

Abstract
We review evidence that sterols can form stoichiometric complexes with certain bilayer phospholipids, and sphingomyelin in particular. These complexes appear to be the basis for the formation of condensed and ordered liquid phases, (micro)domains and/or rafts in both artificial and biological membranes. The sterol content of a membrane can exceed the complexing capacity of its phospholipids. The excess, uncomplexed membrane sterol molecules have a relatively high escape tendency, also referred to as fugacity or chemical activity (and, here, simply activity). Cholesterol is also activated when certain membrane intercalating amphipaths displace it from the phospholipid complexes. Active cholesterol projects from the bilayer and is therefore highly susceptible to attack by cholesterol oxidase. Similarly, active cholesterol rapidly exits the plasma membrane to extracellular acceptors such as cyclodextrin and high-density lipoproteins. For the same reason, the pool of cholesterol in the ER (endoplasmic reticulum) increases sharply when cell surface cholesterol is incremented above the physiological set-point; i.e., equivalence with the complexing phospholipids. As a result, the escape tendency of the excess cholesterol not only returns the plasma membrane bilayer to its set point but also serves as a feedback signal to intracellular homeostatic elements to down-regulate cholesterol accretion.
1. Introduction
Sterols are universal membrane constituents in eukaryotes. Nevertheless, they are not essential to membrane integrity, being rare among prokaryotes and scant in several of the eukaryotic organelles. Rather, they confer important properties on the plasma membrane and the endomembrane organelles with which the plasma membrane is in intermittent continuity [1,2]. Through their interactions with phospholipids and sphingolipids, sterols lower bilayer permeability by reducing the free volume created by the thermal motion of the fatty-acyl chains. For similar reasons, sterols condense bilayers, reduce their fluidity, compressibility and compliance and increase their mechanical strength [3]. As described in Section 2, sterols can enter into stoichiometric complexes with certain membrane phospholipids. These can drive the formation of functionally important bilayer phases, referred to as raft domains [1]. Sterols free of these phospholipid associations have a relatively high tendency to project from the bilayer (escape tendency or activity); this will be discussed in Section 3. Other properties of bilayer sterols in or free of phospholipid complexes will be taken up thereafter.
An early indication that there are two states of cholesterol activity in bilayers (i.e., low and high escape tendency) was the conditionality of the susceptibility of membrane cholesterol to cholesterol oxidase attack. In particular, cholesterol in the outer leaflet of the red blood cell membrane was found to be resistant to this enzyme, while the cytoplasmic leaflet was quite sensitive [4]. It was then shown that this dichotomy corresponded to the asymmetrical composition of the membrane; that is, synthetic vesicles rich in endofacial lipids (namely, unsaturated phosphatidylethanolamines and phosphatidylserines) fostered the oxidation of cholesterol while the exofacial lipids, the more saturated sphingomyelins and phosphatidylcholines, did not [5]. Corresponding differences were later demonstrated for the rate and extent of transfer of cholesterol from synthetic vesicles; that is, sterol efflux was retarded by saturated phospholipid chains, phosphocholine head groups and, most of all, sphingolipids [6,7].
It was also found early on that the susceptibility of the membranes of intact red cells to cholesterol oxidase rose abruptly--from negligible to total—when the cholesterol content was incremented from just below to just above its physiologic level (Figure 1). (Why a slight enrichment caused all of the sterol to become a substrate was a mystery then but is explained below.) Such observations lead to the working hypothesis that underlies this review: Membrane cholesterol forms complexes with certain bilayer phospholipids. These complexes hold the cholesterol in a state of low escape tendency. Cholesterol oxidase acts preferentially on the uncomplexed cholesterol with a high escape tendency. For the same reason, bilayer cholesterol that is not complexed is more readily transferred to acceptors.
Figure 1.

Dependence of the susceptibility of cholesterol to cholesterol oxidase on red cell membrane cholesterol content near its resting level (▲). Replotted from reference 8.
2. Interactions of sterols with phospholipids
The fundamental properties of the phospholipids in bilayers are well understood [9], and their interactions with sterols have been extensively characterized [2,10]. The cholesterol molecule has a rigid, planar steroid nucleus comprised of four fused rings plus a terminal, flexible iso-octyl tail (Figure 2). The sterol molecule aligns roughly parallel to its phospholipid neighbors in bilayers. Its 3-β-hydroxyl group resides in the vicinity of their fatty-acyl carbonyl groups near the aqueous interface. Sterols are not ideal solutes dissolved in the hydrophobic core of the bilayer. Rather, they associate with the more polar lipids with varied strengths of interaction [11]. What matters most in these associations is the length of the non-polar chains of the polar lipids, the (un)saturation of their chains (i.e., the number, position and orientation [cis versus trans] of their double bonds), and the size and structure of their polar head groups [10]). The smooth α-face of the sterol nucleus makes favorable van der Waals contacts with the saturated fatty-acyl chains of phospholipids down to about their tenth methylene group. Phospholipids with phosphocholine head groups interact more strongly with sterols than those with smaller head groups such as phosphatidylethanolamine and phosphatidylserine. Sterols prefer sphingomyelin to phosphatidylcholine; since these two classes of phospholipid have identical phosphocholine head groups, the difference is attributable to the longer and more saturated fatty-acyl tails of sphingolipids as well as to hydrogen bonding between the hydroxyl moiety of the sterol and the polar atoms in the lipid head group.
Figure 2.
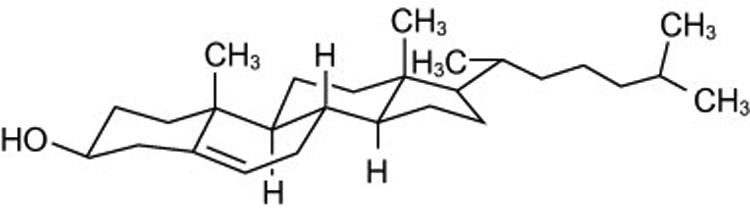
The chemical structure of cholesterol. Note that the α-face of the nucleus (facing down) is "smooth" while the β-face (facing up) is made "rough" by the projection of methyl groups from carbons 10 and 13. Courtesy of Mark E. Duban.
By themselves, phospholipids bearing two long and saturated fatty-acyl substituents typically form a close-packed, ordered (solid or gel) phase below their melting temperature. Their chains are well oriented toward the central plane of the membrane. Interaction with sterols reorganizes such bilayers into a distinctive liquid-ordered phase. In this state, the fatty-acyl chains remain aligned and extended parallel to the sterol nucleus, but the phospholipid molecules now have the high rotational and diffusional mobilities seen in fluid bilayers. Sterol associations are optimal for phospholipids bearing fatty acids of around 17 methylene units (i.e., spanning the length of the extended sterol molecule) and are less pronounced for longer and shorter chains as well as for unsaturated chains [10,12]. At the same time, the irregular β-face of the sterol nucleus complements and constrains the otherwise disordered cis-unsaturated fatty-acyl chains abundant in biological membranes. Opposing these ordering influences are the vacancies that sterols impose near the bilayer center; these arise because the length of the nucleus of the sterols is less than that of the neighboring phospholipid tails. Furthermore, their iso-octyl chains are highly mobile. The net outcome is that sterols confer an intermediate fluidity upon biological membranes [1].
2.1 Sterols condense and order phospholipids
Membranes composed of phospholipids bearing two saturated chains take up sterols more readily than their unsaturated counterparts. An exothermic enthalpy change drives these interactions, estimated in one study to be ~9 Kcal/mole phospholipid [13]. The phospholipid molecules are condensed by their association with the sterols; that is, the net volume of membranes composed of these mixtures is less than that of its separate components [14–17].
This is because the nucleus of sterol molecules constrains trans-gauche isomerizations of the carbon-carbon bonds in the otherwise fluid fatty-acyl chains of the adjacent phospholipids. This chain stiffening causes the bilayer to contract laterally and to thicken. Sterols also can straighten the tilt angle of the phospholipids, serving to further reduce the overall molecular cross section of the membrane. Concomitantly, the bilayer becomes mechanically stronger and less permeable to water and polar solutes [1].
2.2. Phase behavior of sterol-phospholipid mixtures
Homogeneous phospholipid bilayers undergo melting transitions from a well-ordered solid to a disordered liquid phase at a characteristic temperature and pressure [17,18]. Mixtures of two phospholipids can similarly undergo demixing into solid and liquid phases rich in the high melting and low melting constituents at a characteristic temperature, pressure and composition [14,18,19]. Every constituent in the bilayer will then partition between these phases according to their relative affinity for each [20]. The coexisting phases will coalesce into a homogeneous mixture at a high enough temperature and/or pressure.
Under certain conditions, the addition of sterols can drive the separation of phospholipid mixtures into two phases, just as described above. One of the phases will be enriched in the sterol plus the more strongly interacting phospholipids. Typically, these are the phospholipids with large head groups and saturated fatty-acyl chains of intermediate length; phosphatidylcholines and, especially, sphingolipids [1,18,19,21–24]. This sterol-rich phase will be the more condensed and ordered, while the excluded bilayer will be a disordered liquid. Through the use of an intercalated order-sensing dye, phase separation in sterol-containing phospholipid membranes can be visualized in the fluorescence microscope [18]. The minor phase typically appears as dots drifting randomly through the membrane continuum (i.e., the dominant phase that maintains membrane continuity). The circular contours of the minor domains and their ability to diffuse and coalesce show that both the islands and the continuum that surrounds them are fluids.
Immiscibility is more readily demonstrated in monolayers than in bilayers of a comparable composition, at least in part because the low surface pressures attainable in the former promote phase separation [25]. Nevertheless, monolayer and bilayer systems are in good qualitative agreement with respect to the ability of sterols to drive phase separation [26]. In contrast, cholesterol-dependent phase behavior has been difficult to demonstrate directly in biological membranes under physiological conditions. Domains may be present but too small to visualize by ordinary fluorescence microscopy. This hypothesis is supported by simulations using synthetic bilayer vesicles [27]. Small rafts might signify that the interfacial energy (line tension) driving phase separation is quite low [22], perhaps because of the vast heterogeneity of the hundreds of different lipid species in a typical plasma membrane as well as interference from integral proteins [28,29]. Micro-domains in natural membranes might also be composed of specific protein assemblies with sterols and phospholipids [30].
Despite a wealth of valuable data, the structure, composition, size, heterogeneity and functions of biological rafts remain open questions [24,31,32]. Low-density fragments of plasma membranes have been isolated from detergent dispersions or mechanical homogenates by flotation in sucrose density gradients. These are characteristically enriched in cholesterol and, to a lesser degree, sphingolipids. However, their composition varies with the mode of preparation (and particularly with diverse detergent treatments), so that the original cellular form of such domains is uncertain [31]. Nevertheless, various membrane signaling protein activities are cholesterol dependent in situ [33–35]. Furthermore, detergent-resistant membrane fragments replete in cholesterol are, in fact, enriched in proteins assigned to rafts on the basis of their behavior in the parent cells [36]. The cholesterol-dependence of membranous structures such as caveolae and of processes like endocytosis suggest strongly that the sterol plays an important role in the lateral organization of membranes in vivo [37,38].
2.3. Stoichiometric complexes of sterols and phospholipids
That sterols associate with phospholipids and thereby condense, order and otherwise alter bilayers has been appreciated for decades [14,17]. However, the formation of well-defined stoichiometric complexes of sterols and phospholipids was only recently postulated [39]. The argument for this “stoichiometric complex model” is as follows: Monolayers of saturated phospholipids like dimyristoyl- and dipalmitoylphosphatidylcholine exist in homogeneous liquid-disordered phases at low surface pressures and temperatures above their melting points. This homogeneity gives way to the appearance of two coexisting phases as cholesterol is added [13,40]. (Phase separation is reported by the partition of an intercalated fluorescent indicator in such studies [18].) That the contours of the discrete domains are round suggests that both of the phases are liquids, and the distribution of the reporter dye suggests that the minor phase that grows with cholesterol content (i.e., the cholesterol-rich phase) is more ordered than the bulk continuum.
A phase diagram depicting the interaction of a phospholipid monolayer with cholesterol (represented by its non-oxidizable counterpart, dihydrocholesterol) is presented in the left panel of Figure 3. The data points mark the critical surface pressures at which multiple phases merge into a continuum, as judged by fluorescence microscopy. Under the chosen conditions, the phase behavior driven by the sterol has an unexpectedly complex concentration dependence. The resistance of two-phase immiscibility to increasing surface pressures increases with sterol content up to a mole fraction of approximately 0.33. Raising the sterol level in the monolayer then weakens the phase partition until phase separation almost disappears at a sterol mole fraction of ~0.4 (vertical arrow in Figure 3). Surprisingly, as cholesterol levels are increased beyond this trough (cusp), a two-phase, liquid-liquid immiscibility pattern reemerges. However, at these high sterol levels, the partition of the indicator dye is reversed: the discrete domains are now more liquid than the continuum.
Figure 3.

Phase behavior of monolayers of dimyristoylphosphatidylcholine and (dihydro)cholesterol at 13 °C. Left panel: the points represent the transition pressures at which the two coexisting liquid phases become a single phase. (Squares are data using cholesterol; circles are data using dihydrocholesterol. The filled symbols denote striped patterns often seen close to phase transitions while the open symbols simply show the disappearance of phase heterogeneity.) Right panel: the points are the rate constants for the transfer of dihydrocholesterol from a single-phase monolayer to β-cyclodextrin at a surface pressure of 20 mN/m, calculated from the area loss over time. Dihydrocholesterol is used as a good substitute for cholesterol in such studies to minimize sterol oxidation. From reference 13.
These findings are interpreted to signify that when the phospholipid is in excess, early on, most of the sterol molecules associate with the phospholipids to form complexes [13]. Because the complexes are sparingly miscible with the liquid-disordered phospholipid continuum, they separate as more ordered liquid phases. At some characteristic intermediate sterol content (to be interpreted shortly), most of the phospholipid molecules and most of the sterol molecules are associated with one another so that, at the cusp in the curve, the film is predominantly composed of complexes with only small amounts of each uncomplexed lipid species dissolved therein. Consequently, immiscibility is low and a single phase dominates. Additional sterol exceeds the capacity of the phospholipids in the monolayer to form complexes, so that the excess then forms a more ordered sterol-rich continuum in which the complexes are dispersed as the more disordered minor phase. This makes sense because monolayers of cholesterol are typically condensed solids even at low surface pressures.
According to this interpretation, as the sterol is increased, the monolayer passes from a mixed liquid composed of a minor phase rich in complexes dispersed in a phospholipid-rich continuum to a system in which the minor phase is made of complexes and the continuum is composed of excess sterol [39]. In other words, the putative phospholipid-sterol complexes are poorly soluble in both the phospholipid-rich and the sterol-rich phases that dominate at the extremes of composition. The cusp marked by the arrow in Figure 3 then represents the transition point at which the complexes have reached a maximum and both free sterol and free phospholipids are at a minimum. It follows that the sterol/phospholipid mole ratio at the cusp point gives the stoichiometry of the complexes. To emphasize the ordering of the associations of the sterols and phospholipids, these assemblies have been called "condensed complexes" [40]. However, we shall refer to them as stoichiometric complexes to highlight that feature. We do not expect to see a cusp point in vesicles, as we do in monolayers, since sterol molecules by themselves cannot form stable bilayers.
It has been shown that phosphatidylcholine, phosphatidylserine, phosphatidylethanolamine and sphingomyelin all form such sterol complexes when their fatty-acyl chains are of appropriate length and saturation [41]. The mole fraction of cholesterol at the cusp point for various phospholipid types is in the range of 25–50 mole percent, indicating that the phospholipid:sterol mole ratios in these complexes range from 3:1 to 1:1. For example, the apparent stoichiometry in complexes of sphingomyelins bearing various fatty amide chains is two per cholesterol [42]. The net stoichiometry of a complex need not be an integer nor a small number nor does that number reveal the size of the complexes. Rather, the complexes may be cooperatively-assembled, short-lived (microseconds to milliseconds) oligomers of varied size and proportions [39]. For example, dimyristoylphosphatidylcholine-cholesterol complexes were estimated to have a stoichiometry of 3:2 based on a cusp point at ~40% sterol (Figure 3, left panel) [13]. A molecular dynamics simulation of this mixture suggested the presence of roughly comparable amounts of 1:1 and 2:1 complexes, matching the net stoichiometry observed experimentally [43]. In another study, complexes of two sphingolipids and one sterol molecule appeared to cluster into larger units, each containing 3–5 of such complexes; this would suggest 9–15 lipid molecules per cluster [42]. It has also been shown that various unnatural mixtures of dimethylphosphatidylethanolamine, myristic acid and lysophosphatidylethanolamine can mimic the interaction of phosphatidylethanolamine with sterols. That is, they formed monolayers with phase diagrams that resembled Figure 3, manifesting two critical point maxima separated by a cusp point that corresponded to a stoichiometry of 4.0–4.6 acyl chains per cholesterol [44].
Similar to their effect on monolayers, sterols also promote the formation of large liquid-ordered domains in giant unilamellar bilayer vesicles containing a high melting and a low melting phospholipid species [18]. This behavior has been accurately modeled in terms of the thermodynamics of formation of condensed complexes [45]. Molecular dynamics simulations also support the concept of association of sterols with phospholipids [43,46,47]. Furthermore, such modeling suggests that these interactions can extend laterally beyond the complexes themselves, thereby ordering adjacent phospholipids over several molecular diameters [48]. An implication is that cholesterol need not associate with every phospholipid molecule in the plasma membrane in order to influence bilayer attributes such as passive permeability to solutes.
2.4. Sterol complexes are the basis of liquid-ordered domains
We accept the premise that sterols interact preferentially with phospholipids bearing saturated chains and large polar head groups to form stoichiometric complexes that are poorly miscible with the liquid-disordered continuum of non-interacting bilayer lipids [14,39]. If partially immiscible with the bulk phase, such complexes will form condensed and ordered liquid phases. Raising the temperature or the surface pressure of two-phase phospholipid-cholesterol mixtures abolishes this phase separation at a critical point, as illustrated by the data in the left panel of Figure 3. However, conditions that impose miscibility do not necessarily dissociate the complexes; instead, they may simply promote their dissolution in the continuum [13,14,26]. Thus, the absence of visible phase separation (raft formation) in biological membranes does not militate against the existence of sterol-phospholipid complexes.
Some of the sterols that promote liquid-ordered phases in vitro are cholesterol, ergosterol, epicholesterol, 25-hydroxycholesterol and dihydrocholesterol. Sterols deficient in this capacity include lanosterol, androstenolone, coprostanol, cholestane and cholest-4-en-3-one (i.e., cholestenone) [26,49,50]. There is evidence that sterols that fail to promote liquid-liquid phase separation nevertheless form phospholipid complexes that are presumably miscible with the bilayer continuum (Ratajczak, M.K., Steck, T.L., Lee, K-Y. C. and Lange, Y., unpublished data).
3. Membrane sterol escape tendency (activity)
A given molecular species in a lipid monolayer diffuses in-plane and partitions according to its relative affinity for different lateral phases. Similarly, a bilayer constituent has a certain potential to flip from one leaflet to the other as well as to redistribute to acceptors in the adjacent aqueous compartment. Every molecule in each phase of the two bilayer leaflets will thus have its own tendency (fugacity) to partition laterally, distribute transversely and transfer outwardly. We are interested here in the last of these three propensities: the tendency of cholesterol to escape from the surface of a bilayer. We take this potential to be a reflection of the strength of its interactions with its local environment (see Section 2.4 in ref. [39]). For ease of expression, we shall refer to the escape tendency of a membrane sterol as its activity. For a rigorous treatment of principles, see ref. [51].
We have a mental picture of the escape tendency (activity) of bilayer lipids such as sterols. In particular, molecules are driven by thermal energy to project sporadically from the membrane surface into the aqueous phase [52–54]. This bobbing motion will reflect a balance, among other factors, of its entropy-driven tendency to diffuse away, the weak associations holding it within the membrane, the hydrophobic exclusion of its non-polar surface from water as well as the hydration of its polar atoms. One can imagine that each molecular species has its own dynamic equilibrium distribution of flickering projections, characterized by the frequency and extent of its partial excursions. The activity of a sterol species at a membrane surface will therefore manifest its profile of projection into the aqueous phase. One way to interpret the poor interactions of membrane sterols at sub-threshold concentrations with cholesterol oxidase (Figure 1) and cyclodextrin (Figure 3, right panel) would be that they are restrained by complexation with the phospholipids and would therefore have low bobbing activity compared to the subset of molecules that are free of such associations in the liquid-disordered phase.
3.1. The source of high-activity cholesterol in bilayers
Radhakrishnan and McConnell provided experimental evidence that uncomplexed sterol molecules--namely, those in excess of the association capacity of the bilayer phospholipids-- have a relatively high chemical activity (fugacity) [39]. Their quantitative thermodynamic modeling gave additional support to the argument that the chemical activity of the cholesterol in stoichiometric complexes is low compared to that of the free sterol [45]. In our view, this is because uncomplexed sterol molecules have a higher profile of projection into the aqueous compartment.
Huang, Buboltz and Feigenson (like Radhakrishnan and McConnell) also observed an abrupt change in the behavior of the cholesterol in phospholipid vesicles as its abundance was increased to high levels [55]. In particular, they demonstrated defined maxima for the solubility of cholesterol in bilayers of phosphatidylcholine, beyond which microcrystals of the sterol grew over a period of several days. They interpreted this behavior to signify that phospholipid head groups can shield sterol molecules from contact with the aqueous phase. That is, the phosphocholine moiety can provide a molecular umbrella that protects the hydrophobic edge of the sterol nucleus from an energetically unfavorable exposure to water. Sterol molecules in excess of these protective phospholipid head groups will be driven by the hydrophobic effect to cluster in a lower energy state: microcrystals. During the many hours required for this segregation process, the excess sterol would be metastable and have a high escape tendency. As observed, bilayers composed of phospholipids with smaller head groups (e.g., phosphatidylethanolamine) will have a lower capacity for sterols, presumably because they provide less of a shield. The umbrella hypothesis therefore offers a mechanistic explanation for the observed limits on the solubility of sterols in phospholipid bilayers and the consequent high chemical activity of their excess [56].
The umbrella mechanism is, in essence, physical rather than chemical. That is, it postulates no associations between the sterol and phospholipid molecules. Rather, it is the hydrophobic effect acting upon unshielded sterols that drives their activity and, ultimately, crystallization. Monte Carlo simulations not only support the proposed general mechanism of sterol solubility in bilayers and the precise solubility limits found for various phospholipids but also offer an explanation of the observed membrane condensation and reduction of bilayer permeability by sterols [56]. Furthermore, sterol molecules might assume a regular spacing from one another so as to maximize their protection by phospholipid head groups and thereby minimize the free energy penalty incurred by their exposure to water [57]. The umbrella mechanism is also consistent with the lack of structural specificity observed for a wide variety of compounds that displace cholesterol from phospholipids (an issue discussed in Section 5). For example, the length of the fatty-acyl chains of the phospholipids does not appear to affect the capacity of the membrane to solubilize cholesterol [55]. While some studies have not revealed microcrystals in bilayers containing high fractions of cholesterol, sufficient time may not have been allowed for their formation.
Nevertheless, the balance of current evidence may tilt toward the stoichiometric complex model; i.e., weak chemical interactions between sterols and phospholipids. For one thing, this model but not the head-group umbrella accounts for behavior such as documented in the left panel of Figure 3 and for the molecular basis of liquid-ordered domains. Observed fatty-acyl chain length effects and the favorable enthalpy of association for sterol-phospholipid complex formation also implicate van der Waals interactions among nonpolar moieties rather than a hydrophobic driving force [13,41]. Furthermore, the detergent-resistance of rafts suggests that these lipid partners form persistent (albeit, weak) non-covalent associations unlikely to be stabilized simply by the umbrella effect.
However, the stoichiometric complex mechanism and the umbrella mechanism are not mutually exclusive. Rather, much of the data are consistent with both models. For example, both formulations account for the poor attack of cholesterol oxidase on mixed bilayers containing phospholipids with head groups that would shield sterols [5,57]. Furthermore, each mechanism could drive the condensation of phospholipid-sterol mixtures [58]. Both models serve the present argument by predicting an equivalence point in activity when phospholipids are titrated with sterols as well as the high activity of the excess free sterol monomers beyond this point.
4. Experimental evidence for high-activity cholesterol
While sterols prefer to form liquid-ordered complexes with propitious phospholipids, they will also populate liquid-disordered phases, driven there by weakly positive interactions with those phospholipids or, entropically, because the phospholipids capable of strong associations are occupied [59]. The free molecules are less constrained than their complexed counterparts and should therefore have a high (bobbing) activity. Evidence supporting this premise is summarized below.
4.1. Kinetics of cholesterol exit from membranes
The rate of transfer of a sterol from a membrane to an acceptor has been used as a reasonable indicator of its escape tendency where the back reaction and other variables are constant or immaterial. In particular, both the rate of transfer and the equilibrium partition to the acceptor should increase acutely as the sterol is titrated past its equivalence point with the donor phospholipid. The fraction of cholesterol in the donor compartment in most transfer studies has been kept at or below this equivalence point, so that the predicted transition to high cholesterol activity was not in evidence. However, where transfer rates have been studied as a function of cholesterol concentration in the critical range, the anticipated sharp transition has been observed both in monolayers and red cell membranes (see the right panel of Figure 3 and references 54, 59 and 60).
The escape tendency (activity) concept predicts that sterol derivatives of higher polarity than cholesterol will exit monolayers and membranes that much faster. This has been observed [60,61]. Also, as predicted, the rate and extent of transfer from various vesicles and monolayers vary inversely with the known strength of association of sterols with phospholipids of different acyl-chain saturation and polar head group size [1,6,10,62,63]. In particular, sphingomyelins retard cholesterol exit to cyclodextrin most strongly, while phospholipids with mismatched fatty-acyl chains (favoring formation of liquid-disordered phases) favor its transfer [60,64]. Introducing lysophosphatidylcholine into red cell membranes reduces the rate of cholesterol exit [54]; presumably, the extra membrane phospholipid molecules complex free cholesterol and thereby lower its activity.
It has been proposed that sterol transfer is mediated by its spontaneous desorption into the aqueous medium followed by its collisional capture by an acceptor and the diffusion of this soluble complex from the unstirred aqueous layer surrounding the membrane [7,65,66]. An alternative mechanism is that sterols constantly project partially (bob) from the bilayer but rarely escape; rather, acceptors capture them during favorable collisions with the cell surface [52,53,67]. Given that the energy of activation for the transfer process, typically >20 Kcal/mole, is roughly that expected for nonpolar molecules containing 25 or more carbon atoms, cholesterol molecules are presumably captured by the acceptor when they project almost entirely into the aqueous phase. The salient point here is that sterol excursion, whether by intermittent bobbing or complete escape, is the critical, activity-driven first step in the transfer process.
The half-time of cholesterol transfer from biological membranes ranges from seconds to several hours, depending on the nature of the donors and acceptors [6,52,62,65,67,68]. An analysis by Haynes et al. of [3H]cholesterol transfer from a CHO cell line to cyclodextrin suggested that there were two kinetic pools of plasma membrane cholesterol in addition to a small, non-transferable (presumably intracellular) compartment [69]. It is possible that the minor plasma membrane pool (~25%) that exited with a half-time of ~15 seconds was uncomplexed, active and located in a liquid-disordered environment. The major, slow pool (~75%; half-time ~21 min) might then correspond to sterol molecules sequestered in phospholipid complexes. Furthermore, the slow pool replenished the fast pool with a half-time of ~25 min, suggesting that it may have exited the membrane mostly by way of the fast compartment. The fast pool also appeared to expand at the expense of the slow pool when cells were manipulated in ways that might have increased the uncomplexed (active) sterol population. That is, treating cells with sphingomyelinase or phospholipase C, cell disruption, the isolation of the plasma membrane as fragments, and treating cells so as to scramble the asymmetry of their plasma membrane bilayer all increased the relative size of the rapidly-exiting pool [69]. The last of these variables presumably involved redistributing low-affinity cytoplasmic-surface phospholipids to the exofacial leaflet (see Section 4.3).
4.2. Cholesterol oxidase susceptibility of membrane cholesterol
It appears that the rate of attack by the enzyme, cholesterol oxidase, can serve as an indicator of the activity of membrane cholesterol. It is well established that the susceptibility of sterols to cholesterol oxidase is high in mild detergent solutions and in homogeneous monolayers but is limited and even undetectable in certain phospholipid mixtures and in biological membranes [5,57,70–73]. A simple interpretation of these phenomena is that the complexation of sterols by phospholipids reduces their activity compared to that of the free sterol in liquid-disordered phases. That is, cholesterol oxidase readily accesses membrane sterol molecules with a high bobbing profile. Structural studies have shown that this enzyme associates with the surface of the bilayer rather than penetrating into its nonpolar core. Projecting sterol molecules can just reach the catalytic cavity of the periphally-disposed enzyme [74,75]. The catalytic rate of cholesterol oxidase can therefore report on the constraints imposed by various phospholipids on the escape tendency (bobbing activity) of the sterols [57]. Similarly, the divergent efficacy of cholesterol oxidases from different species could reflect subtle variations in the way they access the substrate as it transiently extends from the plasma membrane [76].
This hypothesis is supported by evidence that cholesterol is a better substrate for the oxidase when not membrane-bound and when in membranes made of phosphatidylserine than of phosphatidylcholine or sphingomyelin [5]. Likewise, we can understand why saturated phospholipids reduce cholesterol oxidation rates compared to their unsaturated counterparts, and sphingomyelin constrains oxidation more than phosphatidylcholine [77]. In particular, various phosphatidylcholine species protect cholesterol from the oxidase best when the lengths of their chains are matched to that of the sterol; namely, 14–17 methylene units [78]. Likewise, treating red cells with lysophosphatidylcholine inhibits the oxidation of their membrane cholesterol [79]; presumably, the extra membrane phospholipid complexes with (or shields) free cholesterol, thereby reducing its accessibility to the probe. For the same reason, fibroblast cholesterol can be made a better substrate by treating the cells with sphingomyelinase [73]. There are a variety of other treatments that promote cholesterol oxidase attack but cannot now be rigorously interpreted; they too could report on cholesterol bobbing activity [79]. One of these treatments is exposure to very low ionic strength buffers; perhaps charge repulsions reduce close-packing in the negatively-charged bilayer and thereby increase the quotient of active cholesterol [80].
In both artificial bilayers and intact cells cholesterol oxidase susceptibility rises acutely when the mole fraction of cholesterol is increased beyond a defined threshold (Figure 1) [8,54,70,79]. This makes sense if sterols in excess of the phospholipid complexation capacity have a high activity. Furthermore, these findings suggest that plasma membrane cholesterol naturally matches the capacity of the phospholipids, presumably driven there by homeostatic mechanisms that sense excess cholesterol from its escape tendency (see Section 6). In that the cholesterol in resting plasma membranes is refractory to cholesterol oxidase, the pool of uncomplexed sterol in equilibrium with the complexes would appear to be normally very small--and the bobbing profile of the complexed cholesterol quite low. Nevertheless, the ready displacement of cholesterol from complexes by small amphipaths such as 1-octanol suggests that the strength of its association with phospholipids might not be strong after all (see Section 5.3).
Why do small increments in cholesterol above the threshold lead to oxidation of the entire cholesterol pool rather than just that increment? The answer probably comes from the observation that the enzyme reaction product, cholestenone, remains in the membrane and itself promotes the oxidation of cholesterol. The product would therefore feed back positively on the reaction, as is also suggested by the oxidation time course which accelerates all the way to completion [70,79]. Thus, the all-or-none action of cholesterol oxidase seems to involve the ability of the reaction product, cholestenone, to displace cholesterol from its association with phospholipids, thereby promoting its activity and enzyme susceptibility (Section 5.3) [54,70,75,81].
4.3. Cholesterol activity in the two bilayer leaflets
Cellular membrane bilayers have a characteristic transverse asymmetry in the distribution of their phospholipids. In particular, the outer leaflet of the plasma membrane is rich in phosphatidylcholine and sphingolipids, while the leaflet facing the cytoplasm is rich in phosphatidylethanolamine, phosphatidylserine and other, mostly anionic, phospholipids [82]. An asymmetry in the unsaturation of their fatty-acyl chains runs parallel: the lipids enriched in the outer surface have an abundance of saturated chains, while the inner surface species are characteristically poly-unsaturated [83]. Given that the phospholipids characteristic of the exofacial surface associate with sterols several times more strongly than do those enriched in the endofacial leaflet [10,20,84], we might expect that the mole fraction of the sterol will be significantly higher in the outer than the inner leaflet of a given plasma membrane bilayer. While several studies have pursued this issue, it remains unresolved.
The tendency of plasma membrane cholesterol to escape to an aqueous acceptor will not necessarily be the same at the two membrane surfaces, even though cholesterol is in rapid diffusional equilibrium between the leaflets [52,67]. Similarly, the sterol in lateral domains at equilibrium in the same leaflet can show different escape tendencies to acceptors – see Section 3.0 and the discussion of Haynes et al. in Section 4.1. This is because the fugacities setting the sterol equilibrium distribution between the bilayer leaflets or between phases in a given leaflet are not the same as that determining the escape tendency (projection profile) of sterol molecules at the aqueous interface.
These considerations lead to the prediction that the sterol activity in the outer leaflet of the plasma membrane will be significantly lower than on the cytoplasmic side. This hypothesis has been tested by determining whether scrambling the transverse distribution of the phospholipids in the plasma membrane alters the activity of the cholesterol in the outer leaflet. When the phospholipids of the two leaflets of plasma membrane bilayers were allowed to equilibrate by activating the membrane scramblase through an elevation in cytoplasmic Ca++, the susceptibility of cholesterol to cholesterol oxidase and its rate of capture by extracellular cyclodextrin both increased [69,85]. A plausible inference is that the movement of the less avid inner-leaflet phospholipids to the outer leaflet reduced its sterol complexation and thereby raised the activity of its cholesterol.
5. Cholesterol mimics
5.1. Sterol specificity
Konrad Bloch argued that cholesterol in animal cells and ergosterol in fungi are best suited among sterol species to serve the needs of the plasma membrane bilayer [1,86,87]. His criteria were principally functional; in particular, their ability to decrease membrane permeability and fluidity. We have already noted that these physical properties correlate with the ability of sterols to form condensed, liquid-ordered phases with phosphocholine-bearing phospholipids. In comparison, Bloch considered lanosterol, the first sterol in the biosynthetic pathway, to be stereochemically ill-suited for plasma membrane service. Bloch saw evolutionary advances in the intermediates along the biosynthetic pathway between lanosterol and cholesterol. The "progressive improvement" in "membrane fitness" highlighted by Bloch involved the "streamlining" of the silhouette of the sterol nucleus through the elimination of protruding methyl groups.
The suitability of a variety of sterols and steroids to promote condensed liquid-disordered phases has recently been compared using monolayer films and bilayer vesicles. The data are generally in line with Bloch’s hypothesis. They show that, compared to cholesterol, androstenolone, coprostanol, cholestane and cholestenone have weak or even negative effects on domain formation [26,49,50,88]. The same is true for lanosterol and succeeding sterol intermediates: dihydrolanosterol, desmosterol, zymosterol, and zymostenol [89]. That lanosterol in particular is inferior to cholesterol in many respects has been carefully documented [87]. Furthermore, several cholesterol analogues were shown to fail as cholesterol substitutes in sustaining the growth of cultured CHO cells [90]. Nevertheless, the issue of the uniqueness of cholesterol has recently become more complicated.
While the studies just mentioned document the particular ability of cholesterol to promote the formation of condensed liquid-ordered phases, these publications also provide evidence that various sterols (biosynthetic intermediates and otherwise) are as good as and sometimes better than cholesterol at building rafts [26,49,50,88,89,91]. In this set are ergosterol, 25-hydroxycholesterol, lathosterol, epicholesterol, 7-dehydrocholesterol and dihydrocholesterol. On functional grounds, β-sitosterol and camposterol probably can be added to this list [90]. Such mimics have been referred to as "membrane-active sterols" precisely because they replicate the behavior of cholesterol in driving phase separation [1]. At the very least, these findings argue against a tight structural fit between cholesterol and the polar lipids with which it complexes best.
Indeed, one study showed that lanosterol (at the bottom of Bloch’s list) substituted for cholesterol in maintaining the viability of cultured cells over several days [92]. While a later report found lanosterol to be toxic [90], this could have been a metabolic effect rather than a shortcoming of the physical properties of the membrane. More directly challenging to Bloch’s hypothesis are findings that a synthetic isomer, enantiomeric to cholesterol at each of its chiral centers, nevertheless substituted for it both in physical tests and in its ability to promote cell growth [90,93]. (The failure of enantiomeric cholesterol to support the long-term growth of C. elegans in another study could reflect a metabolic rather than a structural requirement [94].) Furthermore, a recent molecular dynamics simulation suggested that removal of the two projecting methyl groups that make the β-face of the steroid nucleus "rough" and therefore less able to associate with saturated fatty-acyl chains had the paradoxical effect of weakening rather than strengthening sterol-phospholipid associations [16]. In contrast, another molecular dynamics study concluded that the rough and smooth surfaces of the sterol were critical to how it organized the bilayer phospholipids laterally [47]. Finally, a lack of specificity in the interaction of cholesterol with sphingomyelin, considered to be its strongest membrane partner, has been inferred from detailed fluorescence studies [58]. Rather than specific associations, it has been suggested that congregation to minimize hydrophobic mismatch between the sterol and alkyl chains could be a driving force in their association [10,12]. Thus, how the molecular features of cholesterol confer its fitness remains an open issue.
5.2. Cholesterol surrogates
It now seems clear that non-sterol intercalators can substitute for cholesterol. For example, removing a portion of the cholesterol in the human erythrocyte membrane leads to cell lysis, perhaps by increasing its passive permeability to osmotic solutes. 25-hydroxycholesterol reduces this cell lysis, apparently by substituting for the native sterol [81]. Surprisingly, amphipaths as disparate as 1-octanol and short-chain ceramide and diglyceride analogues also prevent lysis. Indeed, ceramides have a higher affinity than cholesterol for ordered phases of 1-palmitoyl-2-oleoyl-phosphatidylcholine [95]. It also appears that ceramides can form condensed complexes with phospholipids like sphingomyelin [96,97]. Thus, the more general premise has been advanced that ceramides can not only form gel-like domains with raft-forming phospholipids in vivo, but also elaborate them into platforms upon which signaling proteins can carry out informational functions [98,99]. In any case, it appears that sterols can associate with phospholipids without great structural specificity, so that their function in stabilizing the membrane can be fulfilled by widely divergent membrane intercalators.
5.3. Agents that displace cholesterol from phospholipids
If amphipaths can substitute for sterols to form phospholipid complexes, they should also be able to compete cholesterol out of such complexes. This hypothesis can explain the ability of a variety of ceramides to potentiate the lytic action of cholesterol-directed cytolysins [100]. Along the same lines, short-chain ceramide and diglyceride analogs, as well as 1-octanol, all potentiate the lysis of red blood cells by saponin [81]. It would seem that these diverse agents can displace the sterol from its complexes, thereby promoting the association of the free (active) species with the lytic agent.
Further support for this hypothesis is the evidence that various ceramides and diglycerides not only displace cholesterol from liquid-ordered systems but also can substitute for the sterol [95,101]. Similarly, ceramide and diglyceride analogues, as well as 1-octanol, both substitute for red cell membrane cholesterol and displace it from phospholipids, as indicated by their ability to increase its sensitivity to cholesterol oxidase and the rate of its transfer to cyclodextrin [54,81]. That the agents actually increased free membrane cholesterol was verified by the demonstration that lysophosphatidylcholine reversed the cholesterol oxidase susceptibility imposed by these agents.
Various other non-sterol amphipaths have recently been shown to displace cholesterol from sphingomyelin-rich bilayer domains [102]. Furthermore, 1-hexadecanol forms condensed, liquid-ordered complexes with monolayers of dimyristoylphosphatidylcholine; these have a stability and stoichiometry similar to that formed by cholesterol [103]. This long-chain alcohol also displaces cholesterol from the phospholipid and promotes its activity, as judged by the greatly increased rate of transfer of the cholesterol to cyclodextrin in its presence. The case can therefore be made that at least 1-alkanols can occupy the sterol site in phospholipid membranes and mimic its properties [104].
It is striking that so many unrelated amphipaths can substitute for and displace cholesterol from complexes, given the specificities often seen for sterol-phospholipid interactions. These findings are consistent with early NMR studies showing that the rate of bilayer cholesterol axial rotation is significantly faster than that of the surrounding phospholipid molecules, implying the absence of tight complexation [2]. Thus, it may be that sterols evolved to be weak ligands, even for the phospholipid subclass with which they interact best. Other properties may have been optimized instead, and weak complexation may even have benefits. For example, high sterol levels could serve to keep xenobiotic amphipaths from associating with plasma membrane phospholipids and thereby discourage their entry into the cytoplasm. The study of a broad range of amphipaths should provide a better understanding of the requirements for the association of membrane intercalators with phospholipids; this would also help define the structural basis for phospholipid–sterol interactions. Substitution for or displacement of cholesterol from liquid-ordered domains could provide tools for such an analysis.
6. Cholesterol activity and cellular cholesterol homeostasis
Cholesterol circulates throughout the body and is transformed in various tissues to oxysterols, bile acids and steroid hormones or is excreted without modification [105]. The body balance is ultimately maintained by cellular sensors and effectors. Either a deficit in or an excess of cholesterol appears to be deleterious [106,107]. Accordingly, cells perceive the abundance of their cholesterol and maintain it at an endogenously-specified level by means of multiple homeostatic mechanisms. We have found for example that over a long period of cultivation the cholesterol content of human fibroblasts varied by only about 9% (n=34); furthermore, much of this spread must have been experimental error.
Cholesterol is typically most abundant in the plasma membrane and in the endomembrane pathways with which it shares lipids through vesicle traffic [108]. These endomembrane organelles include endosomes, lysosomes, the endocytic recycling compartment and distal Golgi elements [109]. The mitochondria, ER and other organelles not in extensive contact with the cell surface have little cholesterol. For example, the magnitude of the ER pool in resting human fibroblasts may be as low as 0.5% of the total [54,110]. Nevertheless, it is in the mitochondria and the ER that sterols are converted to oxysterols, steroid hormones and ecdysones and that the homeostatic effector proteins reside.
The paired concepts of stoichiometric complexes and the high activity of excess cholesterol underlie a general model for maintaining cell (primarily, plasma membrane) cholesterol at a physiological set-point [54,59]. According to this formulation, the abundance of cholesterol in plasma membranes matches the capacity of their phospholipids to form complexes. This is because cholesterol exceeding this equivalence point has a relatively high activity and can therefore readily exit the plasma membrane for other compartments. The recipient compartments are both extracellular and intracellular. The cytoplasmic membranes serve not merely as a buffer or reservoir compartment for the active excess but also play an active role in cholesterol homeostasis by reading their cholesterol content as a signal. In particular, a rise in intracellular pools would inform imbedded regulatory proteins of cholesterol overload and thereby elicit a reduction in cholesterol accretion until the plasma membrane cholesterol returned to its physiological set point.
This hypothesis postulates that the cholesterol level in at least some cytoplasmic membranes will be set in response to the preponderant plasma membrane pool. Perhaps a jump in plasma membrane cholesterol will, by passive partitioning, drive an increase in the intracellular membrane cholesterol level until the chemical potential equalizes among these compartments [59]. The low levels of cholesterol normally found in intracellular membranes such as ER and mitochondria would then reflect the known weak affinity for sterols of their phospholipids compared to those abundant in the plasma membrane, especially in its exofacial leaflet [10,11,62]. An alternative hypothesis is that sterols are pumped against their diffusional gradients so as to metabolically tune their concentration in each organelle. One line of reasoning that suggests an active process is that the partition coefficients for cholesterol among defined phospholipids span no more than a ~10-fold range [11]. But, if >50% of the cholesterol in fibroblasts is in the plasma membrane and ~0.5% in the ER, and these membrane compartments have a comparable abundance of phospholipid, their overall phospholipid partition coefficients would have to differ by about two orders of magnitude for passive equilibration to be the determinant of cholesterol distribution. Since this value seems high, we must consider that ER cholesterol might have been underestimated, since the assay might sample only a subset of ER vesicles (see section 6.1). At this point, accurate estimates of the cholesterol content of the various organelles, free of the usual cross-contamination by plasma membrane fragments, are still needed so that this issue can receive a definitive study.
There is other evidence that the cholesterol is not in equilibrium among the cellular membranes. In particular, the magnitude of the ER cholesterol pool can be increased and decreased by treating fibroblasts with protein kinase C directed agents [111]. Similarly, acute responses of ER cholesterol to small changes in plasma membrane cholesterol could be tuned by cytoplasmic proteins. For example, the hyper-sharp inactivation of hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase) by small increments in plasma membrane cholesterol would seem to involve enzymatic conversion of the active fraction of the sterol to oxysterols [112]. It is therefore conceivable that the bobbing of active cholesterol is mechanistic in its exit from membranes, but that proteins that capture and shuttle the projecting sterol molecules are modulated so as to affect the steady-state transfer rates and distributions of the cholesterol.
Cholesterol has been shown to circulate bidirectionally between the plasma membrane and the organelles [110,113]. Mediation or facilitation (as opposed to passive desorption and aqueous diffusion) is likely, given the fact that half-times of cytoplasmic transfer are in the range of 10–20 minutes, about an order of magnitude faster than observed for flux from cell surfaces to plasma lipoprotein acceptors [65,110,113, 114]. (This implies that the movement of cholesterol among the intracellular compartments is capable of mediating homeostatic responses on a time scale of minutes.) The aforementioned intracellular transfer rate is still perhaps two orders of magnitude slower than sterol movement from the cell surface to the non-physiological acceptor, cyclodextrin [52]. This suggests that, along with cholesterol activity, the efficiency of collision of acceptors with the donor membrane, rather than spontaneous sterol desorption, is rate-limiting physiologically. The transport mechanism appears not to be vesicular [119,120], and various shuttle protein candidates have been studied [108,115–118].
6.1. Endoplasmic reticulum cholesterol
The metabolic basis for cholesterol homeostasis rests in great measure with a battery of sterol-sensing regulatory proteins located in the ER [117,121]. In particular, a rise in the level of ER cholesterol is rapidly countered through increased cholesterol esterification by acyl CoA-cholesterol acyltransferase. Furthermore, excess cholesterol can, within minutes, initiate the inactivation of HMG-CoA reductase, the ER enzyme that limits the rate of sterol biosynthesis [54]. A slower, more powerful response to rising cell cholesterol is the inhibition of the expression of a set of genes encoding proteins that mediate cholesterol biosynthesis and endocytosis. As long as cell cholesterol is abundant, a transcription factor, called the sterol regulatory element binding protein or SREBP, is held in an inactive state in the ER because an associated protein, Scap, is occupied by cholesterol. SREBP retention is also promoted by a second ER protein, Insig, when it is liganded with oxysterols. Oxysterols are presumably synthesized in the ER and mitochondria in response to elevations in their substrate, cholesterol [122]; we would suggest that their synthesis could depend on the active excess of plasma membrane cholesterol that moves to the cytoplasm.
In this manner, cholesterol accretion is regulated by ER cholesterol and/or oxysterol derivatives of cholesterol. The premise would be that these pools are set in proportion to the activity of surplus plasma membrane cholesterol. Even though cholesterol synthesis is completed in the ER and cholesterol also arrives there following the hydrolysis of cholesterol ester stores, a brisk and massive circulation of cholesterol to and from the plasma membrane would continuously sweep out the relatively small ER compartment and peg its size to the activity of the excess plasma membrane cholesterol. (Otherwise, if cholesterol generated locally were to accumulate in the ER, it would miscue the homeostatic effectors as to cellular cholesterol abundance.) According to the stoichiometric complex hypothesis, variations in plasma membrane cholesterol below the equivalence point (i.e., the physiological cholesterol level) would have little effect on ER pool size because plasma membrane sterol activity would remain basal. However, increases in plasma membrane cholesterol above the physiological set-point would evoke a sharp rise in the ER pool.
A test of this hypothesis is shown in Figure 4. ER cholesterol was estimated using a runoff assay in which homogenates were incubated with [14C]oleoyl Coenzyme A to allow all of the cholesterol in membrane fragments that contained the ER enzyme, acyl CoA-cholesterol acyltransferase, to be labeled by [14C]fatty-acylation [123]. The plateau values of the radioactivity in the cholesterol esters synthesized during a prolonged incubation would give a measure of the cholesterol present in those fragments. Plasma membrane cholesterol in intact cells was varied with cyclodextrin prior to homogenization, and the effect on ER cholesterol then determined (Figure 4). It was found that depleting cell cholesterol below its physiological rest value reduced the ER pool somewhat [124]. Raising cell cholesterol above the ambient set point caused the ER pool to increase sharply. In this example, a change in cell surface cholesterol of 10% elicited a rise in ER cholesterol level of about 3-fold; thus small variations near the set-point can lead to large responses in the homeostatic direction. The half-time of such responses was 10–20 minutes.
Figure 4.

Variation of ER cholesterol with cell cholesterol. Human fibroblasts were briefly exposed to different cholesterol-cyclodextrin mixtures to modify their cell (mostly, plasma membrane) cholesterol level. ER pool size was determined 45–60 min later (see Section 6.1). The unmodified control values are the 1.0/1.0 points (▲). Re-plotted from reference 110.
Support for the proposed mechanism of ER cholesterol regulation comes from the observation that sphingomyelinase treatment of intact cells rapidly increased the size of the ER cholesterol pool and, concomitantly, the rate of cholesterol esterification. Presumably, the destruction of sphingomyelin reduced the capacity of the plasma membrane to associate with sterols [123,125]; furthermore, as discussed in Section 5.3, the ceramide digestion product would serve to displace and therefore activate plasma membrane cholesterol (see Section 5.3). Conversely, diverting cell cholesterol to lysosomes reduced the ER pool size [124]. The same was true of treating fibroblasts with lysophosphatidylcholine; in this case, the extra plasma membrane phospholipid presumably complexed free cholesterol [54].
It was suggested above that various intercalating amphipaths can activate plasma membrane cholesterol by competitively displacing it from phospholipid complexes. The high activity of the free sterol should then drive some of it to organelle membranes. In keeping with this premise, we found that exposing cells to the putative displacing agents, 25 hydroxycholesterol and 1-octanol, rapidly elevated ER cholesterol levels [54,110,123].
The rate of cholesterol esterification by the ER enzyme, acyl CoA-cholesterol acyltransferase, is known to vary with the size of the local cholesterol pool [117]. It is therefore not surprising that the dose-response curve for the dependence of esterification on plasma membrane cholesterol varies in parallel with ER cholesterol itself and that both have a sharp threshold at the physiological set-point of plasma membrane cholesterol (compare Figure 5 with Figure 4) [110]. Consistent with the active cholesterol hypothesis, digesting fibroblast plasma membrane sphingomyelin with sphingomyelinase so as to deplete the sterol complexing capacity of the bilayer increased cholesterol esterification rates and inhibited the activity of HMG-CoA reductase [123,126]. (As mentioned above, activation of plasma membrane cholesterol through its displacement by the ceramide hydrolysis product of sphingomyelin presumably contributes to this effect.)
Figure 5.

Effect of cell cholesterol on its esterification in vivo. Human fibroblasts were briefly exposed to different cholesterol-cyclodextrin mixtures to adjust their cell (mostly, plasma membrane) cholesterol level before the assay of the rate of cholesterol esterification in intact cells over the following 2.5 hours. The solid triangles represent unmodified controls for the 3 experiments marked by different symbols. From reference 110.
HMG-CoA reductase is another homeostatic, cholesterol-sensing enzyme in the ER. The proteosomal destruction of this enzyme is effected by a sterol-sensitive ubiquitin-driven pathway [121]. Incrementing plasma membrane cholesterol modestly above its physiologic set-point causes a dramatic fall in the activity of this enzyme (Figure 6) [54]. Treating cells with 1-octanol, ceramide or diglyceride to displace plasma membrane cholesterol from associated phospholipids and thereby activate it, also brings about the rapid loss of HMG-CoA reductase activity [81]. This behavior is in keeping with the proposed hypothesis. However, the pathway is more complex in this case. It is 27-hydroxycholesterol rather than cholesterol itself that stimulates proteolysis, so that (active) intracellular cholesterol must presumably be processed by mitochondria to bring about the feedback inhibition of ER HMG-CoA reductase [112]. Presumably, conversion to potent oxysterols is a way to amplify small signals from excess cholesterol for the implementation of homeostatic responses [127].
Figure 6.

Effect of cell cholesterol on the activity of HMG-CoA reductase. Expression of the enzyme was induced in human fibroblasts by overnight cholesterol starvation. The cells were then briefly exposed to different cholesterol-cyclodextrin mixtures to adjust their cell (mostly, plasma membrane) cholesterol level, and enzyme activity was assayed in homogenates an hour later. The unmodified control values are plotted at the 1.0/1.0 point . From reference 54.
6.2. Mitochondrial transformations of sterols
We recently observed that the production of 27-hydroxycholesterol by the mitochondria of intact fibroblasts rises sharply with small increments in plasma membrane cholesterol [Lange, Y., Ory, D.S, Lanier, M.H., Ye, J. and Steck, T.L., unpublished data]. It makes sense that the oxysterol, as a representative of cholesterol abundance, should be produced in proportion to the level of cholesterol activity above its physiological threshold. It would be important to ascertain if steroidogenesis in the mitochondria of endocrine cells is similarly rate-limited by the availability of cholesterol. Setting mitochondrial cholesterol levels at all times to the active excess of cholesterol could serve to prevent its consumption in that organelle from depleting plasma membrane cholesterol below its equivalence point with the phospholipids.
6.3. Cholesterol export to high-density plasma lipoproteins
The cells of the body share their cholesterol, both that endogenously synthesized and that ingested in the diet, through a battery of circulating lipoprotein carriers [105]. The blood stream also conveys excess cholesterol from scavengers like arterial wall macrophages to the liver for disposal [107,128]. Could the activity of plasma membrane cholesterol play a role in the transfer of cell cholesterol to these plasma proteins?
Certainly, in situ, the level of cholesterol in plasma membranes would be buffered at the physiological level by passive equilibration with the plasma lipoproteins in which the cells are bathed. The simplest mechanism would be for cholesterol to pass from compartments with higher to those with lower chemical activity, with or without mediation [65,68]. Running parallel to this passive route are management mechanisms like the ingestion of low-density lipoproteins via receptor-mediated endocytosis and active lipid export to nascent high-density lipoproteins (HDL). The latter process is mediated by members of the extensive family of integral plasma membrane ATP-binding cassette transport proteins or ABC pumps. Their ancestors presumably evolved to drive the expulsion of extraneous nonpolar molecules from the surface membranes of free-living unicellular organisms. However, in animals, ABCA1 transfers cell surface cholesterol plus phospholipids to the HDL precursor protein, apolipoprotein A-I (apoA-I), while ABCG1 adds cholesterol to mature HDL particles [128].
The process by which ABCA1 builds HDL from apoA-I has been elusive [129]. Among various proposed molecular mechanisms are the following: a) Since the lipid binding pocket of ABCA1 is open to the cytoplasmic leaflet, it could capture cholesterol there and transport it to the outer surface using the energy of ATP hydrolysis. Weighing against this concept is evidence that cholesterol is abundant at the outer leaflet and can diffuse rapidly across the bilayer [67], making transbilayer pumping an unnecessary drain on energy. b) ABCA1 might export phospholipids to apoA-I; the sterol would then follow secondarily [130]. Given that the ATP binding domain and putative substrate entry site of ABCA1 are oriented toward the endofacial leaflet, one might expect that cytoplasmic phospholipids would preferentially be transported to apoA-I. This hypothesis is undercut by the observation that, although ABCA1 might promote the transfer of cytoplasmic phospholipids to the cell surface, nascent HDL particles have a preponderance of exofacial lipid types [131–133]. c) It has been suggested that, because HDL particles are enriched in cholesterol, export draws selectively from sterol rich domains (rafts or caveolae). Countering that premise is evidence that ABCA1 is not localized in rafts and, in fact, can use ATP to disperse them [134,135]. Furthermore, the apoA-I recipient congregates in nonraft regions, apparently bound to ABCA1. d) ABC-type pumps could activate cholesterol by driving its partial projection; this would then promote its collisional capture by extracellular acceptors [136]. Consistent with a role for increased plasma membrane cholesterol activity is the finding that ABCA1 both increases the susceptibility of the cell surface to cholesterol oxidase and the esterification of cholesterol in the ER [137]. e) ABCA1 might transfer bites of plasma membrane to nascent HDL particles [138]. This concept is consistent with the fact that phospholipids from both leaflets are represented in newly-formed HDL particles [131]. It is suggested that ABCA1 uses the energy of ATP hydrolysis to pump cytoplasmic phospholipids to the outer surface, thereby bending the bilayer outward. The strain energy in the acutely curved membrane could then foster the binding of apoA-I and the solubilization of the bilayer segment by the detergent-like properties of this protein [138]. Additional cholesterol would presumably follow.
Whatever the export mechanism and pathway utilized by ABCA1, cells might seek to protect their integrity by limiting their export of plasma membrane cholesterol to the physiological optimum. As argued above, this level could be set at the equivalence point of the plasma membrane phospholipid pool if ABCA1 were only able to transfer active plasma membrane cholesterol. It is relevant that lowering the abundance of plasma membrane sphingomyelin promotes cholesterol export by ABCA1 [139], given that it increases the activity of plasma membrane cholesterol (see Section 3) [10,123,140,141]. Furthermore, synthetic ceramides, believed to displace cholesterol from phospholipid complexes and thereby activate it (Section 5.2), also promote the export of cholesterol by ABCA1 [142,143]. That ABCA1 resides in liquid-disordered regions of the plasma membrane rather than in rafts is also consistent with its preferential export of uncomplexed (high activity) cholesterol [135]. Of additional relevance is the observation that ABCA1 promotes the translocation of phosphatidylserine to the exofacial leaflet of the plasma membrane [133], since this too would increase the activity of cell surface cholesterol [85]. A different (slower and more complex) system by which cholesterol export could be capped at the plasma membrane equivalence point would be through regulation of the expression of ABCA1 by 27-hydroxycholesterol [122], provided that the production of this oxysterol turns out to reflect the active excess of cholesterol, as discussed above.
Whether or not ABCA1 prefers high activity plasma membrane cholesterol as its substrate, it is worth considering that its paralog, ABCG1, might do so, given that its substrate is simply cholesterol [129]. Similarly, scavenger receptor BI (SR-BI), another plasma membrane lipid transfer protein, might facilitate downhill cholesterol exit by increasing the activity of the sterol. Evidence for such a function comes from the ability of SR-BI to increase the susceptibility of plasma membrane cholesterol to cholesterol oxidase and cyclodextrin [65]. SR-BI could operate upon caveolae by dissociating their cholesterol molecules from phospholipid complexes, thereby creating an active pool susceptible for export. Finally, it might be that the synthesis and secretion of mature LDL particles by the epithelial cells of the gut and liver [128] is also limited by their quotient of excess, high-activity cholesterol.
7. Concluding comments
This review has considered evidence for and consequences of the ability of plasma membrane cholesterol to exist in low and high activity states. Figure 7 summarizes this behavior. There is by now overwhelming evidence that sterols like cholesterol associate preferentially with phosphocholine phospholipids bearing saturated chains and less strongly with various other membrane lipid species. The evidence is also strong that the basis for this association is the formation of weak, stoichiometric sterol-phospholipid complexes. These complexes are sufficiently immiscible in the sterol-poor, liquid-disordered continuum that they separate as condensed, liquid-ordered phases. We highlighted here the behavior of the fraction of cholesterol that accumulates when the capacity of plasma membrane phospholipids to form complexes is exceeded. This uncomplexed excess of cholesterol is presumed to be mostly dissolved in the low-affinity liquid-disordered phase of the bilayer. Characteristic of these relatively free sterol molecules would be their high activity or tendency to escape the bilayer.
Figure 7.

The active cholesterol hypothesis. Most of the plasma membrane cholesterol (submerged yellow molecules) is complexed with phospholipids and therefore has low escape tendency (activity). Excess cholesterol can bob more freely into the aqueous space and has relatively high activity (projecting red molecules). Similarly, intercalating amphipaths can displace cholesterol from its complexes, thereby activating it. Projecting sterol molecules can interact with extracellular reactants such as cholesterol oxidase, cyclodextrin, and high density lipoproteins. Active cholesterol also circulates through the cytoplasm to the endoplasmic reticulum and mitochondria, where it can serve as a metabolic reactant or a homeostatic signal effecting feedback down-regulation through its esterification, the inhibition of its biosynthesis and the expression of genes for cholesterol accretion. See text for details.
The high activity of excess plasma membrane cholesterol provides a mechanism for regulating cell cholesterol because it signals that the cell surface is replete with cholesterol. The active sterol molecules continuously and transiently project from the bilayer into the aqueous phase to a far greater degree than their complexed counterparts. As a result, they can readily be transferred from the bilayer by collision with extracellular and intracellular acceptors. An intracellular sterol transport system sets the magnitude of the ER cholesterol pool in response to this high-activity cholesterol signal. A battery of proteins reads the level of the ER cholesterol pool and modulates the accretion of cholesterol accordingly. This feedback loop helps to keep the plasma membrane cholesterol pool set at the equivalence point dictated by the complexing phospholipids; this is presumably the optimal level of cell surface cholesterol. Thus, it would appear that short-term regulatory responses to increments in cell cholesterol involve an upstream signal in the form of plasma membrane cholesterol activity; a rapid response in the size of the sterol pools in the ER and, presumably, mitochondria; immediate regulation of the activity of several downstream effectors of cholesterol accretion; and long-term homeostatic feedback adjustments of cholesterol accretion.
High activity plasma membrane cholesterol might also be selectively transferred to extracellular acceptors, such as plasma HDL, both unassisted and with the help of plasma membrane lipid transfer proteins. This redistribution would help to return cell surface cholesterol to its physiological optimum: the complexing capacity of the plasma membrane phospholipids. One can also speculate that the biosynthesis of steroids, bile acids and/or oxysterols could be pegged to the active cholesterol in excess of the plasma membrane phospholipid set-point so as to avoid potentially deleterious depletion of plasma membrane cholesterol.
Two simple experimental methods to gauge plasma membrane cholesterol activity have emerged: the susceptibility of cholesterol to cholesterol oxidase attack and the kinetics of its transfer to the aqueous acceptor, cyclodextrin. Physiological indicators of the rapid redistribution of active cell surface cholesterol to the cell interior are increases in ER cholesterol and the consequent stimulation of cholesterol esterification and inactivation of HMG-CoA reductase. Amphipaths that mimic, substitute for and/or displace plasma membrane cholesterol are handy tools to test the role of cholesterol activity (escape tendency) in its transfer to intracellular and extracellular compartments and to probe the contacts between phospholipids and sterols that promote their complexation.
The proposed role of sterol activity in cholesterol homeostasis has therapeutic implications. If the activity of plasma membrane cholesterol could be elevated pharmacologically, it should increase intracellular cholesterol, including the ER pool. This effect would be interpreted by the cell’s homeostatic elements as a sign of cholesterol overload, prompting it to reduce cholesterol biosynthesis. This could lead to a reduction in the body burden of the sterol. Such possibilities provide impetus for the further study of the activity of plasma membrane cholesterol.
Acknowledgments
The authors are grateful to Arun Radhakrishnan, Michael Phillips, Will Prinz and, especially, John Silvius for their valuable comments on this manuscript. Support to Y.L. was from NIH grant HL 28448.
Abbreviations
- ER
endoplasmic reticulum
- HDL
high-density lipoprotein
- HMG-CoA reductase
hydroxy-3-methylglutaryl coenzyme A reductase
- SR-BI
scavenger receptor class B type I
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barenholz Y. Sphingomyelin and cholesterol: From membrane biophysics and rafts to potential medical applications. Subcell Biochem. 2004;37:167–215. doi: 10.1007/978-1-4757-5806-1_5. [DOI] [PubMed] [Google Scholar]
- 2.Yeagle PL. Cholesterol and the cell membrane. Biochim Biophys Acta. 1985;822:267–287. doi: 10.1016/0304-4157(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 3.Bloom M, Evans E, Mouritsen O. Physical properties of the fluid lipid-bilayer component of cell membranes. A perspective. Q. Rev. Biophys. 1991;24:293–397. doi: 10.1017/s0033583500003735. [DOI] [PubMed] [Google Scholar]
- 4.Gottlieb MH. The reactivity of human erythrocyte membrane cholesterol with a cholesterol oxidase. Biochim Biophys Acta. 1977;466:422–428. doi: 10.1016/0005-2736(77)90335-2. [DOI] [PubMed] [Google Scholar]
- 5.Patzer EJ, Wagner RR, Barenholz Y. Cholesterol oxidase as a probe for studying membrane organisation. Nature. 1978;274:394–395. doi: 10.1038/274394a0. [DOI] [PubMed] [Google Scholar]
- 6.Bar LK, Barenholz Y, Thompson TE. Dependence on phospholipid composition of the fraction of cholesterol undergoing spontaneous exchange between small unilamellar vesicles. Biochemistry. 1987;26:5460–5465. doi: 10.1021/bi00391a037. [DOI] [PubMed] [Google Scholar]
- 7.Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim Biophys Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 8.Lange Y, Cutler HB, Steck TL. The effect of cholesterol and other intercalated amphipaths on the contour and stability of the isolated red cell membrane. J Biol Chem. 1980;255:9331–9337. [PubMed] [Google Scholar]
- 9.Cevc G, Marsh D. Phospholipid bilayers : Physical principles and models. New York: Wiley; 1987. [Google Scholar]
- 10.Ohvo-Rekila H, Ramstedt B, Leppimaki P, Slotte JP. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41:66–97. doi: 10.1016/s0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- 11.Niu SL, Litman BJ. Determination of membrane cholesterol partition coefficient using a lipid vesicle-cyclodextrin binary system: Effect of phospholipid acyl chain unsaturation and headgroup composition. Biophys J. 2002;83:3408–3415. doi: 10.1016/S0006-3495(02)75340-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMullen T, McElhaney RN. Physical studies of cholesterol-phospholipid interactions Curr. Opin. Colloid Interface Sci. 1996;1:83–90. [Google Scholar]
- 13.Radhakrishnan A, McConnell HM. Thermal dissociation of condensed complexes of cholesterol and phospholipid. J. Phys. Chem. B. 2002;106:4755–4762. [Google Scholar]
- 14.McConnell HM, Vrljic M. Liquid-liquid immiscibility in membranes. Annu Rev Biophys Biomol Struct. 2003;32:469–492. doi: 10.1146/annurev.biophys.32.110601.141704. [DOI] [PubMed] [Google Scholar]
- 15.Hung WC, Lee MT, Chen FY, Huang HW. The condensing effect of cholesterol in lipid bilayers. Biophys J. 2007;92:3960–3967. doi: 10.1529/biophysj.106.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rog T, Pasenkiewicz-Gierula M, Vattulainen I, Karttunen M. What happens if cholesterol is made smoother: Importance of methyl substituents in cholesterol ring structure on phosphatidylcholine-sterol interaction. Biophys. J. 2007;92:3346–3357. doi: 10.1529/biophysj.106.095497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips MC. The physical state of phospholipids and cholesterol in monolayers, bilayers, and membranes. In: Danielli JF, Rosenberg MD, Cadenhead DA, editors. Progress in Surface and Membrane Science. New York: Academic Press; 1972. pp. 139–221. [Google Scholar]
- 18.Veatch SL, Keller SL. Seeing spots: Complex phase behavior in simple membranes. Biochim Biophys Acta. 2005;1746:172–185. doi: 10.1016/j.bbamcr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 19.London E. How principles of domain formation in model membranes may explain ambiguities concerning lipid raft formation in cells. Biochim Biophys Acta. 2005;1746:203–220. doi: 10.1016/j.bbamcr.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Silvius JR. Partitioning of membrane molecules between raft and non-raft domains: Insights from model-membrane studies. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2005;1746:193–202. doi: 10.1016/j.bbamcr.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Silvius JR, Nabi IR. Fluorescence-quenching and resonance energy transfer studies of lipid microdomains in model and biological membranes (review) Mol Membr Biol. 2006;23:5–16. doi: 10.1080/09687860500473002. [DOI] [PubMed] [Google Scholar]
- 22.Silvius J. Lipid microdomains in model and biological membranes: How strong are the connections? Q Rev Biophys. 2005;38:373–383. doi: 10.1017/S003358350600415X. [DOI] [PubMed] [Google Scholar]
- 23.Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 24.Edidin M. The state of lipid rafts: From model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 25.Stottrup BL, Stevens DS, Keller SL. Miscibility of ternary mixtures of phospholipids and cholesterol in monolayers, and application to bilayer systems. Biophys J. 2005;88:269–276. doi: 10.1529/biophysj.104.048439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stottrup BL, Keller SL. Phase behavior of lipid monolayers containing DPPC and cholesterol analogs. Biophys J. 2006;90:3176–3183. doi: 10.1529/biophysj.105.072959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silvius JR. Fluorescence energy transfer reveals microdomain formation at physiological temperatures in lipid mixtures modeling the outer leaflet of the plasma membrane. Biophys. J. 2003;85:1034–1045. doi: 10.1016/S0006-3495(03)74542-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feigenson GW. Phase behavior of lipid mixtures. Nat Chem Biol. 2006;2:560–563. doi: 10.1038/nchembio1106-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yethiraj A, Weisshaar JC. Why are lipid rafts not observed in vivo? Biophys J. 2007 doi: 10.1529/biophysj.106.101931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hancock JF. Lipid rafts: Contentious only from simplistic standpoints. Nat Rev Mol Cell Biol. 2006;7:456–462. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pike LJ. Rafts defined: A report on the keystone symposium on lipid rafts and cell function. J. Lipid Res. 2006;47:1597–1598. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Pike LJ. Lipid rafts: Heterogeneity on the high seas. Biochem. J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 34.Hoessli DC, Ilangumaran S, Soltermann A, Robinson PJ, Borisch B, Nasir Ud D. Signaling through sphingolipid microdomains of the plasma membrane: The concept of signaling platform. Glycoconj J. 2000;17:191–197. doi: 10.1023/a:1026585006064. [DOI] [PubMed] [Google Scholar]
- 35.Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol. 2006;2:584–590. doi: 10.1038/nchembio834. [DOI] [PubMed] [Google Scholar]
- 36.Foster LJ, de Hoog CL, Mann M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. PNAS. 2003;100:5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prinz W. Cholesterol trafficking in the secretory and endocytic systems. Semin Cell Dev Biol. 2002;13:197–203. doi: 10.1016/s1084-9521(02)00048-4. [DOI] [PubMed] [Google Scholar]
- 38.Silvius JR. Role of cholesterol in lipid raft formation: Lessons from lipid model systems. Biochim Biophys Acta. 2003;1610:174–183. doi: 10.1016/s0005-2736(03)00016-6. [DOI] [PubMed] [Google Scholar]
- 39.McConnell HM, Radhakrishnan A. Condensed complexes of cholesterol and phospholipids. Biochim Biophys Acta. 2003;1610:159–173. doi: 10.1016/s0005-2736(03)00015-4. [DOI] [PubMed] [Google Scholar]
- 40.Radhakrishnan A, McConnell HM. Condensed complexes of cholesterol and phospholipids. Biophys J. 1999;77:1507–1517. doi: 10.1016/S0006-3495(99)76998-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keller SL, Radhakrishnan A, McConnell HM. Saturated phospholipids with high melting temperatures form complexes with cholesterol in monolayers. J. Phys. Chem. B. 2000;104:7522–7527. [Google Scholar]
- 42.Radhakrishnan A, Li XM, Brown RE, McConnell HM. Stoichiometry of cholesterol-sphingomyelin condensed complexes in monolayers. Biochim Biophys Acta. 2001;1511:1–6. doi: 10.1016/s0005-2736(01)00274-7. [DOI] [PubMed] [Google Scholar]
- 43.Pandit SA, Bostick D, Berkowitz ML. Complexation of phosphatidylcholine lipids with cholesterol. Biophys. J. 2004;86:1345–1356. doi: 10.1016/S0006-3495(04)74206-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okonogi TM, Radhakrishnan A, McConnell HM. Two fatty acids can replace one phospholipid in condensed complexes with cholesterol. Biochim Biophys Acta. 2002;1564:1–4. doi: 10.1016/s0005-2736(02)00451-0. [DOI] [PubMed] [Google Scholar]
- 45.Radhakrishnan A, McConnell H. Condensed complexes in vesicles containing cholesterol and phospholipids. Proc Natl Acad Sci U S A. 2005;102:12662–12666. doi: 10.1073/pnas.0506043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiu SW, Jakobsson E, Mashl RJ, Scott HL. Cholesterol-induced modifications in lipid bilayers: A simulation study. Biophys J. 2002;83:1842–1853. doi: 10.1016/S0006-3495(02)73949-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandit SA, Khelashvili G, Jakobsson E, Grama A, Scott HL. Lateral organization in lipid-cholesterol mixed bilayers. Biophys. J. 2007;92:440–447. doi: 10.1529/biophysj.106.093864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khelashvili GA, Pandit SA, Scott HL. Self-consistent mean-field model based on molecular dynamics: Application to lipid-cholesterol bilayers. J Chem Phys. 2005;123:34910–34923. doi: 10.1063/1.1943412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu X, London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry. 2000;39:843–849. doi: 10.1021/bi992543v. [DOI] [PubMed] [Google Scholar]
- 50.Beattie ME, Veatch SL, Stottrup BL, Keller SL. Sterol structure determines miscibility versus melting transitions in lipid vesicles. Biophys J. 2005;89:1760–1768. doi: 10.1529/biophysj.104.049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewis GN, Randall M. Thermodynamics. 2d ed. New York: McGraw-Hill; 1961. [Google Scholar]
- 52.Steck TL, Ye J, Lange Y. Probing red cell membrane cholesterol movement with cyclodextrin. Biophys J. 2002;83:2118–2125. doi: 10.1016/S0006-3495(02)73972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steck TL, Kezdy FJ, Lange Y. An activation-collision mechanism for cholesterol transfer between membranes. J Biol Chem. 1988;263:13023–13031. [PubMed] [Google Scholar]
- 54.Lange Y, Ye J, Steck TL. How cholesterol homeostasis is regulated by plasma membrane holesterol in excess of phospholipids. Proc Natl Acad Sci U S A. 2004;101:11664–11667. doi: 10.1073/pnas.0404766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang J, Buboltz JT, Feigenson GW. Maximum solubility of cholesterol in hosphatidylcholine and phosphatidylethanolamine bilayers. Biochim Biophys Acta. 1999;1417:89–100. doi: 10.1016/s0005-2736(98)00260-0. [DOI] [PubMed] [Google Scholar]
- 56.Huang J, Feigenson GW. A microscopic interaction model of maximum solubility of holesterol in lipid bilayers. Biophys J. 1999;76:2142–2157. doi: 10.1016/S0006-3495(99)77369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ali MR, Cheng KH, Huang J. Assess the nature of cholesterol-lipid interactions through the chemical potential of cholesterol in phosphatidylcholine bilayers. Proc Natl Acad Sci U S A. 2007;104:5372–5377. doi: 10.1073/pnas.0611450104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holopainen JM, Metso AJ, Mattila JP, Jutila A, Kinnunen PK. Evidence for the lack of a specific interaction between cholesterol and sphingomyelin. Biophys J. 2004;86:1510–1520. doi: 10.1016/S0006-3495(04)74219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radhakrishnan A, McConnell HM. Chemical activity of cholesterol in membranes. Biochemistry. 2000;39:8119–8124. doi: 10.1021/bi0005097. [DOI] [PubMed] [Google Scholar]
- 60.Ohvo H, Slotte JP. Cyclodextrin-mediated removal of sterols from monolayers: Effects of terol structure and phospholipids on desorption rate. Biochemistry. 1996;35:8018–8024. doi: 10.1021/bi9528816. [DOI] [PubMed] [Google Scholar]
- 61.Lange Y, Ye J, Strebel F. Movement of 25-hydroxycholesterol from the plasma membrane to the rough endoplasmic reticulum in cultured hepatoma cells. J Lipid Res. 1995;36:1092–1097. [PubMed] [Google Scholar]
- 62.Leventis R, Silvius JR. Use of cyclodextrins to monitor transbilayer movement and differential lipid affinities of cholesterol. Biophys J. 2001;81:2257–2267. doi: 10.1016/S0006-3495(01)75873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lund-Katz S, Laboda HM, McLean LR, Phillips MC. Influence of molecular packing and phospholipid type on rates of cholesterol exchange. Biochemistry. 1988;27:3416–3423. doi: 10.1021/bi00409a044. [DOI] [PubMed] [Google Scholar]
- 64.Ramstedt B, Slotte JP. Interaction of cholesterol with sphingomyelins and acyl-chain-matched phosphatidylcholines: A comparative study of the effect of the chain length. Biophysical Journal. 1999;76:908–915. doi: 10.1016/S0006-3495(99)77254-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rothblat GH, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. Cell cholesterol efflux: Integration of old and new observations provides new insights. J Lipid Res. 1999;40:781–796. [PubMed] [Google Scholar]
- 66.Yancey PG, Bortnick AE, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- 67.Hamilton JA. Fast flip-flop of cholesterol and fatty acids in membranes: Implications for membrane transport proteins. Current Opinion in Lipidology. 2003;14:263–271. doi: 10.1097/00041433-200306000-00006. [DOI] [PubMed] [Google Scholar]
- 68.Rothblat GH, de la Llera-Moya M, Favari E, Yancey PG, Kellner-Weibel G. Cellular cholesterol flux studies: Methodological considerations. Atherosclerosis. 2002;163:1–8. doi: 10.1016/s0021-9150(01)00713-4. [DOI] [PubMed] [Google Scholar]
- 69.Haynes MP, Phillips MC, Rothblat GH. Efflux of cholesterol from different cellular pools. Biochemistry. 2000;39:4508–4517. doi: 10.1021/bi992125q. [DOI] [PubMed] [Google Scholar]
- 70.Ahn KW, Sampson NS. Cholesterol oxidase senses subtle changes in lipid bilayer structure. Biochemistry. 2004;43:827–836. doi: 10.1021/bi035697q. [DOI] [PubMed] [Google Scholar]
- 71.Lange Y, Matthies HJ. Transfer of cholesterol from its site of synthesis to the plasma membrane. J Biol Chem. 1984;259:14624–14630. [PubMed] [Google Scholar]
- 72.Pal R, Barenholz Y, Wagner RR. Effect of cholesterol concentration on organization of viral and vesicle membranes. Probed by accessibility to cholesterol oxidase. J Biol Chem. 1980;255:5802–5806. [PubMed] [Google Scholar]
- 73.Slotte JP, Hedstrom G, Rannstrom S, Ekman S. Effects of sphingomyelin degradation on cell cholesterol oxidizability and steady-state distribution between the cell surface and the cell interior. Biochim Biophys Acta. 1989;985:90–96. doi: 10.1016/0005-2736(89)90108-9. [DOI] [PubMed] [Google Scholar]
- 74.Chen X, Wolfgang DE, Sampson NS. Use of the parallax-quench method to determine the position of the active-site loop of cholesterol oxidase in lipid bilayers. Biochemistry. 2000;39:13383–13389. doi: 10.1021/bi001407j. [DOI] [PubMed] [Google Scholar]
- 75.Ghoshroy KB, Zhu W, Sampson NS. Investigation of membrane disruption in the reaction catalyzed by cholesterol oxidase. Biochemistry. 1997;36:6133–6140. doi: 10.1021/bi962190p. [DOI] [PubMed] [Google Scholar]
- 76.El Yandouzi EH, Le Grimellec C. Cholesterol heterogeneity in the plasma membrane of epithelial cells. Biochemistry. 1992;31:547–551. doi: 10.1021/bi00117a035. [DOI] [PubMed] [Google Scholar]
- 77.Mattjus P, Slotte JP. Does cholesterol discriminate between sphingomyelin and phosphatidylcholine in mixed monolayers containing both phospholipids? Chem Phys Lipids. 1996;81:69–80. doi: 10.1016/0009-3084(96)02535-2. [DOI] [PubMed] [Google Scholar]
- 78.Mattjus P, Hedstrom G, Slotte JP. Monolayer interaction of cholesterol with phosphatidylcholines: Effects of phospholipid acyl chain length. Chemistry and Physics of Lipids. 1994;74:195–203. [Google Scholar]
- 79.Lange Y, Matthies H, Steck TL. Cholesterol oxidase susceptibility of the red cell membrane. Biochim Biophys Acta. 1984;769:551–562. doi: 10.1016/0005-2736(84)90053-1. [DOI] [PubMed] [Google Scholar]
- 80.Radhakrishnan A, McConnell HM. Critical points in charged membranes containing cholesterol. Proc Natl Acad Sci U S A. 2002;99:13391–13396. doi: 10.1073/pnas.212522699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lange Y, Ye J, Steck TL. Activation of membrane cholesterol by displacement from phospholipids. J Biol Chem. 2005;280:36126–36131. doi: 10.1074/jbc.M507149200. [DOI] [PubMed] [Google Scholar]
- 82.Quinn PJ. Plasma membrane phospholipid asymmetry. Subcell Biochem. 2002;36:39–60. doi: 10.1007/0-306-47931-1_3. [DOI] [PubMed] [Google Scholar]
- 83.Myher J, Kuksis A, Pind S. Molecular species of glycerophospholipids and sphingomyelins of human erythrocytes: Improved method of analysis. Lipids. 1989;24:396–407. doi: 10.1007/BF02535147. [DOI] [PubMed] [Google Scholar]
- 84.Wang TY, Leventis R, Silvius JR. Fluorescence-based evaluation of the partitioning of lipids and lipidated peptides into liquid-ordered lipid microdomains: A model for molecular partitioning into "Lipid rafts". Biophys J. 2000;79:919–933. doi: 10.1016/S0006-3495(00)76347-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lange Y, Ye J, Steck TL. Scrambling of phospholipids activates red cell membrane cholesterol. Biochemistry. 2007;46:2233–2238. doi: 10.1021/bi6023397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bloch KE. Sterol structure and membrane function. CRC Crit Rev Biochem. 1983;14:47–92. doi: 10.3109/10409238309102790. [DOI] [PubMed] [Google Scholar]
- 87.Miao L, Nielsen M, Thewalt J, Ipsen JH, Bloom M, Zuckermann MJ, Mouritsen OG. From lanosterol to cholesterol: Structural evolution and differential effects on lipid bilayers. Biophys. J. 2002;82:1429–1444. doi: 10.1016/S0006-3495(02)75497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu X, Bittman R, Duportail G, Heissler D, Vilcheze C, London E. Effect of the structure of natural sterols and sphingolipids on the formation of ordered sphingolipid/sterol domains (rafts). Comparison of cholesterol to plant, fungal, and disease-associated sterols and comparison of sphingomyelin, cerebrosides, and ceramide. J Biol Chem. 2001;276:33540–33546. doi: 10.1074/jbc.M104776200. [DOI] [PubMed] [Google Scholar]
- 89.Megha , Bakht O, London E. Cholesterol precursors stabilize ordinary and ceramide-rich ordered lipid domains (lipid rafts) to different degrees: Implications for the Bloch hypothesis and sterol biosynthesis disorders. J. Biol. Chem. 2006;281:21903–21913. doi: 10.1074/jbc.M600395200. [DOI] [PubMed] [Google Scholar]
- 90.Xu F, Rychnovsky SD, Belani JD, Hobbs HH, Cohen JC, Rawson RB. Dual roles for cholesterol in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:14551–14556. doi: 10.1073/pnas.0503590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang J, Megha , London E. Relationship between sterol/steroid structure and participation in ordered lipid domains (lipid rafts): Implications for lipid raft structure and function. Biochemistry. 2004;43:1010–1018. doi: 10.1021/bi035696y. [DOI] [PubMed] [Google Scholar]
- 92.Buttke TM, Folks TM. Complete replacement of membrane cholesterol with 4,4',14- trimethyl sterols in a human T cell line defective in lanosterol demethylation. J Biol Chem. 1992;267:8819–8826. [PubMed] [Google Scholar]
- 93.Mannock DA, McIntosh TJ, Jiang X, Covey DF, McElhaney RN. Effects of natural and enantiomeric cholesterol on the thermotropic phase behavior and structure of egg sphingomyelin bilayer membranes. Biophys J. 2003;84:1038–1046. doi: 10.1016/S0006-3495(03)74920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crowder CM, Westover EJ, Kumar AS, Ostlund RE, Jr, Covey DF. Enantiospecificity of cholesterol function in vivo. J. Biol. Chem. 2001;276:44369–44372. doi: 10.1074/jbc.C100535200. [DOI] [PubMed] [Google Scholar]
- 95.Ali MR, Cheng KH, Huang J. Ceramide drives cholesterol out of the ordered lipid bilayer phase into the crystal phase in 1-palmitoyl-2-oleoyl-sn-glycero-3- phosphocholine/cholesterol/ceramide ternary mixtures. Biochemistry. 2006;45:12629–12638. doi: 10.1021/bi060610x. [DOI] [PubMed] [Google Scholar]
- 96.Silva LC, De Almeida RFM, Castro BM, Fedorov A, Prieto M. Ceramide-domain formation and collapse in lipid rafts: Membrane reorganization by an apoptotic lipid. Biophysical Journal. 2007;92:502–516. doi: 10.1529/biophysj.106.091876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pandit SA, Chiu S-W, Jakobsson E, Grama A, Scott HL. Cholesterol surrogates: A comparison of cholesterol and 16:0 ceramide in POPC bilayers. Biophys. J. 2007;92:920–927. doi: 10.1529/biophysj.106.095034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 99.Grassme H, Riethmuller J, Gulbins E. Biological aspects of ceramide-enriched membrane domains. Progress in Lipid Research. 2007;46:161–170. doi: 10.1016/j.plipres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 100.Zitzer A, Bittman R, Verbicky CA, Erukulla RK, Bhakdi S, Weis S, Valeva A, Palmer M. Coupling of cholesterol and cone-shaped lipids in bilayers augments membrane permeabilization by the cholesterol-specific toxins streptolysin O and vibrio cholerae cytolysin. J Biol Chem. 2001;276:14628–14633. doi: 10.1074/jbc.M100241200. [DOI] [PubMed] [Google Scholar]
- 101.Megha , London E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J Biol Chem. 2004;279:9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 102.Alanko SM, Halling KK, Maunula S, Slotte JP, Ramstedt B. Displacement of sterols from sterol/sphingomyelin domains in fluid bilayer membranes by competing molecules. Biochim Biophys Acta. 2005;1715:111–121. doi: 10.1016/j.bbamem.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 103.Ratajczak MK, Ko YTC, Lange Y, Steck TL, Lee KYC. Cholesterol displacement from membrane phospholipids by hexadecanol. Biophys. J. 2007;93:2038–2047. doi: 10.1529/biophysj.107.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Griepernau B, Leis S, Schneider MF, Sikor M, Steppich D, Bockmann RA. 1-alkanols and membranes: A story of attraction. Biochim Biophys Acta. 2007;1768:2899–2913. doi: 10.1016/j.bbamem.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 105.Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J. Biol. Chem. 2002;277:3801–3804. doi: 10.1074/jbc.R100057200. [DOI] [PubMed] [Google Scholar]
- 106.Brown MS, Goldstein JL. Suppression of 3-hydroxy-3-methylglutaryl Coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J. Biol. Chem. 1974;249:7306–7314. [PubMed] [Google Scholar]
- 107.Tabas I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J Clin Invest. 2002;110:905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maxfield FR, Menon AK. Intracellular sterol transport and distribution. Curr Opin Cell Biol. 2006;18:379–385. doi: 10.1016/j.ceb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 109.Hao M, Lin SX, Karylowski OJ, Wustner D, McGraw TE, Maxfield FR. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J Biol Chem. 2002;277:609–617. doi: 10.1074/jbc.M108861200. [DOI] [PubMed] [Google Scholar]
- 110.Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;40:2264–2270. [PubMed] [Google Scholar]
- 111.Lange Y, Ye J, Steck TL. Effect of protein kinase C on endoplasmic reticulum cholesterol. Biochem Biophys Res Commun. 2002;290:488–493. doi: 10.1006/bbrc.2001.6156. [DOI] [PubMed] [Google Scholar]
- 112.Lange Y, Ory DS, Ye J, Lanier MH, Hsu F-F, Steck TL. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 2008;283:1445–1455. doi: 10.1074/jbc.M706967200. [DOI] [PubMed] [Google Scholar]
- 113.Wustner D, Mondal M, Tabas I, Maxfield FR. Direct observation of rapid internalization and intracellular transport of sterol by macrophage foam cells. Traffic. 2005;6:396–412. doi: 10.1111/j.1600-0854.2005.00285.x. [DOI] [PubMed] [Google Scholar]
- 114.Soccio RE, Breslow JL. Intracellular cholesterol transport. Arterioscler Thromb Vasc Biol. 2004;24:1150–1160. doi: 10.1161/01.ATV.0000131264.66417.d5. [DOI] [PubMed] [Google Scholar]
- 115.Schneiter R. Intracellular sterol transport in eukaryotes, a connection to mitochondrial function? Biochimie. 2007;89:255–259. doi: 10.1016/j.biochi.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 116.Raychaudhuri S, Im YJ, Hurley JH, Prinz WA. Nonvesicular sterol movement from plasma membrane to er requires oxysterol-binding protein-related proteins and phosphoinositides. J Cell Biol. 2006;173:107–119. doi: 10.1083/jcb.200510084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 118.Wang PY, Weng J, Anderson RG. OSBP is a cholesterol-regulated scaffolding protein in control of ERK 1/2 activation. Science. 2005;307:1472–1476. doi: 10.1126/science.1107710. [DOI] [PubMed] [Google Scholar]
- 119.Baumann NA, Sullivan DP, Ohvo-Rekila H, Simonot C, Pottekat A, Klaassen Z, Beh CT, Menon AK. Transport of newly synthesized sterol to the sterol-enriched plasma membrane occurs via nonvesicular equilibration. Biochemistry. 2005;44:5816–5826. doi: 10.1021/bi048296z. [DOI] [PubMed] [Google Scholar]
- 120.Prinz WA. Non-vesicular sterol transport in cells. Prog Lipid Res. 2007;46:297–314. doi: 10.1016/j.plipres.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 122.Reiss AB, Martin KO, Javitt NB, Martin DW, Grossi EA, Galloway AC. Sterol 27- hydroxylase: High levels of activity in vascular endothelium. J. Lipid Res. 1994;35:1026–1030. [PubMed] [Google Scholar]
- 123.Lange Y, Steck TL. Quantitation of the pool of cholesterol associated with acyl-CoA:cholesterol acyltransferase in human fibroblasts. J Biol Chem. 1997;272:13103–13108. doi: 10.1074/jbc.272.20.13103. [DOI] [PubMed] [Google Scholar]
- 124.Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;40:2264–2270. [PubMed] [Google Scholar]
- 125.Okwu AK, Xu XX, Shiratori Y, Tabas I. Regulation of the threshold for lipoprotein-induced acyl-CoA:cholesterol O-acyltransferase stimulation in macrophages by cellular sphingomyelin content. J Lipid Res. 1994;35:644–655. [PubMed] [Google Scholar]
- 126.Slotte JP, Bierman EL. Depletion of plasma-membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membranes and intracellular cholesterol pools in cultured fibroblasts. Biochem J. 1988;250:653–658. doi: 10.1042/bj2500653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to insig. Proc Natl Acad Sci U S A. 2007;104:6511–6518. doi: 10.1073/pnas.0700899104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Attie AD. ABCA1: At the nexus of cholesterol, HDL and atherosclerosis. Trends Biochem Sci. 2007;32:172–179. doi: 10.1016/j.tibs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 129.Oram JF, Vaughan AM. ATP-binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006;99:1031–1043. doi: 10.1161/01.RES.0000250171.54048.5c. [DOI] [PubMed] [Google Scholar]
- 130.Wang N, Silver DL, Thiele C, Tall AR. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J Biol Chem. 2001;276:23742–23747. doi: 10.1074/jbc.M102348200. [DOI] [PubMed] [Google Scholar]
- 131.Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J. Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 132.Alder-Baerens N, Muller P, Pohl A, Korte T, Hamon Y, Chimini G, Pomorski T, Herrmann A. Headgroup-specific exposure of phospholipids in ABCA1-expressing cells. J. Biol. Chem. 2005;280:26321–26329. doi: 10.1074/jbc.M413993200. [DOI] [PubMed] [Google Scholar]
- 133.Hamon Y, Broccardo C, Chambenoit O, Luciani MF, Toti F, Chaslin S, Freyssinet JM, Devaux PF, McNeish J, Marguet D, Chimini G. ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat Cell Biol. 2000;2:399–406. doi: 10.1038/35017029. [DOI] [PubMed] [Google Scholar]
- 134.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J Biol Chem. 2006;281:36091–36101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- 135.Mendez AJ, Lin G, Wade DP, Lawn RM, Oram JF. Membrane lipid domains distinct from cholesterol/sphingomyelin-rich rafts are involved in the ABCA1-mediated lipid secretory pathway. J Biol Chem. 2001;276:3158–3166. doi: 10.1074/jbc.M007717200. [DOI] [PubMed] [Google Scholar]
- 136.Small DM. Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci U S A. 2003;100:4–6. doi: 10.1073/pnas.0237205100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vaughan AM, Oram JF. ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. J Lipid Res. 2003;44:1373–1380. doi: 10.1194/jlr.M300078-JLR200. [DOI] [PubMed] [Google Scholar]
- 138.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 139.Nagao K, Takahashi K, Hanada K, Kioka N, Matsuo M, Ueda K. Enhanced ApoA-1-dependent cholesterol efflux by ABCA1 from sphingomyelin-deficient Chinese hamster ovary cells. J Biol Chem. 2007;282:14868–14874. doi: 10.1074/jbc.M611230200. [DOI] [PubMed] [Google Scholar]
- 140.Skiba PJ, Zha X, Maxfield FR, Schissel SL, Tabas I. The distal pathway of lipoprotein-induced cholesterol esterification, but not sphingomyelinase-induced cholesterol esterification, is energy-dependent. J Biol Chem. 1996;271:13392–13400. doi: 10.1074/jbc.271.23.13392. [DOI] [PubMed] [Google Scholar]
- 141.Slotte JP. Sphingomyelin-cholesterol interactions in biological and model membranes. Chem Phys Lipids. 1999;102:13–27. doi: 10.1016/s0009-3084(99)00071-7. [DOI] [PubMed] [Google Scholar]
- 142.Ghering AB, Davidson WS. Ceramide structural features required to stimulate ABCA1-mediated cholesterol efflux to apolipoprotein A-1. J. Lipid Res. 2006;47:2781–2788. doi: 10.1194/jlr.M600380-JLR200. [DOI] [PubMed] [Google Scholar]
- 143.Witting SR, Maiorano JN, Davidson WS. Ceramide enhances cholesterol efflux to apolipoprotein A-1 by increasing the cell surface presence of ATP-binding cassette transporter A-1. J. Biol. Chem. 2003;278:40121–40127. doi: 10.1074/jbc.M305193200. [DOI] [PubMed] [Google Scholar]