 was also diagnosed with papillary thyroid carcinoma at the age of 4018.
MANAGEMENT
Sarles et al. recommended annual colonoscopy for screening patients with mixed polyposis due to the risk of malignancy127. Whitelaw et al. recommended colonoscopy every two years for surveillance, since in family SM96, they found one individual that developed 12 tubular adenomas in a two year period. They felt that sigmoidoscopic surveillance played no role in HPMS, since over half of the cancers in family SM96 were proximal to the midtransverse colon152. Rozen et al. also recommended careful screening and polypectomy to prevent the development of CRC121. When surgical management is needed, Sarles et al. proposed that total colectomy was justified with patients who were found to have adenomas on colonoscopy, or when there was a family history that increased the risk of malignancy127.
GENETICS
Jeevaratnam et al. examined a three generation kindred with HMPS, and suggested that the disorder could be caused by a mutation in a mismatch repair gene, furthermore, that the germline mutation seemed to be inherited in an autosomal dominant pattern72. Since then, other authors concur that HMPS appears to be a distinct syndrome, and is inherited in an autosomal dominant fashion18,142,152. In 1996, Thomas et al. performed linkage analysis on the SM96 family. Linkage excluded APC, hMSH2, MLH1, TP53, DCC, and other loci involved in HNPCC142,143,152. The maximum multipoint LOD score was 3.93, found between markers D6S301 and D6S283 located on chromosome 6q. Karyotype analysis combined with FISH revealed no inversion or other gross rearrangements on chromosome 6142. In 1999, Tomlinson et al. performed linkage analysis in family SM1311, where members had the predisposition to develop colorectal adenomas and carcinomas. They had previously found no mutations of the APC gene, or any other known genes that cause colon cancer. They found a LOD score of 3.06 with D15S118, which mapped a new colorectal cancer susceptibility gene to chromosome 15q14-q22, which they called colorectal adenoma and carcinoma (CRAC1)143. Won Sang Park et al. performed high-density LOH with markers spanning chromosome 15q15-q22 in family SM1311, and found a deletion mapping to 15q21.1, where the tumor suppressor gene THBS1 was thought to be the most likely candidate gene108.
In 2003, Jaeger et al. reassessed family SM96 since they felt that the linkage done earlier by Thomas et al. might not be correct, since a member of the family, which did not have the disease haplotype, had developed multiple colorectal adenomas. Updating the affection status of individuals, and performing a genome-wide linkage analysis, chromosome 6 was excluded, and a maximum 2 point LOD score of 3.98 and a multipoint LOD score of 4.67 was found on chromosome 15q13-q2166. In addition, Jaeger et al. compared haplotypes at the CRAC1 locus found by Tomlinson et al. in 1999 to that in SM96 and found that they shared the same alleles between markers D15S1031-D15S118. Furthermore, they examined another Ashkenazi family believed to have HMPS (SM2952), and typing of markers D15S1031-D15S118 at 15q13-q14 showed that all affected members shared the HMPS/CRAC1 haplotype. Therefore, using data from the three families, 2 point and multipoint LOD scores were 5.31 and 7.19 respectively66. However, Peng et al. using the markers D15S1031-D15S118 for the HMPS/CRAC1 haplotype, did not find linkage in the two HMPS families from Singapore111. Later, Cao et al. updated the affection status of family 1 and 2 from Singapore and found linkage with a multipoint LOD score of 4.60 between D10S1696 and D10S1739 on chromosome 10q23, and found the same haplotype from markers on 10q23.1 -10q23.32 segregated with disease in the two families. This area was then screened for mutations by sequencing all coding exons in the PTEN, MINPP1, PCSH21, and BMPR1A genes, and an 11 base pair deletion was found in the proband of family 2 in exon 2 of BMPR1A, which resulted in a frameshift. There were no mutations identified in these genes in affected members of family 1, however18.
Counseling
Physicians should provide genetic counseling, and genetic testing, if there is suspicion of a hamartomatous polyposis syndrome or if another polyposis syndrome is suspected. Since the gene responsible for HMPS has not been clearly identified, these patients should be managed accordingly, and perhaps should be considered for sequencing of JP genes (SMAD4 and BMPR1A) and perhaps HNPCC genes. If the family is large enough, linkage studies using markers from chromosome 15q13-21 should be considered. Although some evidence suggests that medical management with aspirin or COX-2 inhibitors significantly inhibit sporadic colorectal adenoma recurrences and reduce the risk for CRC8, careful screening via colonoscopy, either annually or every two years, and surveillance of all polyps found via polypectomy is the most effective method to date to prevent advanced cancers from developing. When CRC has been identified more aggressive surgical management is indicated, and either a subtotal or total colectomy should be the procedure of choice.
PTEN HAMARTOMA TUMOR SYNDROME (PHTS): COWDEN SYNDROME (CS) and BANNAYAN-RILEY-RUVALBABA SYNDROME (BRRS)
HISTORY
In 1960, Riley and Smith described a mother and her four (of seven) children with macrocephaly and pseudopapilledema. The father of the children did not exhibit any of these traits. They concluded that this syndrome was due to a mutation in a single autosomal gene118. In 1962, Lloyd and Dennis described a 20-year-old female with a lesion of her right breast that was ulcerated and draining. She had mild mental retardation, poor fine motor movements, microstomia, macrocephaly, and multiple hyperkeratotic papillomata over her lips. She underwent bilateral modified simple mastectomies, and pathology demonstrated adenocarcinoma of the right breast. Biopsy from a thyroid mass was consistent with an adenoma. They concluded that the spectrum of findings might be genetically linked, and coined the disease entity as “Cowden’s Syndrome”, named after the patient, Rachel Cowden87. In 1971, Bannayan described a 3-year-old girl with macrocephaly and numerous subcutaneous lesions that ranged from lipolymphangio-hemangiomas to vascular hamartomas. She underwent resection of multiple masses in the thoracic cavity, which all grossly appeared to be adipose tissue. At autopsy, the entire central nervous system was enlarged, with prominent gyri, but no dilation of the ventricles. The thoracic and abdominal cavities had multiple fatty tumors. The entire subserosa of the GI tract had sessile and pedunculated fatty tumors. They felt that this syndrome was related to the one reported by Riley and Smith, since both lipomatosis and hemangiomatosis are hamartomatous malformations4. In 1980, Ruvalcaba, Myhre, and Smith described two patients with macrocephaly, mental retardation, pigmented spotting of the glans penis and shaft, and multiple polyps of the GI tract. Histologically, the polyps were consistent with hamartomas, and they felt that this syndrome differed from PJS122. The cases described by Bannayan, Riley, Ruvalcaba, Myhre, and Smith would be come known as the Bannayan-Riley-Ruvalcaba syndrome (BRRS) in 1992 when Gorlin et al. coined the term, and later would be linked to mutations in the PTEN gene, which also causes CS. Many physicians and researchers now consider BRRS and CS to be a single entity, with a phenotypic spectrum caused by mutations of the PTEN gene, now referred to as the PTEN Hamartoma Tumor Syndrome2,21,93,161.
CLINICAL FEATURES
Sogol et al. reviewed all cases of CS up to the year 1978, and found that out of the 40 cases described, that thyroid disease occurred in 68% (27/40) of CS patients, with the most common thyroid lesion being goiter, which occurred in 45% of patients. Sixty-six per cent of these patients underwent total thyroidectomy. Thyroid cancer was found in only 7.5% of patients with CS (2 patients had follicular carcinoma, and 1 had a mixed papillary-follicular carcinoma). They also found that 55% (22/40) of women had breast disease, however, they did not specify the various types of breast pathology135. In contrast, Starink et al. found that 68% of CS patients had thyroid disase, with only 3% having follicular carcinoma, and 70% of patients had breast disease, with 52% having fibrocystic changes, and 28% ductal adenocarcinoma. Furthermore, 100% of the patients had mucocutaneous lesions, which included facial trichilemmomas (Figure 6), acral keratoses, oral papillomas and fibromas, skin tags, scrotal tongue, AVMs, and lipomas138. Carlson et al. described involvement of the GI system with a review of the literature revealing that 27% of CS patients had hamartomatous polyps in the colorectum19. In contrast, Starink et al. reported that 43% of patients had involvement of the GI tract, with 22% of patients having hamartomatous polyps in the upper GI tract, and 29% in the colorectum138.
Figure 6.
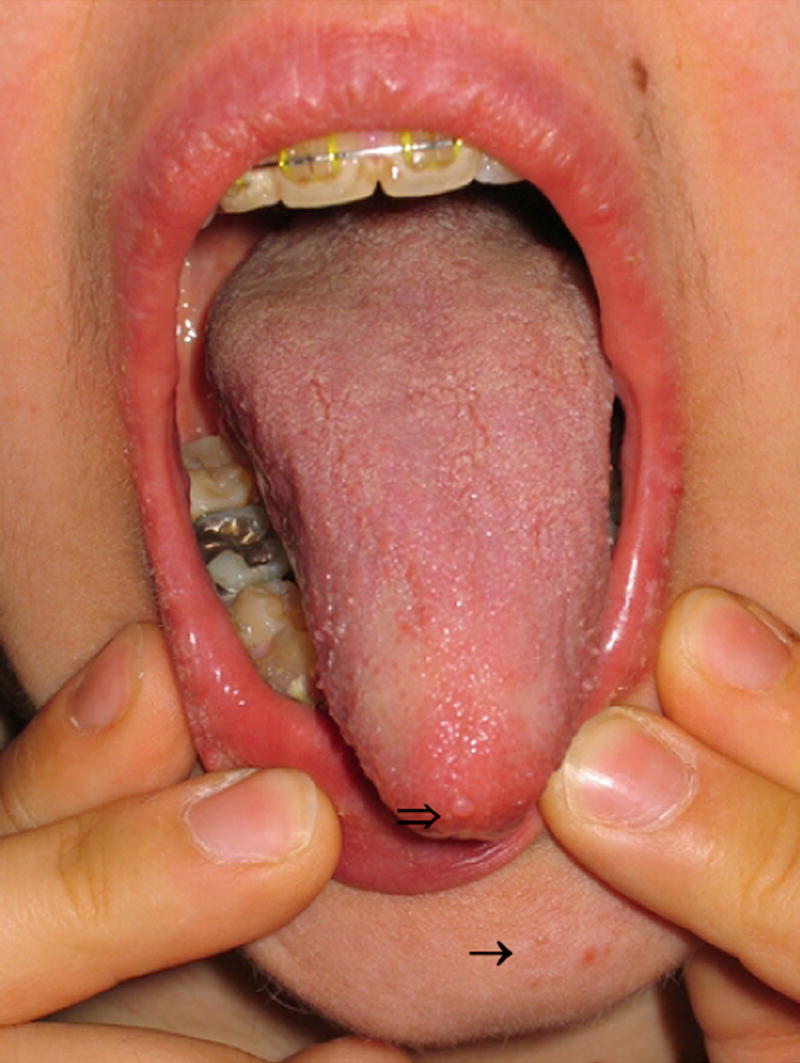
Some of the pathognomonic features of CS, which are facial trichilemmoma (solid black arrow), papillomatosis of the tongue (open arrow), and acral keratoses (reprinted with permission from77).
Albrecht et al. suggested that Lhermitte-Duclos disease (LDD), where individuals have cerebellar hamartomas (gangliocytomas) and mental dullness, is associated or closely linked to CS. In 1991, they published two cases with both CS and LDD, and on review of the literature they found 52 other cases of patients diagnosed with LDD that also had clinical signs of CS. They concluded that patients with LDD might have a broader diagnosis of CS. The two patients they described in addition to having classical clinical signs of CS had vertigo, headaches, visual changes, and ataxia, all due to hydrocephalus secondary to enlarged and thickened cerebellar tonsils, with cerebellar biopsies consistent with LDD1. Since then other authors have included LDD in the diagnosis of CS32,105. The International Cowden Consortium Diagnostic Criteria are divided into 3 categories: 1) pathognomonic criteria, in which individuals are diagnosed with CS if they have facial trichilemmomas, keratosis of the palms or plantar surfaces, oral papillomatous lesions, and mucosal lesions (there must be 6 or more of these lesions, and three or more must be trichilemmomas); 2) major criteria, which include LDD, macrocephaly, breast cancer, and thyroid cancer (especially papillary thyroid cancer); and 3) minor criteria, which include multinodular goiter, mental retardation, gastrointestinal hamartomas, fibrocystic disease of the breast, lipomas, fibromas, and genitourinary tumors or malformations. If the individual does not have any of the pathognomonic criteria, then in order to make the diagnosis of CS, there must be 2 major criteria (where one is either LDD or macrocephaly), or one major and three minor criteria, or 4 minor criteria (Table 1)31.
Table 1.
Diagnostic criteria for Cowden syndrome31.
| Pathognomonic Criteria | Major Criteria | Minor Criteria |
|---|---|---|
|
|
|
Gorlin et al. upon review of the literature for BRRS reported that the macrocephaly with a normal ventricular system is common to all BRRS patients. Sixty percent have downslanting of the palpebral fissures, 15% have strabismus or amlyopia, 35% have Schwalbe lines, the birth weight is usually above the mean as well as body length, which is generally above the 97th percentile. Fifty percent of patients have hypotonia, mild mental retardation, gross motor delay, and speech delay. The motor delay is for the most part transient, and improves with time, but 60% of patients have irreversible myopathic disease. Fifty percent of patients have speckling of the glans penis and shaft, cutaneous angiolipomas, lymphangiomyomas, angiokeratomas, joint hyperextensibility, pectus excavatum, scoliosis, and accelerated growth of the metacarpals, first and second phalanges. Seventy-five percent of patients have subcutaneous lipomas, 10% have hemangiomas, and café-au-lait spots of the trunk and lower extremities. Forty-five percent of patients have hamartomatous polyps, which are limited to the ileum and colon, and can lead to rectal bleeding and intussusception44. In 1999, Marsh et al. noted that BRRS and CS have several clinical features in common, which are Hashimotos’s thyroiditis, vascular malformations, and mental retardation. However, BRRS patients develop pigmented macules of the glans penis and shaft, and delayed motor development, which are not seen in CS patients. Lipomatosis is rarely seen in patients with CS, but is common in BRRS94.
Associated Anomalies
Starink et al. reviewed the CS patients in 1986, and found that 7% of males had bilateral gynecomastia, 3% of males had hydroceles, 20% of females had mestrual irregularities, 19% had ovarian cysts, 5% had leiomyomas of the uterus, and 6% had cysts in the uterus. Macrocephaly was found in 21% of patients, adenoid facies (long, thin face with malar hypoplasia, high-arched palate, which is seen alone in 14% of patients, and a narrow maxillary arch) in 8%, pectus excavatum in 6%, and bone cysts were found in 4%. In terms of the neurological system, 5% had neuromas of cutaneous nerves, 3% had neurofibromas, 3% had meningiomas, and 2% had hearing loss. Some patients have eye involvement, with 3% of patients having cataracts, 2% having angioid streaks, 3% myopia, and 1% congenital blood vessel anomalies138.
HISTOPATHOLOGY
Brownstein et al. described the cutaneous tumors as trichilemmomas, which are benign neoplasms arising from the outer root sheath of hair follicles. Histologically, these are made up of keratinized, palisading cells in the periphery, which are surrounded by a vitreous, thick basement membrane, rich in glycogen. These tumors tend to occur around the mouth, nose, and ears10,11. The acral keratoses found on the palms are hyperkeratotic, and colored or brownish lesions. Oral lesions, which tend to be fibromas, are seen under the tongue, in the lips, mucous membranes, or palate. These lesions are benign, and are formed by a fibrovascular core covered by epithelial cells with no nuclear atypia10.
CANCER PREDISPOSITION
Colorectal
In 1985, Walton et al. described a 36 year old female with CS that was found to have breast cancer incidentally after bilateral prophylactic mastectomies. Her father, who was believed to have CS, died from metastatic colon cancer. This was the first individuals with CS reported to have colon cancer149. Starink et al. found that out of 100 patients with CS, two females had adenocarcinoma of the cecum, and one male of the colon. However, both patients that had cancer in the cecum did not have polyps throughout the colon, therefore they concluded that no meaningful association between colorectal cancer and CS could be made138. In BRRS, colorectal cancer and cancer of the small bowel have not been reported94.
Thyroid and Reproductive Organs
In 1986, Starink et al. reviewed 100 cases of CS, and they found that the most common organs affected with cancer are the thyroid and the female organs. Three per cent had follicular thyroid cancers, 28% had breast adenocarcinoma, 6% had adenocarcioma of the uterus, 3% had carcinoma of the cervix, 2% of the ovary, and 2% of females had transitional cell carcinoma of the renal pelvis. In addition, they found that 3% of males had transitional cell carcinoma of the bladder138. In 1995, Hanssen et al. reported that malignancies of the thyroid were present in 7% of patients, breast cancer was present in 19%, and there was a 45% risk of cancer of the reproductive organs (see Figure 5)49.
In 2001, Frackenthal et al. reported two males with CS that developed breast cancer. One patient was a sporadic case of CS diagnosed at the age of 31, and developed breast cancer at the age of 41. He was found to have a de novo mutation in PTEN. The second patient belonged to a 3 generation CS family with 6 affected members. He developed breast cancer at the age of 43, and also had a PTEN mutation, which segregated with the disease in his family. Both of these patients died from breast cancer in their early fifties33. Longy et al. described a BRRS family with 4 affected individuals, in which the grandmother of the proband was diagnosed with adenocarcinoma of the breast and endometrium at age 53. This was the first report of cancer in a BRRS patient and the authors concluded that it may be warranted to follow BRRS patients for the cancers screened for in CS88. Marsh et al. confirmed that breast cancer and fibroadenoma were also found in patients with BRRS, CS, or patients with overlapping features. They found that patients with truncating mutations in PTEN were at higher risk of developing breast cancer or fibroadenomas94.
MANAGEMENT
Walton et al. felt that periodic contrast studies of the GI tract and colonoscopies should be done to screen CS patients for disease of the upper and lower GI tracts149. Today, the method of choice for screening suspicious nodules of the thyroid gland is by thyroid ultrasound with fine needle aspiration, in addition to TSH and free T4 levels, which should be obtained if one is suspicious of thyroid disease. Williard et al. stated that female patients should perform monthly self breast exams, combined with physician breast exams every 3 months, and mammography every 6–12 months with biopsies of any suspicious lesions153.
Walton et al. described a 36-year-old female with CS who had bilateral oophorectomy at the time of tubal ligation for reportedly incidentally found benign cysts. Colonoscopy was normal, she had facial trichilemmomas, multinodular goiter, facial verruca vulgaris, and mammography showed fibrocystic breast disease bilaterally, but no suspicious areas for cancer. She elected to undergo total thyroidectomy, and prophylactic bilateral simple mastectomy with immediate reconstruction. On pathologic examination, an unexpected small focus of infiltrating ductal carcinoma surrounded by epithelial dysplasia was identified in her right breast. She then had a right axillary dissection, and 1/17 nodes were positive for metastatic disease. They believed that serial mammography should start at the age of 30, with monthly self-breast examinations. They felt that females with CS should be offered prophylactic mastectomy around the fourth decade of life with counseling about the procedure and its indications149. Williard et al. described a 32-year-old female with a 2-month history of a painless mass in her right breast and nipple inversion. She was diagnosed earlier with fibrocystic breast disease, and had had several benign cysts removed from her breasts 10 years earlier. She was diagnosed with CS, and her family had a very high incidence of breast cancer. Her mother died of breast cancer at the age of 42, and she had two maternal aunts that had breast cancer prior to menopause. She was found to have bilateral intraductal and infiltrating ductal carcinoma, and had bilateral modified radical mastectomies, with 7/49 positive nodes on the left, and 10/31 nodes on the right. They felt that female patients with CS should be offered prophylactic bilateral total mastectomy in the third decade, or have monthly self breast exams and every 3 month physician breast exams153.
GENETICS
Cowden’s syndrome is an autosomal dominant inherited disorder20,50,138, and Hanssen et al. suggested that anticipation and imprinting through the female were also part of CS50. In 1996, Nelen et al. performed a linkage-based genome screen in 12 families with CS. They found linage to markers on the long arm of chromosome 10 with a maximum LOD score of 6.67 with the marker D10S215104. In 1997, Liaw et al. sequenced all coding exons of the PTEN gene from 10q22-3 in five CS families, of which four were linked to markers on 10q22-23. They found that two different families had missense mutations in exon 5, a third family had a nonsense mutation in exon 7, and a fourth family had a different nonsense mutations in exon 785. Since then, other authors have confirmed the involvement of PTEN in CS89,106, and germline mutations have been found by sequencing in about 80% of cases85,91. PTEN is a dual-specificity tyrosine phosphatase with homology to the chicken protein tensin and bovine auxilin84. The phosphatase domain is where the active site is located and carries out the enzymatic function, and the C2 domain allows PTEN to bind to the cell membrane where its substrate is located. PTEN dephosphorylates phosphatidylinositol (4,5)-bisphosphate, so that phospholipase C can hydrolyze the phosphodiester link and form inositol triphosphate and diacylglycerol (DAG), which are two important intracellular messengers. PTEN plays an important role in apoptosis, preventing uncontrolled cell growth, and possibly also involved in cell migration and adhesion83,126,133,159.
Also in 1997, Marsh et al. examined two unrelated families with BRRS syndrome, and one was found to have a germline missense mutation in exon 7, and the other a nonsense mutation in exon 6 of the PTEN gene. Both mutations segregated with affected members in each family, and were absent in unaffected members, as well as 100 control subjects. They concluded that BRRS and CS represent the clinical spectrum of the same disease92. Marsh et al. later examined 43 unrelated BRRS patients (32 had features of BRRS, and 11 with mixed features of CS and BRRS) for germline PTEN mutations by sequencing. Twenty-six of the forty-three (60%) individuals had PTEN mutations, 17 of which were substitutions (10 missense, 7 nonsense), 5 were small deletions, 1 a gross deletion of 10q23.3-q24.1, 3 were insertions, and was a one balanced translocation. All exons were involved except 1, 4 and 9, and the mutations segregated within affected members of each family (27 were familial cases and 16 were sporadic). Of the 17 BRRS cases that did not have any mutations found in PTEN, they retained hemizygosity when analyzed with various markers of the PTEN region, which eliminated the likelihood of gross gene deletions. From the 27 familial cases, 11 had clinical symptoms that overlapped between the two syndromes, and 10 of these families had mutations in PTEN. In 37 CS families, the findings were very similar, and there were no significant differences in the spectrum of mutations. Four nonsense mutations were common to both CS and BRRS (Q110X, R130X, R233X, and R335X). The balanced translocation and large deletion were only seen in BRRS patients, and no gross deletions were seen in CS94. Longy et al. confirmed that CS and BRRS syndrome are allelic diseases, and noted that in BRRS syndrome, mutations occurred preferentially in exons 6 and 7, while in CS the spectrum of mutations occur in all exons except 1, 4 and 988.
Zhou et al. evaluated 122 PTEN mutation negative cases of CS and BRRS, and found 3 with deletions involving the entire gene, exon, and exons 1–5. They also found 9 cases with heterozygous germline promoter mutations159. Sarquis et al. sequenced 85 subjects with the CS/BRRS phenotype or suggestive features (65 females and 20 males). Out of the 85 patients, 22 patients did not have PTEN mutations, and 63 had PTEN mutations (43 with known pathogenic PTEN mutations, and 20 with variants). From the 63 patients with PTEN mutations, 28 had mixed phenotypic features of both CS and BRRS, 26 had clear phenotypic features of CS, 1 of BRRS, and 8 could not be classified as either CS or BRRS. They analyzed the full length PTEN mRNA and found 8 novel splice variants (SV) and reduced expression of full-length transcripts compared to controls.
They found that 5 of the 8 novel SV showed differential expression in individuals with CS/BRRS phenotype compared to controls, and differential expression correlated with the distinct phenotypes seen in the various groups (mixed CS/BRRS, CS, and BRRS)128.
Counseling
Higginbottom and Schultz felt that genetic counseling would be important, and could lead to early diagnosis54. Liaw et al. recommended that since PTEN was identified as the gene responsible for CS, individuals at risk should be tested in order to receive more heightened cancer surveillance for tumors of the breast, thyroid and GI tract85,106.
CRONKHITE-CANADA SYNDROME (CCS)
HISTORY
In 1955, Cronkhite and Canada described two female patients that were 42 and 75 years old presenting with several months of diarrhea, vomiting, nausea, and abdominal pain. Several weeks prior to symptoms, loss of hair of the eyebrows and axillary region, diffuse brown discoloration of the face, neck and hands, and onychotrophia were noted. On laboratory examination, the only abnormality found was anemia. They had multiple large polyps in the stomach and duodenum, as well as small sessile and polypoid defects covering the entire colonic mucosa on barium examination. The mucosal pattern throughout seemed more coarse, especially in the jejunum. Biopsies of the gastric and colonic lesions showed histology consistent with benign adenomatous polyps. Both patients died, and an autopsy on one revealed pitting edema that extended from the feet to the costal margins. Epidermal bullae were noted over the hip area, and xanthomas were present bilaterally. The tongue was atrophic and the surface had a brown discoloration. The tip of the index finger was gangrenous, she had ascities as well as a pleural and pericardial effusions. Gross examination of the GI tract showed a normal esophagus, but the gastic and duodenal mucosa was covered with polypoid lesions, and the architecture of the mucosa was distorted. The jejunum and ileum were not as heavily involved, but did have polyps, which were more sessile than in the stomach and duodenum. The large bowel and rectum were also carpeted with polyps. All polyps were consistent with simple adenomatous polyps without evidence of malignancy24.
CLINICAL FEATURES
In 1966, Jarnum and Jensen published their observation on the syndrome now recognized as Cronkhite-Canada syndrome (CCS). In addition to the h features of generalized gastrointestinal polyposis (not affecting the esophagus), alopecia, dermal pigmentation, and atrophy of the nail beds, they found that severe protein losing enteropathy with electrolyte disturbances (hypocalcemia, hypomagnesimia, and hypokalemia) was also part of the clinical syndrome. Another very important observation by Jarnum and Jensen was that histologically, the polyps only showed cystic dilatation of glands with no areas of adenomatous epithelium67. In 1972, Johnson reported that the stomach and large bowel are not carpeted with adenomatous polyps as originally described, but with hamartomatous polyps75, which confirmed the original description of Jarnum and Jensen in 1966. All the above clinical characteristics have been described by multiple authors, as well as the fact that this disease spares the esophagus15,30,103.
In 1995, Goto et al. proposed a classification of the CCS into five groups. Type 1 patients complained of diarrhea as the presenting symptom, type 2 had disguesia (abnormal taste sensation), type 3 complained of abnormal sensation in the mouth accompanied by thirst, type 4 patients had abdominal symptoms other than diarrhea, and in type 5 alopecia was the main symptom. All patients must have gastrointestinal polyposis and hyperpigmentation in order to be diagnosed with CCS, which carries an unfavorable prognosis, with a 5-year mortality rate of 55%45. Daniel et al. described the potential of life threatening gastrointestinal bleeding, intussusception, and rectal prolapse26. Other complications have been described, such as portal vein thrombosis, high titers of antinuclear antibodies, and membranous glomerulonephritis141.
HISTOPATHOLOGY
Egawa et al. identified nests of cancer cells in the gastric mucosa, which were predominantly poorly differentiated with tubular formation as well. The gastric specimen showed typical features of hamartomatous polyps, however, mild infiltration of inflammatory cells, massive submucosal edema (mostly located in the lamina propia), hyperplasia of the foveolar epithelium, and cystic dilation of the mucosal glands was also noted. No adenomatous changes were noted around the cancer cells, which suggested to them that the cancer originated from the gastric mucosa and not from the polyps30. To validate this theory, they stained the tissue for Ki-67 and p53, which showed that only cancer cells had overexpression of these proteins, reinforcing the theory that only these cells have the ability to proliferate without cellular control, and had the mutated p53 protein. There were no intermediate changes in the surrounding tissue30.
CANCER PREDISPOSITION
Colorectal
Egawa et al. reviewed the CCS literature, and found that 9% of patients had colorectal cancer (34 of 374 patients), and 41% of these also had adenomas or adenomatous changes30. In addition, there were multiple reports showing a clear transition from hamartomatous polyps to adenomatous polyps, with some of the adenomatous polyps having evidence of dysplasia and carcinoma. p53 accumulation was seen in the carcinomatous parts of the polyp, but not in the hamartomatous elements30. In 2005, Yashiro et al. reviewed 31 cases of CCS and colorectal cancer reported in the literature, and found that 40% had serrated adenomas, and they hypothesized that not only were CCS patients with serrated adenomas at a higher risk of colorectal cancer, but that this was a precursor lesion155.
Gastric
Egawa et al. described a 52-year-old male with CCS and adenocarcinoma of the stomach arising from the gastric mucosa and not from polypoid lesions. Review of the literature revealed a 5% rate of gastric cancer, seen in 19 out of 374 cases. The mean age of diagnosis was 64 years of age, and 15 were males. Although it seems there is a high risk for gastric cancer, Egawa et al. felt it was hard to make a precise correlation, and that the risk of gastric cancer was low in CCS30.
MANAGEMENT
Since there is no evidence to date of germline mutations predisposing to this condition, and most experts consider CCS a rare, sporadic, and acquired syndrome, there are no genetic tests for screening individuals that meet the diagnostic criteria155. However, due to the high risk of CRC, screening of the stomach, colon, and rectum by endoscopy should be performed. Biopsies taken will aid in identifying dysplastic or adenomatous epithelium, which would lead to early surgical intervention30,141. Egawa et al. recommend total gastrectomy in CCS patients diagnosed with gastric cancer to remove the malignant potential in the rest of the stomach, prevent the high rate of anastomotic leak due to the edema, and to prevent protein loss from the gastric mucosa30. In terms ofcolorectal cancer, the procedure of choice is determined by the location of the lesion, and if the colon is carpeted with polyps, subtotal or total proctocolectomy would be indicated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albrecht S, Haber RM, Goodman JC, et al. Cowden syndrome and Lhermitte-Duclos disease. Cancer. 1992;70:869. doi: 10.1002/1097-0142(19920815)70:4<869::aid-cncr2820700424>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 2.Arch EM, Goodman BK, Van Wesep RA, et al. Deletion of PTEN in a patient with Bannayan-Riley-Ruvalcaba syndrome suggests allelism with Cowden disease. Am J Med Genet. 1997;71:489. [PubMed] [Google Scholar]
- 3.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44:702. doi: 10.1136/jmg.2007.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bannayan GA. Lipomatosis, angiomatosis, and macrencephalia. A previously undescribed congenital syndrome. Arch Pathol. 1971;92:1. [PubMed] [Google Scholar]
- 5.Bartholomew LG, More CE, Dahlin DC, et al. Intestinal polyposis associated with mucocutaneous pigmentation. Surgery, Gynecology, and Obstetrics. 1962;115:1. [PubMed] [Google Scholar]
- 6.Bentley E, Chandrasoma P, Radin R, et al. Generalized juvenile polyposis with carcinoma. American Journal of Gastroenterology. 1989;84:1456. [PubMed] [Google Scholar]
- 7.Bowlby LS. Pancreatic adenocarcinoma in an adolescent male with Peutz-Jeghers syndrome. Hum Pathol. 1986;17:97. doi: 10.1016/s0046-8177(86)80163-0. [DOI] [PubMed] [Google Scholar]
- 8.Brazowski E, Misonzhnick-Bedny F, Rozen P. Cyclooxygenase-2 expression in the hereditary mixed polyposis syndrome. Dig Dis Sci. 2004;49:1906. doi: 10.1007/s10620-004-9591-2. [DOI] [PubMed] [Google Scholar]
- 9.Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007 doi: 10.1136/gut.2006.116913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brownstein MH, Mehregan AH, Bikowski JB, et al. The dermatopathology of Cowden’s syndrome. Br J Dermatol. 1979;100:667. doi: 10.1111/j.1365-2133.1979.tb08070.x. [DOI] [PubMed] [Google Scholar]
- 11.Brownstein MH, Mehregan AH, Bilowski JB. Trichilemmomas in Cowden’s disease. JAMA. 1977;238:26. [PubMed] [Google Scholar]
- 12.Bruwer A, Bargen JA, Kierland RR. Surface pigmentation and generalized intestinal polyposis; (Peutz-Jeghers syndrome) Mayo Clin Proc. 1954;29:168. [PubMed] [Google Scholar]
- 13.Burdick D, Prior JT. Peutz-Jeghers syndrome. A clinicopathologic study of a large family with a 27-year follow-up. Cancer. 1982;50:2139. doi: 10.1002/1097-0142(19821115)50:10<2139::aid-cncr2820501028>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 14.Burkart AL, Sheridan T, Lewin M, et al. Do sporadic Peutz-Jeghers polyps exist? Experience of a large teaching hospital. Am J Surg Pathol. 2007;31:1209. doi: 10.1097/PAS.0b013e3180339944. [DOI] [PubMed] [Google Scholar]
- 15.Burke AP, Sobin LH. The pathology of Cronkhite-Canada polyps. A comparison to juvenile polyposis. Am J Surg Pathol. 1989;13:940. doi: 10.1097/00000478-198911000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Bussey HJ, Veale AM, Morson BC. Genetics of gastrointestinal polyposis. Gastroenterology. 1978;74:1325. [PubMed] [Google Scholar]
- 17.Cantu JM, Rivera H, Ocampo-Campos R, et al. Peutz-Jeghers syndrome with feminizing sertoli cell tumor. Cancer. 1980;46:223. doi: 10.1002/1097-0142(19800701)46:1<223::aid-cncr2820460137>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Cao X, Eu KW, Kumarasinghe MP, et al. Mapping of hereditary mixed polyposis syndrome (HMPS) to chromosome 10q23 by genomewide high-density single nucleotide polymorphism (SNP) scan and identification of BMPR1A loss of function. J Med Genet. 2006;43:e13. doi: 10.1136/jmg.2005.034827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlson GJ, Nivatvongs S, Snover DC. Colorectal polyps in Cowden’s disease (multiple hamartoma syndrome) Am J Surg Pathol. 1984;8:763. doi: 10.1097/00000478-198410000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Carlson HE, Burns TW, Davenport SL, et al. Cowden disease: gene marker studies and measurements of epidermal growth factor. Am J Hum Genet. 1986;38:908. [PMC free article] [PubMed] [Google Scholar]
- 21.Celebi JT, Tsou HC, Chen FF, et al. Phenotypic findings of Cowden syndrome and Bannayan-Zonana syndrome in a family associated with a single germline mutation in PTEN. J Med Genet. 1999;36:360. [PMC free article] [PubMed] [Google Scholar]
- 22.Christian CD, McLoughlin TG, Cathcart ER, et al. Peutz-Jeghers Syndrome Associated with Functioning Ovarian Tumor. JAMA. 1964;190:935. doi: 10.1001/jama.1964.03070230071027. [DOI] [PubMed] [Google Scholar]
- 23.Coburn MC, Pricolo VE, DeLuca FG, et al. Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol. 1995;2:386. doi: 10.1007/BF02306370. [DOI] [PubMed] [Google Scholar]
- 24.Cronkhite LW, Jr, Canada WJ. Generalized gastrointestinal polyposis; an unusual syndrome of polyposis, pigmentation, alopecia and onychotrophia. N Engl J Med. 1955;252:1011. doi: 10.1056/NEJM195506162522401. [DOI] [PubMed] [Google Scholar]
- 25.Cureton E, Kim S. Images in clinical medicine. Peutz-Jeghers syndrome. N Engl J Med. 2007;357:e9. doi: 10.1056/NEJMicm061305. [DOI] [PubMed] [Google Scholar]
- 26.Daniel ES, Ludwig SL, Lewin KJ, et al. The Cronkhite-Canada Syndrome. An analysis of clinical and pathologic features and therapy in 55 patients. Medicine (Baltimore) 1982;61:293. [PubMed] [Google Scholar]
- 27.Desai DC, Neale KF, Talbot IC, et al. Juvenile polyposis. British Journal of Surgery. 1995;82:14. doi: 10.1002/bjs.1800820106. [DOI] [PubMed] [Google Scholar]
- 28.Diamond M. Adenoma of the rectum in children: Report of a case in a thirty month old girl. American Journal of Diseases in Children. 1939;57:360. [Google Scholar]
- 29.Dormandy TL. Gastrointestinal polyposis with mucocutaneous pigmentation (Peutz-Jeghers syndrome) N Engl J Med. 1957;256:1141. doi: 10.1056/NEJM195706132562405. [DOI] [PubMed] [Google Scholar]
- 30.Egawa T, Kubota T, Otani Y, et al. Surgically treated Cronkhite-Canada syndrome associated with gastric cancer. Gastric Cancer. 2000;3:156. doi: 10.1007/pl00011711. [DOI] [PubMed] [Google Scholar]
- 31.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37:828. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eng C, Murday V, Seal S, et al. Cowden syndrome and Lhermitte-Duclos disease in a family: a single genetic syndrome with pleiotropy? J Med Genet. 1994;31:458. doi: 10.1136/jmg.31.6.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fackenthal JD, Marsh DJ, Richardson AL, et al. Male breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet. 2001;38:159. doi: 10.1136/jmg.38.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farmer RG, Hawk WA, Turnbull RB., Jr The Spectrum of the Peutz-Jeghers Syndrome. Report of 3 Cases. Am J Dig Dis. 1963;8:953. doi: 10.1007/BF02232093. [DOI] [PubMed] [Google Scholar]
- 35.Foley TR, McGarrity TJ, Abt AB. Peutz-Jeghers syndrome: a clinicopathologic survey of the “Harrisburg family” with a 49-year follow-up. Gastroenterology. 1988;95:1535. doi: 10.1016/s0016-5085(88)80074-x. [DOI] [PubMed] [Google Scholar]
- 36.Foster DR, Foster DB. Gall-bladder polyps in Peutz-Jeghers syndrome. Postgrad Med J. 1980;56:373. doi: 10.1136/pgmj.56.655.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedl W, Kruse R, Uhlhaas S, et al. Frequent 4-bp deletion in exon 9 of the SMAD4/MADH4 gene in familial juvenile polyposis patients. Genes Chromosomes Cancer. 1999;25:403. [PubMed] [Google Scholar]
- 38.Friedl W, Uhlhaas S, Schulman K, et al. Juvenile Polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Human Genetics. 2002;111:108. doi: 10.1007/s00439-002-0748-9. [DOI] [PubMed] [Google Scholar]
- 39.Gentile AT, Bickler SW, Harrison MW, et al. Common bile duct obstruction related to intestinal polyposis in a child with Peutz-Jeghers syndrome. J Pediatr Surg. 1994;29:1584. doi: 10.1016/0022-3468(94)90225-9. [DOI] [PubMed] [Google Scholar]
- 40.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 41.Giardiello FM, Hamilton SR, Kern SE, et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Archives of Diseases in Childhood. 1991;66:971. doi: 10.1136/adc.66.8.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006;4:408. doi: 10.1016/j.cgh.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Goodman ZD, Yardley JH, Milligan FD. Pathogenesis of colonic polyps in multiple juvenile polyposis. Cancer. 1979;43:1906. doi: 10.1002/1097-0142(197905)43:5<1906::aid-cncr2820430548>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 44.Gorlin RJ, Cohen MM, Jr, Condon LM, et al. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44:307. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 45.Goto A. Cronkhite-Canada syndrome: epidemiological study of 110 cases reported in Japan. Nippon Geka Hokan. 1995;64:3. [PubMed] [Google Scholar]
- 46.Grigioni WF, Alampi G, Martinelli G, et al. Atypical juvenile polyposis. Histopathology. 1981;5:361. doi: 10.1111/j.1365-2559.1981.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 47.Grosfeld JL, West KW. Generalized juvenile polyposis coli. Archives of Surgery. 1986;121:530. doi: 10.1001/archsurg.1986.01400050040005. [DOI] [PubMed] [Google Scholar]
- 48.Haggitt RC, Pitcock JA. Familial juvenile polyposis of the colon. Cancer. 1970;26:1232. doi: 10.1002/1097-0142(197012)26:6<1232::aid-cncr2820260609>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 49.Hanssen AM, Fryns JP. Cowden syndrome. J Med Genet. 1995;32:117. doi: 10.1136/jmg.32.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanssen AM, Werquin H, Suys E, et al. Cowden syndrome: report of a large family with macrocephaly and increased severity of signs in subsequent generations. Clin Genet. 1993;44:281. doi: 10.1111/j.1399-0004.1993.tb03901.x. [DOI] [PubMed] [Google Scholar]
- 51.Heldin C-H, Miyazono K, Ten Dijke P. TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 52.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 53.Hemminki A, Tomlinson I, Markie D, et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nature Genetics. 1997;15:87. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- 54.Higginbottom MC, Schultz P. The Bannayan syndrome: an autosomal dominant disorder consisting of macrocephaly, lipomas, hemangiomas, and risk for intracranial tumors. Pediatrics. 1982;69:632. [PubMed] [Google Scholar]
- 55.Hofting I, Pott G, Schrameyer B, et al. Familiare juvenile polyposis mit vorwiegender magenbeteiligung. Zeitschrift fur Gastroenterologie. 1993;31:480. [PubMed] [Google Scholar]
- 56.Horrilleno EG, Eckert C, Ackerman LV. Polyps of the rectum and colon in children. Cancer. 1957;10:1210. doi: 10.1002/1097-0142(195711/12)10:6<1210::aid-cncr2820100619>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 57.Houlston R, Bevan S, Williams A, et al. Mutations in DPC4 (SMAD4) cause juvenile polyposis syndrome, but only account for a minority of cases. Human Molecular Genetics. 1998;7:1907. doi: 10.1093/hmg/7.12.1907. [DOI] [PubMed] [Google Scholar]
- 58.Howe JR, Bair JL, Sayed MG, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nature Genetics. 2001;28:184. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 59.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5:751. doi: 10.1007/BF02303487. [DOI] [PubMed] [Google Scholar]
- 60.Howe JR, Ringold JC, Hughes J, et al. Direct genetic testing for Smad4 mutations in patients at risk for juvenile polyposis. Surgery. 1999;126:162. [PubMed] [Google Scholar]
- 61.Howe JR, Ringold JC, Summers RW, et al. A gene for familial juvenile polyposis maps to chromosome 18q21.1. American Journal of Human Genetics. 1998;62:1129. doi: 10.1086/301840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 63.Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41:484. doi: 10.1136/jmg.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humphries AL, Jr, Shepherd MH, Peters HJ. Peutz-Jeghers syndrome with colonic adenocarcinoma and ovarian tumors. JAMA. 1966;197:296. [Google Scholar]
- 65.Hutchinson J. Pigmentation of Lips and Mouth. Arch Surg. 1896;7:290. [Google Scholar]
- 66.Jaeger EE, Woodford-Richens KL, Lockett M, et al. An ancestral Ashkenazi haplotype at the HMPS/CRAC1 locus on 15q13-q14 is associated with hereditary mixed polyposis syndrome. Am J Hum Genet. 2003;72:1261. doi: 10.1086/375144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jarnum S, Jensen H. Diffuse gastrointestinal polyposis with ectodermal changes. A case with severe malabsorption and enteric loss of plasma proteins and electrolytes. Gastroenterology. 1966;50:107. [PubMed] [Google Scholar]
- 68.Jarvinen HJ. Juvenile gastrointestinal polyposis. Problems in General Surgery. 1993;10:749. [Google Scholar]
- 69.Jarvinen HJ, Franssila KO. Familial juvenile polyposis coli: Increased risk of colorectal cancer. Gut. 1984;25:792. doi: 10.1136/gut.25.7.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jass JR. Pathology of polyposis syndromes with special reference to juvenile polyposis. In: Utsunomiya J, Lynch HT, editors. Hereditary Colorectal Cancer. Tokyo: Springer-Verlag; 1990. p. 343. [Google Scholar]
- 71.Jass JR, Williams CB, Bussey HJR, et al. Juvenile polyposis-A precancerous condition. Histopathology. 1988;13:619. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 72.Jeevaratnam P, Cottier DS, Browett PJ, et al. Familial giant hyperplastic polyposis predisposing to colorectal cancer: a new hereditary bowel cancer syndrome Genetic mapping of hereditary mixed polyposis syndrome to chromosome 6q Clinical and molecular features of the hereditary mixed polyposis syndrome Hereditary mixed polyposis syndrome: a zebra or a horse dressed in pinstripes A prospective study of the clinical, genetic, screening, and pathologic features of a family with hereditary mixed polyposis syndrome. Cyclooxygenase-2 expression in the hereditary mixed polyposis syndrome [Haplotype and linkage analysis in Chinese hereditary mixed polyposis syndrome] Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis [Hereditary mixed polyposis syndrome. First report in Mexico] Gene symbol: BMPR1A. Disease: Hereditary mixed polyposis syndrome. Accession #Hd0511 [Mixed hereditary polyposis with atypical extracolonic manifestations] J Pathol. 1996;179:20. doi: 10.1002/(SICI)1096-9896(199605)179:1<20::AID-PATH538>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 73.Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of oral mucosa, lips, and digits. The New England Journal of Medicine. 1949;241:993. doi: 10.1056/NEJM194912222412501. [DOI] [PubMed] [Google Scholar]
- 74.Jenne DE, Reimann H, Nezu J-I, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nature Genetics. 1998;18:38. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 75.Johnson JG, Gilbert E, Zimmermann B, et al. Gardner’s syndrome, colon cancer, and sarcoma. Journal of Surgical Oncology. 1972;4:354. doi: 10.1002/jso.2930040406. [DOI] [PubMed] [Google Scholar]
- 76.Jones MA, Hebert JC, Trainer TD. Juvenile polyp with intramucosal carcinoma. Archives of Pathology and Laboratory Medicine. 1987;111:200. [PubMed] [Google Scholar]
- 77.Jornayvaz FR, Philippe J. Mucocutaneous papillomatous papules in Cowden’s syndrome. Clin Exp Dermatol. 2008;33:151. doi: 10.1111/j.1365-2230.2007.02602.x. [DOI] [PubMed] [Google Scholar]
- 78.Kaschula RO. Mixed juvenile, adenomatous and intermediate polyposis coli: report of a case. Dis Colon Rectum. 1971;14:368. doi: 10.1007/BF02553425. [DOI] [PubMed] [Google Scholar]
- 79.Kim IJ, Park JH, Kang HC, et al. Identification of a novel BMPR1A germline mutation in a Korean juvenile polyposis patient without SMAD4 mutation. Clin Genet. 2003;63:126. doi: 10.1034/j.1399-0004.2003.00008.x. [DOI] [PubMed] [Google Scholar]
- 80.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 81.Konishi F, Wyse NE, Muto T, et al. Peutz-Jeghers polyposis associated with carcinoma of the digestive organs. Report of three cases and review of the literature. Dis Colon Rectum. 1987;30:790. doi: 10.1007/BF02554629. [DOI] [PubMed] [Google Scholar]
- 82.Lehur PA, Madarnas P, Devroede G, et al. Peutz-Jeghers syndrome. Association of duodenal and bilateral breast cancers in the same patient. Dig Dis Sci. 1984;29:178. doi: 10.1007/BF01317062. [DOI] [PubMed] [Google Scholar]
- 83.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 85.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 86.Lin JI, Caracta PF, Lindner A, et al. Peutz-Jeghers polyposis with metastasizing duodenal carcinoma. South Med J. 1977;70:882. doi: 10.1097/00007611-197707000-00038. [DOI] [PubMed] [Google Scholar]
- 87.Lloyd KM, 2nd, Dennis M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963;58:136. doi: 10.7326/0003-4819-58-1-136. [DOI] [PubMed] [Google Scholar]
- 88.Longy M, Coulon V, Duboue B, et al. Mutations of PTEN in patients with Bannayan-Riley-Ruvalcaba phenotype. J Med Genet. 1998;35:886. doi: 10.1136/jmg.35.11.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lynch ED, Ostermeyer EA, Lee MK, et al. Inherited mutations in PTEN that are associated with breast cancer, Cowden disease, and juvenile polyposis. American Journal of Human Genetics. 1997;61:1254. doi: 10.1086/301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marignani PA. LKB1, the multitasking tumour suppressor kinase. J Clin Pathol. 2005;58:15. doi: 10.1136/jcp.2003.015255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7:507. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 92.Marsh DJ, Dahia PL, Zheng Z, et al. Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet. 1997;16:333. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]
- 93.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 94.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 95.Massague J. TGFβ signaling: Receptors, Transducers, and Mad proteins. Cell. 1996;85:947. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- 96.Matuchansky C, Babin P, Coutrot S, et al. Peutz-Jeghers syndrome with metastasizing carcinoma arising from a jejunal hamartoma. Gastroenterology. 1979;77:1311. [PubMed] [Google Scholar]
- 97.McAllister AJ, Hicken NF, Latimer RG, et al. Seventeen patients with Peutz-Jehgers syndrome in four generations. Am J Surg. 1967;114:839. doi: 10.1016/0002-9610(67)90403-5. [DOI] [PubMed] [Google Scholar]
- 98.McColl I, Bussey HJR, Veale AMO, et al. Juvenile polyposis coli. Proceedings of the Royal Society of Medicine. 1964;57:896. [PubMed] [Google Scholar]
- 99.McGarrity TJ, Kulin HE, Zaino RJ. Peutz-Jeghers syndrome. The American Journal of Gastroenterology. 2000;95:596. doi: 10.1111/j.1572-0241.2000.01831.x. [DOI] [PubMed] [Google Scholar]
- 100.Mehenni H, Blouin J-L, Radhakrishna U, et al. Peutz-Jeghers syndrome: Confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. American Journal of Human Genetics. 1997;61:1327. doi: 10.1086/301644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Merg A, Lynch HT, Lynch JF, et al. Hereditary colorectal cancer-part II. Curr Probl Surg. 2005;42:267. doi: 10.1067/j.cpsurg.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 102.Morson BC. Some peculiarities in the histology of intestinal polyps. Diseases of the Colon and Rectum. 1962;5:337. doi: 10.1007/BF02616633. [DOI] [PubMed] [Google Scholar]
- 103.Murata I, Yoshikawa I, Endo M, et al. Cronkhite-Canada syndrome: report of two cases. J Gastroenterol. 2000;35:706. doi: 10.1007/s005350070051. [DOI] [PubMed] [Google Scholar]
- 104.Nelen MR, Padberg GW, Peeters EA, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13:114. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 105.Nelen MR, Padberg GW, Peeters EAJ, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nature Genetics. 1996;13:114. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 106.Nelen MR, van Staveren WC, Peeters EA, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- 107.Niimi K, Tomoda H, Furusawa M, et al. Peutz-Jeghers syndrome associated with adenocarcinoma of the cecum and focal carcinomas in hamartomatous polyps of the colon: a case report. Jpn J Surg. 1991;21:220. doi: 10.1007/BF02470912. [DOI] [PubMed] [Google Scholar]
- 108.Park WS, Park JY, Oh RR, et al. A distinct tumor suppressor gene locus on chromosome 15q21.1 in sporadic form of colorectal cancer. Cancer Res. 2000;60:70. [PubMed] [Google Scholar]
- 109.Parker MC, Knight M. Peutz-Jeghers syndrome causing obstructive jaundice due to polyp in common bile duct. J R Soc Med. 1983;76:701. doi: 10.1177/014107688307600814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Payson BA, Moumgis B. Metastasizing carcinoma of the stomach in Peutz-Jeghers syndrome. Ann Surg. 1967;165:145. doi: 10.1097/00000658-196701000-00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peng H, Cao X, Li HH, et al. [Haplotype and linkage analysis in Chinese hereditary mixed polyposis syndrome] Zhonghua Wei Chang Wai Ke Za Zhi. 2005;8:312. [PubMed] [Google Scholar]
- 112.Peutz JL. A very remarkable case of familial polyposis of mucous membrane of intestinal tract and accompanied by peculiar pigmentations of skin and mucous membrane. Nederlands Tijdschrift voor Geneeskunde. 1921;10:134. [Google Scholar]
- 113.Plum N, May AD, Manner H, et al. [Peutz-Jeghers syndrome: endoscopic detection and treatment of small bowel polyps by double-balloon enteroscopy] Z Gastroenterol. 2007;45:1049. doi: 10.1055/s-2007-963345. [DOI] [PubMed] [Google Scholar]
- 114.Pollack JL, Swinton NW. Congenital of the colon with extension to the small intestine and stomach. Lahey Clin Bull. 1955;9:174. [PubMed] [Google Scholar]
- 115.Pyatt RE, Pilarski R, Prior TW. Mutation screening in juvenile polyposis syndrome. J Mol Diagn. 2006;8:84. doi: 10.2353/jmoldx.2006.050072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ramaswamy G, Elhosseiny AA, Tchertkoff V. Juvenile polyposis of the colon with atypical adenomatous changes and carcinoma in situ. Diseases of the Colon and Rectum. 1984;27:393. doi: 10.1007/BF02553009. [DOI] [PubMed] [Google Scholar]
- 117.Ravitch MM. Polypoid adenomatosis of the entire gastrointestinal tract. Annals of Surgery. 1948;128:283. doi: 10.1097/00000658-194808000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Riley HD, Smith WR. Macrocephaly, pseudopapilledema and multiple hemangiomata: a previously undescribed heredofamilial syndrome. Pediatrics. 1960;26:293. [Google Scholar]
- 119.Roth S, Sistonen P, Salovaara R, et al. SMAD genes in juvenile polyposis. Genes Chromosomes Cancer. 1999;26:54. doi: 10.1002/(sici)1098-2264(199909)26:1<54::aid-gcc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 120.Rozen P, Baratz M. Familial juvenile colonic polyposis with associated colon cancer. Cancer. 1982;49:1500. doi: 10.1002/1097-0142(19820401)49:7<1500::aid-cncr2820490732>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 121.Rozen P, Samuel Z, Brazowski E. A prospective study of the clinical, genetic, screening, and pathologic features of a family with hereditary mixed polyposis syndrome. Am J Gastroenterol. 2003;98:2317. doi: 10.1111/j.1572-0241.2003.07714.x. [DOI] [PubMed] [Google Scholar]
- 122.Ruvalcaba RH, Myhre S, Smith DW. Sotos syndrome with intestinal polyposis and pigmentary changes of the genitalia. Clin Genet. 1980;18:413. doi: 10.1111/j.1399-0004.1980.tb01785.x. [DOI] [PubMed] [Google Scholar]
- 123.Sachatello CR. Polypoid diseases of the gastrointestinal tract. J Ky Med Assoc. 1972;70:540. [PubMed] [Google Scholar]
- 124.Sachatello CR, Hahn IL, Carrington CB. Juvenile gastrointestinal polyposis in a female infant: Report of a case and review of the literature of a recently recognized syndrome. Surgery. 1974;75:107. [PubMed] [Google Scholar]
- 125.Sachatello CR, Pickren JW, Grace JT. Generalized juvenile gastrointestinal polyposis. Gastroenterology. 1970;58:699. [PubMed] [Google Scholar]
- 126.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 127.Sarles JC, Consentino B, Leandri R, et al. Mixed familial polyposis syndromes. Int J Colorectal Dis. 1987;2:96. doi: 10.1007/BF01647700. [DOI] [PubMed] [Google Scholar]
- 128.Sarquis MS, Agrawal S, Shen L, et al. Distinct expression profiles for PTEN transcript and its splice variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Hum Genet. 2006;79:23. doi: 10.1086/504392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sassatelli R, Bertoni G, Serra L, et al. Generalized juvenile polyposis with mixed pattern and gastric cancer. Gastroenterology. 1993;104:910. doi: 10.1016/0016-5085(93)91031-c. [DOI] [PubMed] [Google Scholar]
- 130.Sayed MG, Ahmed AF, Ringold JC, et al. Germline SMAD4 or BMPR1A mutations and phenotype of Juvenile Polyposis. Ann Surg Oncol. 2002;9:901. doi: 10.1007/BF02557528. [DOI] [PubMed] [Google Scholar]
- 131.Scott-Conner CEH, Hausmann M, Hall TJ, et al. Familial juvenile polyposis: Patterns of recurrence and implications for surgical management. Journal of the American College of Surgeons. 1995;181:407. [PubMed] [Google Scholar]
- 132.Scully RE. Sex cord tumor with annular tubules a distinctive ovarian tumor of the Peutz-Jeghers syndrome. Cancer. 1970;25:1107. doi: 10.1002/1097-0142(197005)25:5<1107::aid-cncr2820250516>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 133.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 134.Soares J, Lopes L, Vilas Boas G, et al. Wireless capsule endoscopy for evaluation of phenotypic expression of small-bowel polyps in patients with Peutz-Jeghers syndrome and in symptomatic first-degree relatives. Endoscopy. 2004;36:1060. doi: 10.1055/s-2004-826038. [DOI] [PubMed] [Google Scholar]
- 135.Sogol PB, Sugawara M, Gordon HE, et al. Cowden’s disease: familial goiter and skin hamartomas. A report of three cases. West J Med. 1983;139:324. [PMC free article] [PubMed] [Google Scholar]
- 136.Solh HM, Azoury RS, Najjar SS. Peutz-Jeghers syndrome associated with precocious puberty. J Pediatr. 1983;103:593. doi: 10.1016/s0022-3476(83)80595-2. [DOI] [PubMed] [Google Scholar]
- 137.Sommerhaug RG, Mason T. Peutz-Jeghers syndrome and ureteral polyposis. JAMA. 1970;211:120. [PubMed] [Google Scholar]
- 138.Starink TM, van der Veen JP, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. 1986;29:222. doi: 10.1111/j.1399-0004.1986.tb00816.x. [DOI] [PubMed] [Google Scholar]
- 139.Stemper TJ, Kent TH, Summers RW. Juvenile polyposis and gastrointestinal carcinoma. Annals of Internal Medicine. 1975;83:639. doi: 10.7326/0003-4819-83-5-639. [DOI] [PubMed] [Google Scholar]
- 140.Subramony C, Scott-Conner CEH, Skelton D, et al. Familial juvenile polyposis. Study of a kindred: Evolution of polyps and relationship to gastrointestinal carcinoma. Am J Clin Pathol. 1994;102:91. doi: 10.1093/ajcp/102.1.91. [DOI] [PubMed] [Google Scholar]
- 141.Takeuchi Y, Yoshikawa M, Tsukamoto N, et al. Cronkhite-Canada syndrome with colon cancer, portal thrombosis, high titer of antinuclear antibodies, and membranous glomerulonephritis. J Gastroenterol. 2003;38:791. doi: 10.1007/s00535-002-1148-6. [DOI] [PubMed] [Google Scholar]
- 142.Thomas HJW, Whitelaw SC, Cottrell SE, et al. Genetic mapping of the hereditary mixed polyposis syndrome to chromosome 6q. American Journal of Human Genetics. 1996;58:770. [PMC free article] [PubMed] [Google Scholar]
- 143.Tomlinson I, Rahman N, Frayling I, et al. Inherited susceptibility to colorectal adenomas and carcinomas: evidence for a new predisposition gene on 15q14-q22. Gastroenterology. 1999;116:789. doi: 10.1016/s0016-5085(99)70061-2. [DOI] [PubMed] [Google Scholar]
- 144.Tweedie JH, McCann BG. Peutz-Jeghers syndrome and metastasising colonic adenocarcinoma. Gut. 1984;25:1118. doi: 10.1136/gut.25.10.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Veale AMO, McColl I, Bussey HJR, et al. Juvenile polyposis coli. Journal of Medical Genetics. 1966;3:5. doi: 10.1136/jmg.3.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Volikos E, Robinson J, Aittomaki K, et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet. 2006;43:e18. doi: 10.1136/jmg.2005.039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wada K, Tanaka M, Yamaguchi K. Carcinoma and polyps of the gallbladder associated with Peutz-Jeghers syndrome. Dig Dis Sci. 1987;32:943. doi: 10.1007/BF01296719. [DOI] [PubMed] [Google Scholar]
- 148.Walpole IR, Cullity G. Juvenile polyposis: A case with early presentation and death attributable to adenocarcinoma of the pancreas. American Journal of Medical Genetics. 1989;32:1. doi: 10.1002/ajmg.1320320102. [DOI] [PubMed] [Google Scholar]
- 149.Walton BJ, Morain WD, Baughman RD, et al. Cowden’s disease: a further indication for prophylactic mastectomy. Surgery. 1986;99:82. [PubMed] [Google Scholar]
- 150.Watanabe A, Nagashima H, Motoi M, et al. Familial juvenile polyposis of the stomach. Gastroenterology. 1979;77:148. [PubMed] [Google Scholar]
- 151.Westerman AM, Wilson JH. Peutz-Jeghers syndrome: risks of a hereditary condition. Scand J Gastroenterol Suppl. 1999;230:64. doi: 10.1080/003655299750025561. [DOI] [PubMed] [Google Scholar]
- 152.Whitelaw SC, Murday VA, Tomlinson IPM, et al. Clinical and molecular features of the hereditary mixed polyposis syndrome. Gastroenterology. 1997;112:327. doi: 10.1053/gast.1997.v112.pm9024286. [DOI] [PubMed] [Google Scholar]
- 153.Williard W, Borgen P, Bol R, et al. Cowden’s disease. A case report with analyses at the molecular level. Cancer. 1992;69:2969. doi: 10.1002/1097-0142(19920615)69:12<2969::aid-cncr2820691217>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 154.Wilson DM, Pitts WC, Hintz RL, et al. Testicular tumors with Peutz-Jeghers syndrome. Cancer. 1986;57:2238. doi: 10.1002/1097-0142(19860601)57:11<2238::aid-cncr2820571128>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 155.Yashiro M, Kobayashi H, Kubo N, et al. Cronkhite-Canada syndrome containing colon cancer and serrated adenoma lesions. Digestion. 2004;69:57. doi: 10.1159/000076560. [DOI] [PubMed] [Google Scholar]
- 156.Yoshida T, Haraguchi Y, Tanaka A, et al. A case of generalized juvenile gastrointestinal polyposis associated with gastric carcinoma. Endoscopy. 1988;20:33. doi: 10.1055/s-2007-1018122. [DOI] [PubMed] [Google Scholar]
- 157.Young RH, Welch WR, Dickersin GR, et al. Ovarian sex cord tumor with annular tubules: review of 74 cases including 27 with Peutz-Jeghers syndrome and four with adenoma malignum of the cervix. Cancer. 1982;50:1384. doi: 10.1002/1097-0142(19821001)50:7<1384::aid-cncr2820500726>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 158.Young S, Gooneratne S, Straus FH, 2nd, et al. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol. 1995;19:50. doi: 10.1097/00000478-199501000-00007. [DOI] [PubMed] [Google Scholar]
- 159.Zhou XP, Waite KA, Pilarski R, et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet. 2003;73:404. doi: 10.1086/377109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou XP, Woodford-Richens K, Lehtonen R, et al. Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. American Journal of Human Genetics. 2001;69:704. doi: 10.1086/323703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zigman AF, Lavine JE, Jones MC, et al. Localization of the Bannayan-Riley-Ruvalcaba syndrome gene to chromosome 10q23. Gastroenterology. 1997;113:1433. doi: 10.1053/gast.1997.v113.pm9352843. [DOI] [PubMed] [Google Scholar]