

Abstract
Animal heme-containing peroxidases play roles in innate immunity, hormone biosynthesis and the pathogenesis of inflammatory diseases. Using the peroxidase-like domain of Duox1 as a query, we carried out homology searching of the NCBI database. Two novel heme-containing peroxidases were identified in humans and mice. One, termed VPO1 (vascular peroxidase 1), shows highest tissue expression in heart and vascular wall. A second, VPO2, present in humans but not in mice, is 63% identical to VPO1, and is highly expressed in heart. The peroxidase-homology region of VPO1 shows 42% identity to myeloperoxidase (MPO) and 57% identity to insect peroxidase, peroxidasin. A molecular model of VPO1 peroxidase region shows a structure that is highly similar to known peroxidases, including a conserved heme-binding cavity, critical catalytic residues, and a calcium-binding site. Absorbance spectra of VPO1 are similar to lactoperoxidase and covalent attachment of the heme to VPO1 protein was demonstrated by chemiluminescent heme staining. VPO1 purified from heart or expressed in HEK cells is catalytically active and shows a Km for H2O2 of 1.5 mM. When co-expressed in cells, VPO1 can utilize H2O2 produced by Nox enzymes. VPO1 is likely to carry out peroxidative reactions in the vascular system previously attributed exclusively to MPO.
Keywords: Animal heme-containing peroxidase, gene expression, enzyme activity, protein structure, Km, NADPH oxidase
Introduction
In mammals, heme-containing peroxidases catalyze the oxidation of a variety of substrates by H2O2, playing important roles in innate immunity, synthesis of thyroid hormone and extracellular matrix as well as in the pathogenesis of a number of inflammatory diseases such as atherosclerosis. This enzyme family has been found in organisms ranging from C. elegans and Drosophila to mammals. In human, it includes myeloperoxidase (MPO), eosinophil peroxidase (EPO), lactoperoxidse (LPO), thyroid peroxidase (TPO) (reviewed in [1]), Duox1 and Duox2 (dual oxidase) [2]. Duox1 and Duox2 are less well characterized and less closely related to the classical peroxidases. The other members of the group are highly restricted in their tissue distributions. For example, MPO is expressed in neutrophils and mononuclear phagocytes, while EPO is found in eosinophils and TPO is in thyroid [1].
MPO, the most thoroughly studied member of this family, has antimicrobial properties. Klebanoff and others showed that MPO-H2O2-halide systems exhibit broad microbicidal activity against bacteria, fungi and viruses (reviewed in [1]). Leukocytes from patients with inherited MPO defects have impaired fungicidal activity, predisposing them to disseminated candidiasis [3, 4]. LPO plays a similar antimicrobial role in saliva, airways and milk [5, 6].
TPO is a biosynthetic enzyme that plays an important role in iodide organification and iodotyrosine coupling to form iodothyronines; its genetic deficiency causes congenital hypothyroidism [7]. The human TPO gene encodes a 933 amino acid, membrane-bound, glycosylated, heme-containing peroxidase [8].
The present study describes the cloning from humans and mice and initial characterization of a heme-containing peroxidase, which we term VPO1 (Vascular PerOxidase based on its highest tissue expression in heart and vascular wall). A closely related homolog, VPO2, was also identified and is expressed abundantly in human heart, but is not considered in detail here. Both are structurally similar to the insect peroxidase peroxidasin, which was previously described [9]. In addition to its peroxidase domain, VPO1 contains several distinctive domains that are suggestive of participation in protein complexes. VPO1 expressed in live cells can utilize H2O2 generated from co-expressed Nox enzymes to catalyze peroxidative reactions. Some of these Nox enzymes are normally expressed in the same vascular cells where VPO1 is expressed. Thus, VPO1 is a novel heme-containing peroxidase and is likely to participate in H2O2 metabolism and peroxidative reactions in the cardiovascular system.
Materials and Methods
Cells and reagents
HEK293H (Invitrogen, Carlsbad, CA), Cos7 and H9c2 (Rat embryonic myocardium) (American Type Culture Collection, Manassas, VA) cells were maintained in DMEM media (MediaTech, Herndon, VA) with 10% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin. Luminol, hematin, sodium butyrate (NaBu), 3,3’,5,5’-tetramethylbezidine (TMB) and TMB liquid system, 5’-aminobenzohydrazide (ABH), N-cetyl-N,N,N-trimethylammonium bromide (CTAB), horseradish peroxidase (HRP), LPO (from bovine milk), MPO, cytochrome c, hemin, myoglobin, formic acid, dithiothreitol (DTT), bovine serum albumin (BSA) and anti-tubulin monoclonal antibody were purchased from Sigma-Aldrich (St. Louis, MO), Hanks’ solution containing 1.26 mM calcium and 0.91 mM magnesium (HBSS) and HEPES buffer from Invitrogen (Carlsbad, CA), DNA polymerase from Clontech (Clontech, Palo Alto, CA), restriction endonucleases and T4 DNA ligase from New England Biolabs (Beverly, MA), DEAE-Sepharose Fast Flow from GE Healthcare (Piscataway, NJ), chemiluminescent substrate for Western blot and heme staining from Pierce Biotechnology (Rockford,IL), anti-MPO antibody from CALBIOCHEM (Gibbstown, NJ), anti-LPO antibody from Santa Cruz Biotechnology (Santa Cruz, CA).
Identification of VPO1
A BLAST search of EST and genomic databases at the National Center for Biotechnology Information (NCBI) using the peroxidase-like domain of human Duox1 as a query sequence identified a human genomic sequence (GenBank NT_086610) on chromosome 2p25, and human and mouse EST clones such as D86983 and AK122223. The hypothetical protein translated from the genomic DNA and EST sequences had 21% identity to corresponding regions in Duox1 and 42% identity to the peroxidase domain of MPO. Current E values for BLAST search identified of VPO1 in the human genome are 10−106 (with MPO) and 3 × 10−24 (with Duox1).
Identification of VPO2
The same BLAST search as above also identified a human genomic sequence (GenBank NT_008183) on chromosome 8q11 and human EST clones such as CD637080. Chimpanzee has an orthologue (XM_519754) which is predicted by automated computational analysis at NCBI. However, the mouse orthologue could not be identified. The peroxidase region conceptually translated from the genomic DNA and EST sequences had 63% identity to corresponding regions in VPO1, 21% identity in Duox1 and 38% identity in MPO (E=5 × 10−83).
First–strand cDNA synthesis
Total RNA was extracted from cell lines with the mRNA extraction kit following the manufacturer’s protocol (QIAGEN Inc. Valencia, CA). mRNAs were reversely transcribed into first-strand cDNA with Supercript II (Invitrogen) using oligo-dT according the method provided by the manufacturer.
5’-Rapid amplification of cDNA ends (RACE)
To determine the N-terminal sequence of VPO1 and to obtain the full-length sequence, 5’-RACE was carried out using BD™ Marathon-Ready cDNAs of human fetal kidney, spleen, thyroid and testis (Clontech, Palo Alto, CA) as templates using the company’s universal primers, AP1 and AP2 (for nested PCR), and the following primers which were designed based on the EST and genomic sequences: primer 1: 5’-AGTTGCTCTAGAGAGGCAAGTCCC-3’ and primer 2: 5’-TGTGTTCAAGTTCCTCAGCCGCCT-3’ (for nested PCR). The major PCR fragments (about 350 bp) from above four tissues were cut out of the agarose gel and purified by QIAquick® Gel Extraction kit (QIAGEN Inc. Valencia, CA), respectively. The fragments were sequenced using Applied Biosystems model 377 sequencer.
PCR detection of mRNA for human and mouse VPO1
VPO1 expression patterns were determined using human and mouse Multiple Tissue PCR panels (Clontech) with the following primers: human: 5’-ATCGCAAACCTGTCGGGCTGTACC-3’ (forward) and 5’-GCAGTCCTGCCACACCCGGAGGTC-3’ (backward); mouse: 5’-CTGACCAGCATGCATACGCTGTGG-3’ (forward) and 5’-CACTGTGTGGGCCATGGAGAACAG-3’ (backward). PCR parameters were 95°C for 30 s, 62°C for 20 s, 72°C for 1 min, 35 cycles after denaturing for 1 min 30 s at 95°C.
Molecular cloning of the VPO1 cDNAs
The PCR fragments coding the N-terminus of human and mouse VPO1 were amplified from human heart cDNA and mouse 17 day embryo (Clontech, Palo Alto, CA) by using primers to introduce appropriate restriction enzyme sites. These fragments then were subcloned with PCR fragments of the C-terminus of human and mouse VPO1 from respective EST clones into a mammalian expression vector (pcDNA3). The cDNA clones of human Nox1, Nox2, Nox3, Nox4, Nox5, NOXO1, NOXA1, p47phox, p67phox, myc2-Rac1(G12V) were described in [10–12]. All cDNA clones were confirmed by DNA sequencing.
Transient transfection
Plasmids (empty vector or vector carrying cDNA) were transfected into HEK 293H cells using FuGENE 6 (Roche, Indianapolis, IN) according to manufacturer’s instructions. After 24 hr, NaBu (5 mM) and hematin (1 µg/ml) were added and incubated for 24 hrs. For experimental optimization of induction of VPO1 activity, NaBu was varied in the presence of 1 µg/ml hematin. The cells were removed from the well and washed twice with cold HBSS.
Construction of VPO1-stably expressing cell lines
pcDNA3/VPO1 was transfected into HEK 293H cells using FuGENE 6 (Roche, Indianapolis, IN) as described above. After 48 hr, cells were re-plated and G418 (500 µg/ml) was added to the media. Media containing G418 was exchanged every 3 days for 3 weeks. Nine G418-resistant colonies were isolated and grown in 10 cm plates. Peroxidase activity and VPO1 expression were verified by TMB oxidation and Western blotting with anti-VPO1 antibody, respectively. GJ-3 and GJ-4 VPO1-stably expressing cell colonies show the highest VPO1 levels and peroxidative activity.
Measurement of protein concentration
Protein concentration was determined using the Bio-Rad Protein Assay based on the Bradford dye-binding procedure. BSA served as the protein standard.
Suspension culture of VPO1-stably expressing cells
0.5 × 107-cells/ml were grown in spinner bottles with 40 ml of 293 Serum-free Medium (Invitrogen) at 37°C and 8% CO2 with stirring at 100 rpm. After two days, 60 ml of Serum-free Medium was added and the culture was continued for two days. To induce VPO1 expression, NaBu (5 mM) and hematin (1 µg/ml) were added 24 hr before harvesting. Cells were centrifuged at 1000 × g at 4°C for 10 min. VPO1 was purified from the supernatant.
Partial purification of VPO1
300 ml of the supernatant from the suspension culture of VPO1-stable cells was loaded into a 2 × 80 cm column of DEAE Sepharose Fast Flow (GE Healthcare, Piscataway, NJ) at 4°C. The column was washed with 200 ml of 20 mM potassium phosphate, pH 8.0, containing 100 mM NaCl. Sequentially, the column was eluted by a gradient of 400 ml of 100 mM NaCl and 400 ml of 1 M NaCl in the phosphate buffer. Eluent was collected in 15 ml aliquots. The peroxidase activity was monitored by TMB oxidation. The sample with the strongest peroxidase activity was then used for spectroscopy.
UV visible spectra
Spectra of VPO1, LPO, MPO and myoglobin were recorded from 350 nm to 700 nm using an AMINCO DW-2000 spectrophotometer according to the method described in [13]. Reduced spectra were obtained by adding a few crystals of solid sodium dithionite. Pyridine hemochrome spectra were obtained by adding pyridine to the reduced proteins to a final concentration of 2.4 M. Absorption spectra were also recorded in 88% (v/v) formic acid.
Turnover number and Km value
5 µl each of LPO, MPO, hematin, myoglobin, cytochrome c, or partially purified VPO1, were added to 95 µl of TMB solution containing the indicated concentration of H2O2 (Sigma-Aldrich) and the reaction was recorded using a THERMOmax Microplate Reader (Molecular Devices, Sunnyvale, CA) equipped with Softmax Kinetic analysis software. The turnover numbers were calculated based on the formula: ΔA/min=εCTMB and T.N. = CTMB/Cheme (ΔA/min: absorbance changes in a minute; ε: extinction coefficient of TMB, 3.9 × 104mol−1cm−1; CTMB: TMB generated in 1 minute; T.N.: turnover number, min−1 per heme; Cheme: heme content of indicated hemoproteins). For Km determinations, 2 µl of VPO1, hematin, myoglobin or cytochrome c was added into potassium phosphate buffer, pH 5.4 with 100 mM NaCl and 0.2 mM TMB in 100 µl reactions, respectively. The reaction was initiated by added series of H2O2 (4, 8, 16, 32, 64, 128, 256, 512, 1024, 2048 and 4096 µM), respectively. Vmax and Km were obtained using a non-linear least squares fit to the Michaelis-Menten equation. For display purposes, a Lineweaver-Burke plot is also shown. The relative activity of VPO1, hematin, myoglobin and cytochrome c vs. LPO using luminol as substrates is also compared. The reactions were initiated by adding 4 µl of 5 mM H2O2 into 94 µl HBSS with 1 mM luminol and 2 ul of above proteins (range from 3.5 to 5 µM), respectively. The light emission was recorded using FluoStar™ luminometor every 30 second for 20 min.
Homology modeling
A homology molecular model for the VPO1 peroxidase domain was constructed using the INSIGHTII/HOMOLOGY package from Accelrys Software, Inc. (San Diego, CA). Initially, the VPO1 sequence was aligned using CLUSTAL W with the sequences of the solved structural homologues MPO, LPO, and the more distantly related prostaglandin synthase (PGS). Several related sequences including mammalian and invertebrate Duox peroxidase-like protein domains were included in the alignment to provide additional clues. The structures of the basis set proteins were aligned using Homology; the PGS structure had to be aligned manually. The CLUSTAL alignment was edited within INSIGHT to bring structurally equivalent residues of the solved proteins into alignment; in practice, this required some adjustment of the alignment of PGS vs. the two solved peroxidases. Some manual editing was also necessary to align VPO1 with the basis set proteins.
Peroxidase activity assay in cell lysates
Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1mM EDTA) with 0.1% CTAB. 50 µg of protein was added to 100 µl TMB Solution (Sigma). The reaction mixture was incubated at room temperature for 30 min. The absorbance was recorded at 650 nm. Peroxidase activity in the cell lysate was also measured using luminol-based chemiluminescence, slightly modified according as in [9]. 25 µg of protein was mixed with 200 µM of luminol and H2O2 as indicated in 200 µl. Luminescence was immediately quantified using a FluoStar™ illuminometer (BMG Labtech, Durham, NC) and luminescence was recorded every 10 to 50 second for 1 hr upon the numbers of samples.
Peroxidase activity assay in live cells
Because luminol is cell permeable, it can be used to detect peroxidase activity in live cells, essentially according to previous reports [14]. Briefly, for each well in a 96-well plate, 2 × 105 cells (transient-transfected or VPO1 stable cells) in HBSS or 10 mM potassium phosphate (pH 7.4) were mixed with 200 µM luminol and an indicated concentration of H2O2 in a total volume of 200 µl. In some experiments, H2O2 was omitted, demonstrating the utilization of endogenously produced H2O2. In these experiments, Nox enzymes along with their regulatory subunits were cotransfected with pcDNA3/VPO1 into HEK293H cells as described [10–12]. Luminescence was quantified using a FluoStar™ luminometer and luminescence was recorded every 10 to 20 second for 1 hr.
Anti-VPO1 antibody
A region of the VPO1 peroxidase domain with highly predicted antigenicity was chosen using DNAStar™ software (Madison, WI). The peptide (corresponding to residues 1197–1211) was synthesized, purified by reverse phase high performance liquid chromatograph and conjugated with keyhole limpet hemocyanin by Sigma-Genosys (The Woodlands, TX). Anti-VPO1 antibody was raised in rabbit against the conjugated peptide [Sigma-Genosys (The Woodlands, TX)]. Antibody was purified using Protein-G agarose beads according to standard procedures. The specificity of the antiserum was determined by Western blotting using the overexpressed VPO1 and compared with MPO (purified, Sigma-Aldrich), bovine LPO (purified, Sigma-Aldrich), EPO (cell lysat of EPO-transfected HEK293 cells) and TPO (cell lysat of TPO-transfected HEK293 cells). The antibody was specific for VPO1 and did not cross-react with MPO, LPO, TPO or EPO as shown in supplementary data, Figure 1S.
Western blot analysis
Cells or homogenized tissues were lysed in RIPA buffer or RIPA buffer with 0.1% CTAB containing protease inhibitor mixture (Sigma). Lysate (50 µg of protein) was resolved by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes using a Mini Tank Transfer System (Bio-Rad Laboratories, Hercules, CA) at 80 V for 1.5 hr. Proteins were detected using Western blotting, visualized by chemiluminescence (Pierce, SuperSignal West Pico Chemiluminescent Substrate).
Immunohistochemistry
Immunohistochemistry was carried out according to [15] using the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). Sections of mouse carotid arteries were incubated with a 1: 800 dilution of anti-VPO1 antibody and stained with diaminobenzidine tetrahydrochloride (DAB) and hematoxylin. The negative control was treated with PBS instead of primary antibody.
Detection of covalently-bound heme
Covalently-bound heme was detected using a chemiluminescence-based method that detects the peroxidase activity of heme retained in protein subjected to SDS-PAGE and blotting [16]. In brief, approximately 2 µg of myoglobin, cytochrome c and 1 µg of partially purified VPO1 were prepared in loading buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 0.5% bromphenol blue and 20% glycerol). The samples were run on 15% SDS-PAGE and transferred to PVDF membrane. The membrane was washed three times with 1 X PBS and covered with Pierce’s chemiluminescent substrates (luminol/enhancer) for 5 min. The membrane was exposed to X-ray film to detect luminescence. The same membrane was subject to Coomassie’s Blue staining according to standard procedures.
RESULTS
Identification and cloning of human and mouse VPO1
Fig. 1A shows a dendrogram of human heme-containing peroxidase family including VPO1 and VPO2, described herein (only the peroxidase domains are compared). VPO1 and VPO2 were initially identified by BLAST searching of the NCBI EST and genomic database using the peroxidase domain of human Duox1 as a probe; analysis of genome databases shows that the VPO1 gene is located on chromosome 2p25 while VPO2 is on 8q11. VPO1 and VPO2 are approximately 63% identical, and fall within a subfamily that also contains MPO, EPO, LPO and TPO. They are more distantly related to the Duox peroxidase-like domains, but are closely related both in sequence and domain structure to the previously described insect peroxidase peroxidasin [9] (see Discussion).
Fig. 1.


Identification of VPO1. A. Shown is a dendrogram showing the similarity based on sequence identity among the peroxidase domains of human heme-containing peroxidases. B. Shown are results from 5’-RACE of VPO1 from four human tissues. 5’-RACE was carried out as described in Materials and Methods. The 350 bp bands (arrow) from the four tissues were extracted from the gel and verified by DNA sequencing. M is DNA size marker and its size is shown on left. C. Shown are predicted domain or motif structures of VPO1, VPO2 and other heme-containing peroxidases. D. Alignment of amino acid sequences of the peroxidase domains of the human heme-containing peroxidases. Filled circles indicate conserved residues that in MPO are predicted to form heme binding cavity[24]. The residues of axial and distal histidine are at 1074 and 827, respectively. The highly conserved sites for binding to calcium are indicated by filled square (Asp828) and the horizontal superior line. All these conserved residues are shown in homology model of VPO1 (Fig.2).
Herein, we focus on the identification and characterization of VPO1 since it appears to be more highly expressed than VPO2 in adult vascular tissues. VPO1 represents the complete sequence of a gene product that was previously hypothetically identified by Mitchell et al [17] as a homolog of the interleukin 1 receptor antagonist using subtractive hybridization and EST sequence assembly. Mitchell et al [18] reported the gene (termed it Melanoma-associated Gene 50 or MG50), which is located on chromosome 2p25.3, was overexpressed in melanoma. However, the authors did not determine the 5’-end nucleotide of the hypothetical sequence in the assembly. In the present study, the complete sequence includes 5’-end was determined by the 5-RACE (see below). Horikoshi et al. [19] reported a short form of this gene, which contains approximately half sequence of the complete sequence of VPO1, and included the peroxidase region and VWC domain. This sequence may represent an alternative splicing form of VPO1. This short form was induced by p53 in colon cancer cells and was therefore termed p53 response gene 2 (PRG2). As shown in Fig. 1B, 5’ RACE from four human tissues (fetal kidney, spleen, testis, and thyroid) showed a major band at 350 bp, and these fragments were verified as 5’ region of VPO1 by automatic sequencing. This region was also confirmed in mouse heart and 17-day mouse embryonic tissue (data not shown). This region includes an ATG initiation codon; the open read frame (TCGGCCatgG) that corresponds to a classical Kozak consensus sequence (GCCRCCatgG), where R represents purine [20]. During the course of our studies, an updated computational sequence (GenBank accession number XM_056455.6) shows a gene with a predicted protein sequence identical to the VPO1 sequence reported here. The entire VPO1 amino acid sequence is also identical to a sequence contained in the KIAA0230 clone, which was originally isolated from a cDNA library from a human myeloid cell line enriched for relatively long cDNA transcripts [21]. Thus, the reported sequence represents the major form of VPO1 in multiple tissues although other minor N-terminal splicing forms cannot be ruled out. This VPO1 sequence is deposited in GenBank with accession number EF090903. VPO2 was identified by a similar strategy, and the sequence is deposited with accession number EU170240.
By alignment, human and mouse VPO1 show 91% identity at the amino acid level, similar to the 86% identity shared by human and mouse MPO. Analysis of the gene sequence (GenBank) shows that the VPO1 gene is about 110 kb and contains 23 exons and 22 introns. Interestingly, VPO1 is located tail-to-tail with TPO at 2p25. The domain structure of VPO1 and VPO2 is shown in Fig. 1C, and indicates that the peroxidase domain located in the C-terminal half of the molecule. At 165 kDa, the peroxidase is predicted to be much larger than MPO (89 kDa, precursor form) and the other classical peroxidases. The N-terminus contains a signal peptide sequence as predicted by the SignalP server (Technical University of Denmark). MPO also has a signal peptide sequence; a portion of this enzyme is secreted while the majority is retained in cells [22]. The N-terminal half of VPO1 is comprised of multiple predicted subdomains including: 1) leucine-rich repeats (LRRs); 2) leucine-rich repeat N-terminal or C-terminal domains, which are often located N- or C-terminal to LRRs; 3) immunoglobulin C-2 type domains. VPO1 also contains a von Willebrand factor type C domain (VWC) at the extreme C-terminus following the peroxidase domain. These structural domains (except for the peroxidase domain) have been found in other proteins to mediate protein-protein interactions, but the binding partners for VPO1 are unknown.
The peroxidase domain of VPO1 has 42%, 41%, 39%, 41%, 21% and 19% identity to the corresponding region of MPO, EPO, LPO, TPO, Duox1 and Duox2, respectively. Fig. 1D shows the alignment of the peroxidase domain of the human heme-containing peroxidase family. Like MPO, EPO, LPO and TPO, VPO1 contains the catalytically-important conserved axial ligand and distal histidine residues as well as many conserved residues in the heme binding cavity known to participate in heme binding in other peroxidases. Thus, the heme-binding and catalytic site of VPO1 is similar to the other classical members of this family.
While the present study is the first to describe and characterize mammalian VPO1, in fact the gene is also widely distributed among animal species including C.elegans, Drosophila, cow, and monkey. Nelson et al. [9] reported a Drosophila homolog, peroxidasin (referring to peroxidase in insect), which functions in the biogenesis of extracellular matrix during embryogenesis of hematocytes (see Discussion). VPO1 and VPO2 have similarly predicted domain structures, implying evolutionary conservation and perhaps orthologous biological function.
Molecular model of VPO1
Fig. 2 shows a molecular model of the VPO1 peroxidase domain, constructed as described in Materials and Methods. The model reveals 10 conserved cysteine residues that formed five cys-cys cross bridges in the other peroxidases (except for prostaglandin synthase, PGS), conserved proximal histidine (axial heme ligand) and distal histidine [23], conserved amino acid residues that bind to the heme propionate group (Asp830, Arg1071 and Arg1161) [24], and two conserved acidic residues which in LPO and MPO are covalently linked to heme vinyl groups (Asp826 and Glu980) [24]. However, the third position, which is covalently linked to a heme vinyl group in MPO (Met409) and contributes to its unique Soret peak at 430 nm, is not conserved in VPO1, which instead contains Gln981 at the corresponding position. This is consistent with the Soret peak of VPO1 at 410 nm (see spectrum of VPO1, Fig.5). In addition, four conserved residues (corresponding to residue numbers 334 and 338–340 in MPO) and the conserved aspartic acid residue (residue number 262 in MPO) form a high affinity calcium-binding site, a common feature of most animal peroxidases (but not PGS). A large number of other residues within the structural core of the VPO1 molecule were highly conserved for presumed structural reasons. The presence of a large number of important conserved residues over much of the length of the sequence lends confidence in the general accuracy of structural model.
Fig. 2.
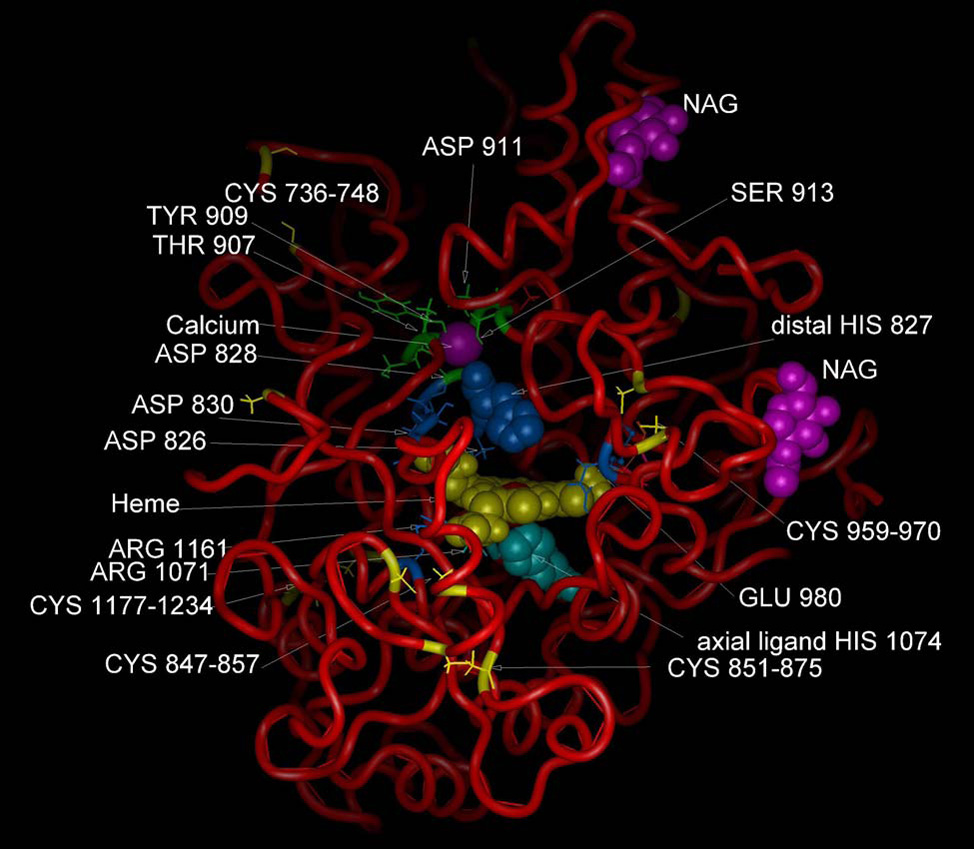
Predicted molecular structure of VPO1. A structural homology model was constructed as described in Materials and Methods, and utilized LPO, MPO and PGS as a basis set. Heme is indicated in yellow. Calcium ion is shown in purple and predicted residues binding to calcium are Asp828, Thr907 Tyr909 and Asp911. There are two putative N-acetylglucosamine (NAG) sugar moieties shown. Residues comprising the conserved heme-binding cavity are shown. These include conserved axial and distal histidines (1074 and 827, respectively), residues cross-linking to the vinyl groups of heme (Asp826 and Glu980) and residues interacting with the heme propionate group (Asp830, Arg1071,and Arg1161) [24]. The highly conserved Cys-Cys disulfides are also indicated.
Fig. 5.

UV-visible absorbance spectra of human partially purified full-length VPO1. A. Spectra of expressed, partially purified VPO1 are shown, including oxidized VPO1, oxidized VPO1 in pyridine (pyr), reduced VPO1 in pyridine, and oxidized VPO1 in 88% formic acid. B. Reduced spectra of pyridine hemochrome. Reduced spectra of pyridine hemochrome were obtained by addition of pyridine to 2.4 M and a few crystals of sodium dithionite. The reduced spectrum showed a split Soret band with features at 393 and 413 nm. The presence of the symmetrical Soret peak of VPO1 in above conditions provides evidence for a native environment for the heme. C. Spectra from 450 nm to 700 nm of B.
Coordinate assignment was carried out using MPO and LPO structures and to a lesser extent using PGS. Over 98% of the structure could be assigned directly from the basis set. Three short surface loops differed by one residue from the basis set structure and required loop replacement from the Protein Data Bank (PDB). One five-residue insertion in a surface loop remote from the access channel required similar treatment. These are the only regions of low confidence. Minor relaxation of side chains and loops using DISCOVER (Accelrys Software) produced an energetically reasonable structure with no serious steric overlaps.
The VPO1 peroxidase domain shares the common core structure of the three basis set proteins (MPO, LPO and PGS). The fold forms a helical cage with an unusually wide ‘slot’ providing access to the catalytic site. A small amount of beta structure is present, primarily at one edge of the fold. The extreme C-terminal region, covering about 50 residues, is conserved among LPO, MPO and VPO1, but not with PGS, and forms a characteristic mammalian peroxidase C-terminal motif including short helical regions and a conserved cys-cys bond.
The common core appears to contain at least 25 conserved helical regions characteristic of the mammalian peroxidases. LPO structures have these helices and three very short (four or five residues) additional regions defined as helical; MPO has the twenty five conserved helices and one very short helix that LPO lacks. While small differences in Ramachandran angles and backbone H bonding exist, the main chain in these proteins follows a similar path in these regions and the helical character of the four short nonconserved helical regions is of secondary importance. The very small amount of beta structure appears conserved.
Highly conserved secondary structural features include two long helices that form a twisted helical hairpin structure that provides the spine of the proteins’ fold. Three additional well-conserved helices run across the faces of the heme. These helices and immediately adjacent loop regions supply the distal and proximal histidines, propionate group ligands, and many of the hydrophobic residues that line the heme pocket. The heme pocket and catalytic site have an unusually wide access channel, suggesting that a variety of substrate structures may gain access to the active site.
The Ca+2 binding site is well conserved among these peroxidases [25]. Three of the Ca+2 ligands are supplied within a compact loop region as shown in the alignment, but the fourth ligand is located within one of the regions which is involved in heme binding and catalytic site conformation. The ligand is directly adjacent to the distal histidine and close to propionate group ligands and one of the acidic groups that forms a covalent bond with a vinyl group on the heme periphery. This indicates strong coupling between the binding of calcium and the conformation of the heme pocket and catalytic site.
Tissue distribution
RT-PCR (Fig. 3A) revealed VPO1 mRNA expression in multiple adult and embryonic tissues including heart, liver, lung, pancreas and placenta. Lesser expression was observed in brain and skeletal muscle. In addition, VPO1 message is expressed in spleen, testis and thyroid (Fig. 3A). VPO1 message is also present in mouse embryo (7 to 17 days). Because for some proteins, protein expression differs considerably from mRNA expression, western blot analysis was carried out using Protein-G purified anti-VPO1 antibody. As shown in Fig. 3B, VPO1 protein was abundant in mouse heart and H9c2 cells, but was not detected in other tissues, indicating that mRNA expression does not correspond exactly to protein expression. Using immunohistochemistry, significant levels of VPO1 were also detected in vascular wall, both in endothelium and smooth muscle (Fig. 3C). These data indicate that VPO1 protein is highly expressed in the cardiovascular system. Thus, we refer to the protein as VPO1 or “Vascular PerOxidase 1”, conforming to the convention of naming mammalian heme-containing peroxidases according to their major tissue expression.
Fig. 3.

Tissue distribution of VPO1. A. RT-PCR was used to detect VPO1 in a variety of tissues as indicated using human- and mouse-specific PCR primers as described in Materials and Methods. The identity of the PCR fragment from human heart and mouse 17 day embryo was confirmed by DNA sequencing. B. A Western blotting was carried out as described in Materials and Methods using Protein-G purified anti-VPO1 polyclonal antibody in mouse (left); and in the rat myocardium cell line H9c2 (right). The lower panel uses anti-tubulin antibody as loading control. C. Immunohistochemistry of mouse carotid artery. Immunohistochemistry was performed as described in Materials and Methods. In the left panel primary antibody is omitted. The primary antibody used in the right panel was the same anti-VPO1 antibody as in B.
Measurements of VPO1 activity
To determine whether the cloned VPO1 has peroxidase activity, full-length VPO1 was transfected into HEK293H cells. After 48 hr incubation, the cells were harvested and lysed in RIPA/CTAB buffer. The peroxidase activity was detected by TMB oxidation. However, activity initially was indistinguishable from vector control. Because VPO1, like other members of this family, is a heme-containing protein, the lack of activity may have been due to inadequate incorporation of heme into VPO1 apoprotein. Therefore, 1 µg/ml of hematin was incubated with VPO1-transfected HEK293H cells for 24 hr. However, the peroxidase activity increased only slightly, and VPO1 protein expression was as low as measured by Western blot (data not shown). NaBu is an inhibitor of histone deacetylases and can selectively induce the expression of certain genes [26], both in transient transfection and stable cells [27, 28]. Fig. 4A shows that the VPO1-dependent TMB oxidation was effectively reconstituted in VPO1-transfected HEK293H cells in the presence of both hematin and NaBu. The dependence of peroxidase activity on hematin and NaBu in cells stably expressing VPO1 was the same as in the transiently transfected cells (data not shown).
Fig. 4.

Reconstitution of VPO1 activity. A. Effect of NaBu and hematin on cellular peroxidase activity. Empty vector (1) or expression vector encoding VPO1 (2) was transfected into HEK 293H cells as described in Materials and Methods. The cells were cultured in 5 mM of NaBu and 1 µg/ml of hematin at 37%, 5% CO2 for 24 hrs before harvest. The TMB oxidation was carried out using lyzed cells. Error bars show the range of three independent experiments. Inset shows a Western blot indicating VPO1 expression in one of the experiment. B. VPO1-stably expressing cells were incubated with 5 mM NaBu and 1 µg/ ml hematin for indicated time. Western blotting was carried out to determine the VPO1 expression levels using anti-VPO1 antibody while the same membrane was blotted with anti-tubulin antibody as a loading control. C. Optimization of NaBu concentration. Cells were grown to 80% confluence, and 1 µg/ml hematin plus the indicated concentration of NaBu were added to the media and cells were cultured for an additional 24 hrs. TMB oxidation was carried out as described in A. GJ-3 and GJ-4 are two cell colonies that stably-expressing VPO1 and are derived from HK293H cells as described in Materials and Methods. D. Peroxidase activity in living cells. Luminol-based chemiluminescence was measured in living cells as described in Materials and Methods at the indicated concentrations of exogenously added H2O2. Data are representative of three independent experiments. RLU refers to relative light units. E. Inhibition of VPO1-dependent peroxidation of TMB by ABH. HEK293H and GJ-4 cells were incubated with 5 mM NaBu and 1 µg/ ml hematin for 24 hrs before harvesting. TMB oxidation was measured as described in A except that the indicated concentration of ABH was added to the incubation.
While neither hematin nor NaBu alone significantly increase VPO1 activity, NaBu alone induced VPO1 protein expression about five fold (Fig 4B). In the presence of both hematin and NaBu, VPO1 activity increased significantly implying that significant VPO1 apoprotein expression is induced by NaBu, but exogenous heme is needed to optimally reconstitute active holoenzyme (Fig.4B). Fig. 4C shows that VPO1 activity is dependent on NaBu in a dose-dependent manner in the presence of hematin. The optimal concentration of NaBu for VPO1 expression was found to be 5 mM; at higher concentrations, NaBu caused significant apoptosis after 24 hr, consistent with previous reports [29]. The effect of these supplements was time dependent, showing optimal induction at 24 hr. The longer incubation, even at 5 mM NaBu, induced apoptosis (data not shown). As shown in Fig. 4D, VPO1 activity in live cells was H2O2-dose dependent. As shown in Fig. 4E, the VPO1-dependent peroxidase activity was inhibited in a dose-dependent manner by ABH, a broadly specific inhibitor of heme-containing peroxidases. Taken together, these data establish the in vivo activity of the novel heme-containing peroxidase VPO1.
Spectral characterization and covalent heme binding in VPO1
Fig. 5A shows the native ferriheme UV-visible absorbance spectrum of partially purified oxidized VPO1 as well as VPO1 in formic acid, pyridine and the reduced form in pyridine. The ferric enzyme has a sharp Soret maximum at 410 nm. The reduced form of VPO1 in pyridine is compared with those of MPO, LPO (containing a covalently-bound heme) and myoglobin (containing non-covalently bound heme) in Figs. 5B, with the alpha and beta bands expanded in Fig. 5C. UV-visible absorbance spectral properties of the homologous peroxidase perxidasin were not previously reported.
The spectrum of the reduced form of VPO1 in pyridine was similar to that of LPO under the same conditions and differed significantly from that of myoglobin (Fig. 5B and 5C). As in LPO, there is a split Soret band with features at 393 and 413 nm. Because of slight turbidity in the VPO1 preparation, the latter peak is visible as a shoulder. The alpha band of the reduced pyridine complex of VPO1 is significantly blue-shifted compared to the corresponding myoglobin-derived complexes (550 versus 554 nm, respectively) (Fig. 5C). In contrast, the LPO complex has a red-shifted alpha band, with the peak at 559 nm. The reduced MPO pyridine hemochrome spectrum is radically red shifted (peak reported at 580 to 586 nm in various preparations [30, 31] compared to LPO. The atypical spectra of LPO and MPO are due to covalent attachment of the heme to the polypeptide, which prevents the complete removal of protein-heme interactions in the pyridine hemochrome experiment, and allows the protein to perturb what would otherwise be identical to a hematin-pyridine spectrum. The covalent attachment is observed in crystal structures of LPO and MPO, and involves acidic amino acid residues that are covalently linked to peripheral heme vinyl groups. These acidic residues are conserved in VPO1 making similar attachments likely. Thus, the most probable interpretation of the split Soret bands in VPO1 is that there are several forms of the enzyme in the sample representing covalently and possibly non-covalently-bound hemes.
To investigate directly whether heme was covalently bound to VPO1 protein, protein was subjected to SDS-PAGE and blotted, and covalently-associated heme was detected using a chemiluminescence method as in [16]. Using this method, hemoproteins containing non-covalently bound heme such as myoglobin are not detected because heme is lost during SDS-PAGE. In contrast, heme is retained by hemoproteins containing covalently bound heme. As previously reported [16, 32], heme is detected in the control protein, cytochrome c (Fig. 6, upper panel). As expected, no heme was detected in myoglobin, despite abundant protein staining (Fig. 6, lower panel). Heme was detected in VPO1 following SDS-PAGE and blotting as with cytochrome c (Fig. 6). This indicates that at least a portion of VPO1 contains covalently bound heme, consistent with the molecular model showing conservation of two of the three residues that in MPO form a covalent bond with the heme.
Fig. 6.

VPO1 covalently binds to heme. The experimental procedures are described as in Materials and Methods. The blot in the upper panel was developed with Pierce’s chemiluminescent substrate and chemiluminescent signal was detected by exposure to X-ray film. After extensive washing, the same blot was stained with Coomassie’s Blue and destained using standard procedures (lower panel). 1. myoglobin; 2. cytochrome c; 3. VPO1. Molecular size scale is shown on the left.
The low spin species with peaks at ~417 nm (yielding the Soret shoulder at 413 nm), 550 nm (alpha band), and 520 nm (beta band) is a six coordinate low spin complex which may be bis-pyridine or a mixed ligand complex with pyridine and a protein-derived ligand (e.g., histidine). The 393 nm band is typical of a high-spin Soret feature and may represent a heme population with only one axial ligand, almost certainly pyridine. The likely cause of the heterogeneity is the existence of different levels of peroxide driven covalent linkage to the acidic residues mentioned above. Molecules with no links, one link and two links may be simultaneously present.
To obtain an extinction coefficient for VPO1, the enzyme was dissolved in an 88% solution of formic acid. The heme spectrum of VPO1 in formic acid was almost identical to the spectrum of LPO under the same conditions [13] with a Soret peak at about 397 nm (Fig. 5A). Using LPO in formic acid as a standard, we calculated the concentration of VPO1 heme in solution and obtained an extinction coefficient for the ferric Soret band of native VPO1 as approximately 112 ±10 mM−1cm−1, the same within experimental error as the Soret band extinction coefficient of LPO. Using heme concentrations obtained from this extinction coefficient, the alpha band of the reduced pyridine hemochrome of VPO1 is calculated to have an extinction coefficient of around 22 mM−1cm−1, very similar to that of the corresponding band for LPO [33]. This provides independent confirmation of the extinction coefficient. Because the effects of turbidity are less at longer wavelengths, the use of this band is preferable for calculating concentration from the pyridine hemochrome in turbid samples.
Based on the heme content calculated using the above extinction coefficients, and the protein amount in preparations, the best preparations of VPO1 can be calculated to have a heme content of 1.64 nmol/mg protein, compared with a theoretical heme content of 6.0 nmol/mg protein based on a molecular weight of 165,274 Daltons. Thus, preparations of VPO1 can be estimated to be approximately 30% pure, consistent with SDS-PAGE showing numerous smaller bands in addition to the predicted 165 kDa band. This less-than-theoretical purity may also reflect a significant content of VPO1 apoprotein. Extensive attempts to further purify the material were not successful and resulted in loss of most of the activity (data not shown)
Turnover number and catalytic parameters of VPO1
Based upon the heme extinction coefficient and activity towards oxidation of TMB and luminol, we determined the turnover number (mols substrate oxidized per min per mol heme) of VPO1, and compared it to the peroxidase activities of LPO, hematin, myoglobin and cytochrome c (Table 1). The turnover number of VPO1 with TMB is much higher than that of hematin, myoglobin and cytochrome c, and is inactivated by heat, in contrast to the heat stability of pseudoperoxidase activities of hematin, myoglobin and cytochrome c. The activity towards TMB, while quite significant, was considerably lower (about 0.44%) than that of LPO (Table 1). Likewise, VPO1 activity was considerably higher towards luminol than the pseudoperoxidase activities of hematin, myoglobin and cytochrome c, but lower (about 4%) than the activity of LPO (Table 2).
Table 1. Turnover number of VPO1.
Turnover number (min− per heme) is measured by TMB oxidation and calculated as in Materials and Methods. Data are the representative of three independent experiments.
| Hemoprotein | Native | Heat inactivated |
|---|---|---|
| VPO1 | 1235 | 38 |
| LPO | 2.8 × 105 | 224 |
| Hematin | 288 | 278 |
| Myoglobin | 155 | 92 |
| Cytochrome c | 408 | 463 |
Table 2. Relative activity comparing with LPO.
Relative activity is expressed as percentage of LPO activity. Luminol oxidation was carried out in HBSS solution with 1 mM luminol and 100 µM H2O2.RLU was recorded using a FluoStar™ luminometer. Data are the average of three independent experiments.
| Component | Luminol assay (Relative %) |
|---|---|
| LPO | 100 |
| VPO1 | 4.0 ± 0.2 |
| Hematin | 0.5 ± 0.1 |
| Myoglobin | 0.6 ± 0.1 |
| Cytochrome c | 1.0 ± 0.1 |
It is not clear whether the lower turnover number of VPO1 compared with LPO reflects the intrinsic catalytic activity of the native enzyme, whether this is due to the purification which might remove important activators, or if this results as an artifact of the recombinant overexpression system. Interestingly, peroxidasin also shows a low peroxidase activity (about 2–5% compared with ovoperoxidase, MPO and LPO) [9]. VPO1 activity was also tested at a range of calcium concentrations from 100 nM to 1 mM, which was without significant effect on the activity (data not shown). In addition, the pH was varied from 3.6 to 8.0, and the optimal pH of 6.0 (data not shown) was used in these experiments. As discussed above, recombinant partially purified VPO1 exists in multiple states, which probably reflects various modes of heme linkage to the protein; it is possible that such inhomogeneity might contribute to the lower turnover number of VPO1 vs. LPO if some of these states have low activity. This is typical of animal peroxidases [e.g., as described in [13]]. While the activity of VPO1 in cells appears to be considerable (see Fig. 4), it has not yet been possible to calculate an in situ turnover number. Thus, whether other co-factors or protein-binding partners up-regulate VPO1 activity remains to be elucidated.
The Km value (1.5 mM ) of VPO1 for H2O2 was determined using a non-linear least squares fit of H2O2 concentration versus rate data (Fig. 7), using TMB as a substrate as described in Materials and Methods, while the Km for MPO and LPO are 0.5 mM and 0.2 mM, respectively [34, 35]. The data indicate that the Km of VPO1 is about 3-fold and 7.5-fold higher than MPO and LPO, respectively.
Fig. 7.

Lineweaver-Burk plot of TMB oxidation by VPO1. Inverse rate data were plotted as a function of the inverse of H2O2 concentration. The Km for H2O2 using TMB as a substrate was determined as described in Materials and Methods. Data are the average of three independent experiments.
Cellular VPO1 can utilize H2O2 generated from Nox enzymes
Sources of cellular H2O2 reportedly include the mitochondrial electron transfer chain, xanthine oxidase (XO), and NADPH-oxidases (Noxes). The Nox enzymes have recently gained attention as the major sources of ROS under a variety of physiological and pathological situations [36]. Most enzymes of the Nox family form H2O2 by generating superoxide, which dismutates to form H2O2. Nox2 is the predominant source of H2O2 in phagocytes, but accumulating evidence now points to Nox enzymes as a predominant source of H2O2 in many non-phagocytic cells such as the cells of vascular walls [37]. Luminol, a cell-permeant substrate for heme-containing peroxidases, has been extensively used to investigate MPO activity in live polymorphonuclear leukocytes, and to detect Nox-dependent H2O2 generation in the presence of HRP. We previously used luminol oxidation in the presence of HRP to demonstrate that the various Nox enzymes, when co-transfected with their cognate regulatory subunits, generate H2O2 in cells [10–12]. To determine whether VPO1 utilizes H2O2 generated by cellular expressed Noxes, VPO1 was co-transfected with the various Nox enzymes together with their respective regulatory subunits. In this assay, exogenous HRP was omitted so that the luminol oxidation would occur solely from endogenous peroxidase activity. As shown in Fig. 8, Noxes alone or VPO1 alone produced only a small rate of luminol oxidation. However, the signal greatly increased in VPO1 transfected cells when any of the Nox enzymes were present in an active form. Thus, VPO1 can utilize H2O2 generated in cells by expressed Nox1, Nox2, Nox3, Nox4 and Nox5. These data suggest that VPO1 may catalyze peroxidations in the cardiovascular system by utilizing H2O2 generated by Nox enzymes.
Fig. 8.

VPO1 utilizes H2O2 generated by Nox enzymes. Plasmids encoding the indicated Noxes or Nox regulatory proteins were transfected into HEK293H cells as described in Materials and Methods. After 24 hr, NaBu and hematin were added as in Fig. 4 and cells were allowed to continue in culture for an additional 24 hrs. Luminol-based chemiluminescence was then detected in the absence of added H2O2.The data are representative of three independent experiments. For the Nox2 group, the cells were treated with 200 nM phorbol 12-myristate 13-acetate at 37°C for 10 min prior to the assay.
DISCUSSION
Animal containing peroxidases are conserved in organisms ranging from invertebrates to humans. The structural conservation of the peroxidase domain and localized expression patterns are consistent with their fundamental tissue-specific biological functions. TPO, for example, catalyzes the iodination of thyroid hormone, while MPO, LPO and EPO all have an antimicrobial activity resulting from oxidation of microbial biomolecules. While the function of VPO1 is not yet known, one can speculate that there is a role in the cardiovascular system, where the VPO1 protein is highly expressed. (This is in contrast to its mRNA, which shows a fairly widespread expression, consistent with an earlier report [19].) Such a role in the cardiovascular system could involve defense against microbes that enter or colonize the vasculature; an analogous role is seen with LPO in the salivary gland and bronchi where it has an antimicrobial effect in the alimentary and upper airway systems [38]. Alternatively, VPO1 may carry out peroxidative reactions in the vascular system that were previously attributed exclusively to MPO, and these might participate in the development of atherosclerosis. A role in modification of extracellular matrix proteins is also possible, based on the analogous role of the VPO1 homolog peroxidasin in insects [9].
VPO1 and VPO2, which are similar to peroxidasin in molecular structure [9], are unique among the members of the mammalian peroxidase family in the structure of their N-termini. VPO1 contains multiple motifs that in other proteins participate in protein-protein interactions and protein-lipid interactions. The C-terminus also contains a potential protein-protein interaction motif not present in other peroxidases. The LRR regions are widely distributed amphpathic motif that frequently mediate protein-protein and protein-lipid interactions, e.g., as in cell adhesion of fibronectin-leucine-rich transmembrane protein [39]. LRR also mediate the binding of enzymes to regulatory proteins such as the interaction between adenylyl cyclase and Ras [40]. The immunoglobulin constant chain domains are ~100 amino acids in length and are related to an extracellular domain found in both class I and class II major histocompatibility complex (MHC) alpha and beta chains. Immunoglobulin-like domains are typically involved in protein-protein and protein-ligand interactions, as with vascular cell adhesion molecule-1 (VCAM-1) binding to integrins increasing leukocyte adhesion to endothelium [41]. In addition, the VWC domain is found in multidomain protein/multifunctional proteins, and is thought to mediate complex formation and to participate in numerous biological events including cell adhesion, migration, and signal transduction such as bone morphogenetic protein signaling modulated by chordin and chordin-like 2 [42]. In contrast to VPO1 and VPO2, other heme-containing peroxidases do not possess obvious motifs for protein-protein interactions. Such LRR, immunoglobulin constant chain domains and VWC domain are also present in peroxidasin [9]. The latter protein was reported as a trimer, based on the evidence of its electron micrographs and native SDS-PAGE, so it is possible that these regions participate in homotrimerization of VPO1 and VPO2. Alternatively, these regions may mediate binding of the peroxidase to specific target biomolecules, and this may act to direct peroxidase in reactions with towards specific substrates. Such protein-protein interactions might mediate activation or targeting of the peroxidase activity under specific biological conditions such as inflammation or infection. It is currently unknown whether activation of VPO1 occurs under some conditions, but this is consistent with the fact that VPO1 as well as peroxidasin have a relatively low peroxidative activity in vitro (Table 1, 2 and [9]).
VPO1 when expressed in HEK293H cells exhibited significant activity towards the cell-permeant substrate luminol, and utilized both exogenously added H2O2 and H2O2 generated from any of the expressed Nox enzymes tested. When partially purified, the enzyme exhibited peroxidative activity towards both luminol and TMB, both general substrates for peroxidases. The activity, while low compared with several other peroxidases such as MPO and LPO, is significant and much higher compared with the pseudoperoxidase activities of hematin and several non-peroxidase hemoproteins such as myoglobin and cytochrome c. Furthermore, the VPO1 peroxidase activity is destroyed by heating, in contrast with the pseudoperoxidase activities of other non-peroxidase hemoproteins. Finally, characteristic features of peroxidase heme binding sites/active sites are preserved based on sequence homology and molecular modeling. Many other features, including multiple conserved Cys-Cys disulfides, a structural calcium-binding site, and amino acid residues that bind to the heme propionate side chains are also conserved. Therefore, VPO1 is clearly a true peroxidase, although it remains to be resolved whether the relatively low peroxidase activity of the partially purified enzyme is due to loss of activity during purification, peroxidative inactivation of the heme, missing regulatory factors, or other unknown factors.
The question arises as to the origin of H2O2 in the vasculature to support VPO1-dependent peroxidations. In a number of cases, Nox/Duox enzymes provide H2O2 (secondarily formed by dismutation of superoxide) for various functionally paired peroxidases. MPO is functionally coupled to Nox2-derived H2O2 in phagocytes while TPO and LPO utilize H2O2 produced by Duox in thyroid and lung, respectively [38, 43]. It is therefore reasonable to ask whether specific Nox partner(s) also produce H2O2 for use by VPO1. Nox1, Nox2, Nox4 and Nox5 are distributed in blood vessel wall and myocardium [44–48], and Nox enzymes appear to be the major source of ROS in vasculature [45, 48]; their tissue distribution in vasculature is similar to that of VPO1 (Fig. 3). The present studies (Fig. 8) show that VPO1 can utilize H2O2 generated from expressed Nox enzymes to support its H2O2-dependent luminol oxidizing activity.
In summary, we report the identification of mammalian VPO1 and VPO2, novel animal heme-containing peroxidases, and the initial characterization of VPO1. VPO1, orthologous to peroxidasin has a classical heme-containing peroxidase domain that shows significant homology and structural similarity to LPO and MPO. VPO1 protein is primarily expressed in the cardiovascular system and exhibits constitutive peroxidase activity. In addition, the data suggest that VPO1 can utilize H2O2 generated by Nox enzymes in live cells.
Supplementary Material
Fig. 1S. Specificity of anti-VPO1 antibody. Western blotting was carried out to determine the specificity of the anti-VPO1 antibody. Blotting was also carried out using commercially available antibody to MPO (Anti-MPO) and LPO (Anti-LPO). Lanes contain the following proteins or cell lysates: 1. control HEK293 cell lysates; 2. GJ-4 (VPO1-expressing HEK293 cells) cell lysates; 3. Purified human MPO (59kDa); 4. Purified bovine LPO (79 kDa); 5. Lysates of HEK293 cells expressing human EPO (34kDa); 6. Lysates of HEK293 cells expressing human TPO (105kDa). Molecular size scale is shown at the left. Bars on the right indicate the respective proteins recognized by the antibodies in the panels. Peroxidase activity was confirmed in cell lysates of lane 2, 5 and 6. Anti-LPO antibody also recognizes MPO (dot in the right of the blot). The lowest panel uses anti-tubulin antibody as loading control.
Acknowledgements
This work was supported by the American Heart Association grant 0635122N, NIH grants RO1 HL086836 and CA 105116. J. C. S is supported from the Neel endowment and P. J. P is supported by NIH grants HL55425, HL079207, HL28982 and the Fund for Henry Ford Hospital. P. J. P is an Established Investigator of the American Heart Association.
Abbreviations
- ROS
reactive oxygen species
- Duox
dual oxidase
- EST
Expressed Sequence Tags
- NaBu
sodium butyrate
- TMB
3,3’,5,5’-tetramethylbezidine
- ABH
5’-aminobenzohydrazide
- CTAB
Ncetyl-N,N,N-trimethylammonium bromide
- PBS
phosphate buffered saline
- SDS-PAGE
sodium dodecylsulfate-polyacrylamide gel electrophoresis
- PVDF
polyvinylidene difluoride
- HRP
horseradish peroxidase
- LPO
lactoperoxidase
- MPO
myeloperoxidase
- TPO
thyroid peroxidase
- EPO
eosinophil peroxidase
- BSA
bovine serum albumin
- DTT
dithiothreitol
- PDB
Protein Data Bank
- RT-PCR
reverse transcription-polymerase chain reaction
- bp
base pair
- RLU
relative light units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 2.Edens WA, Sharling L, Cheng G, Shapira R, Kinkade JM, Lee T, Edens HA, Tang X, Sullards C, Flaherty DB, Benian GM, Lambeth JD. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J Cell Biol. 2001;154:879–891. doi: 10.1083/jcb.200103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salmon SE, Cline MJ, Schultz J, Lehrer RI. Myeloperoxidase deficiency. Immunologic study of a genetic leukocyte defect. The New England journal of medicine. 1970;282:250–253. doi: 10.1056/NEJM197001292820505. [DOI] [PubMed] [Google Scholar]
- 4.Lehrer RI, Cline MJ. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: the role of myeloperoxidase in resistance to Candida infection. J Clin Invest. 1969;48:1478–1488. doi: 10.1172/JCI106114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansson-Rahemtulla B, Rahemtulla F, Baldone DC, Pruitt KM, Hjerpe A. Purification and characterization of human salivary peroxidase. Biochemistry. 1988;27:233–239. doi: 10.1021/bi00401a035. [DOI] [PubMed] [Google Scholar]
- 6.Gerson C, Sabater J, Scuri M, Torbati A, Coffey R, Abraham JW, Lauredo I, Forteza R, Wanner A, Salathe M, Abraham WM, Conner GE. The lactoperoxidase system functions in bacterial clearance of airways. Am J Respir Cell Mol Biol. 2000;22:665–671. doi: 10.1165/ajrcmb.22.6.3980. [DOI] [PubMed] [Google Scholar]
- 7.Bakker B, Bikker H, Vulsma T, de Randamie JS, Wiedijk BM, De Vijlder JJ. Two decades of screening for congenital hypothyroidism in The Netherlands: TPO gene mutations in total iodide organification defects (an update) J Clin Endocrinol Metab. 2000;85:3708–3712. doi: 10.1210/jcem.85.10.6878. [DOI] [PubMed] [Google Scholar]
- 8.Kimura S, Kotani T, McBride OW, Umeki K, Hirai K, Nakayama T, Ohtaki S. Human thyroid peroxidase: complete cDNA and protein sequence, chromosome mapping, and identification of two alternately spliced mRNAs. Proc Natl Acad Sci U S A. 1987;84:5555–5559. doi: 10.1073/pnas.84.16.5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson RE, Fessler LI, Takagi Y, Blumberg B, Keene DR, Olson PF, Parker CG, Fessler JH. Peroxidasin: a novel enzyme-matrix protein of Drosophila development. Embo J. 1994;13:3438–3447. doi: 10.1002/j.1460-2075.1994.tb06649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. The Journal of biological chemistry. 2006 doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 11.Cheng G, Ritsick D, Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. The Journal of biological chemistry. 2004;279:34250–34255. doi: 10.1074/jbc.M400660200. [DOI] [PubMed] [Google Scholar]
- 12.Cheng G, Lambeth JD. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. The Journal of biological chemistry. 2004;279:4737–4742. doi: 10.1074/jbc.M305968200. [DOI] [PubMed] [Google Scholar]
- 13.Bolscher BG, Plat H, Wever R. Some properties of human eosinophil peroxidase, a comparison with other peroxidases. Biochimica et biophysica acta. 1984;784:177–186. doi: 10.1016/0167-4838(84)90125-0. [DOI] [PubMed] [Google Scholar]
- 14.Johansson A, Dahlgren C. Characterization of the luminol-amplified light-generating reaction induced in human monocytes. J Leukoc Biol. 1989;45:444–451. doi: 10.1002/jlb.45.5.444. [DOI] [PubMed] [Google Scholar]
- 15.Weaver M, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol. 2006;290:H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 16.Feissner R, Xiang Y, Kranz RG. Chemiluminescent-based methods to detect subpicomole levels of c-type cytochromes. Analytical biochemistry. 2003;315:90–94. doi: 10.1016/s0003-2697(02)00658-9. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell MS, Kan-Mitchell J, Minev B, Edman C, Deans RJ. A novel melanoma gene (MG50) encoding the interleukin 1 receptor antagonist and six epitopes recognized by human cytolytic T lymphocytes. Cancer Res. 2000;60:6448–6456. [PubMed] [Google Scholar]
- 18.Weiler SR, Taylor SM, Deans RJ, Kan-Mitchell J, Mitchell MS, Trent JM. Assignment of a human melanoma associated gene MG50 (D2S448) to chromosome 2p25.3 by fluorescence in situ hybridization. Genomics. 1994;22:243–244. doi: 10.1006/geno.1994.1374. [DOI] [PubMed] [Google Scholar]
- 19.Horikoshi N, Cong J, Kley N, Shenk T. Isolation of Differentially Expressed cDNAs from p53-Dependent Apoptotic Cells: Activation of the Human Homologue of the Drosophila Peroxidasin Gene. Biochemical and Biophysical Research Communications. 1999;261:864–869. doi: 10.1006/bbrc.1999.1123. [DOI] [PubMed] [Google Scholar]
- 20.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 21.Nagase T, Seki N, Ishikawa K-i, Ohira M, Kawarabayasi Y, Ohara O, Tanaka A, Kotani H, Miyajima N, Nomura N. Prediction of the Coding Sequences of Unidentified Human Genes. VI. The Coding Sequences of 80 New Genes (KIAA0201-KIAA0280) Deduced by Analysis of cDNA Clones from Cell Line KG-1 and Brain. DNA Res. 1996;3:321–329. doi: 10.1093/dnares/3.5.321. [DOI] [PubMed] [Google Scholar]
- 22.Bradley PP, Christensen RD, Rothstein G. Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood. 1982;60:618–622. [PubMed] [Google Scholar]
- 23.Fenna R, Zeng J, Davey C. Structure of the green heme in myeloperoxidase. Arch Biochem Biophys. 1995;316:653–656. doi: 10.1006/abbi.1995.1086. [DOI] [PubMed] [Google Scholar]
- 24.Fiedler TJ, Davey CA, Fenna RE. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 A resolution. The Journal of biological chemistry. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 25.Zeng J, Fenna RE. X-ray crystal structure of canine myeloperoxidase at 3 A resolution. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- 26.Bordin M, D'Atri F, Guillemot L, Citi S. Histone Deacetylase Inhibitors Up-Regulate the Expression of Tight Junction Proteins. Mol Cancer Res. 2004;2:692–701. [PubMed] [Google Scholar]
- 27.Gorman CM, Howard BH, Reeves R. Expression of recombinant plasmids in mammalian cells is enhanced by sodium butyrate. Nucleic acids research. 1983;11:7631–7648. doi: 10.1093/nar/11.21.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Backliwal G, Hildinger M, Kuettel I, Delegrange F, Hacker DL, Wurm FM. Valproic acid: a viable alternative to sodium butyrate for enhancing protein expression in mammalian cell cultures. Biotechnology and bioengineering. 2008;101:182–189. doi: 10.1002/bit.21882. [DOI] [PubMed] [Google Scholar]
- 29.Hague A, Manning AM, Hanlon KA, Huschtscha LI, Hart D, Paraskeva C. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int J Cancer. 1993;55:498–505. doi: 10.1002/ijc.2910550329. [DOI] [PubMed] [Google Scholar]
- 30.Kooter IM, Moguilevsky N, Bollen A, Sijtsema NM, Otto C, Dekker HL, Wever R. Characterization of the Asp94 and Glu242 mutants in myeloperoxidase, the residues linking the heme group via ester bonds. European journal of biochemistry / FEBS. 1999;264:211–217. doi: 10.1046/j.1432-1327.1999.00606.x. [DOI] [PubMed] [Google Scholar]
- 31.Davis JC, Averill BA. Isolation from bovine spleen of a green heme protein with properties of myeloperoxidase. The Journal of biological chemistry. 1981;256:5992–5996. [PubMed] [Google Scholar]
- 32.Vargas C, McEwan AG, Downie JA. Detection of c-type cytochromes using enhanced chemiluminescence. Analytical biochemistry. 1993;209:323–326. doi: 10.1006/abio.1993.1127. [DOI] [PubMed] [Google Scholar]
- 33.Morrison M, Bayse GS. Catalysis of iodination by lactoperoxidase. Biochemistry. 1970;9:2995–3000. doi: 10.1021/bi00817a010. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki K, Yamada M, Akashi K, Fujikura T. Similarity of kinetics of three types of myeloperoxidase from human leukocytes and four types from HL-60 cells. Arch Biochem Biophys. 1986;245:167–173. doi: 10.1016/0003-9861(86)90201-8. [DOI] [PubMed] [Google Scholar]
- 35.Uguz MT, Ozdemir H. Purification of bovine milk lactoperoxidase and investigation of antibacterial properties at different thiocyanate mediated. Prikl Biokhim Mikrobiol. 2005;41:397–401. [PubMed] [Google Scholar]
- 36.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free radical biology & medicine. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akasaki T, Ohya Y, Kuroda J, Eto K, Abe I, Sumimoto H, Iida M. Increased expression of gp91phox homologues of NAD(P)H oxidase in the aortic media during chronic hypertension: involvement of the renin-angiotensin system. Hypertens Res. 2006;29:813–820. doi: 10.1291/hypres.29.813. [DOI] [PubMed] [Google Scholar]
- 38.Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. Faseb J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- 39.Karaulanov EE, Bottcher RT, Niehrs C. A role for fibronectin-leucine-rich transmembrane cell-surface proteins in homotypic cell adhesion. EMBO reports. 2006;7:283–290. doi: 10.1038/sj.embor.7400614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Field J, Xu HP, Michaeli T, Ballester R, Sass P, Wigler M, Colicelli J. Mutations of the adenylyl cyclase gene that block RAS function in Saccharomyces cerevisiae. Science (New York, N.Y. 1990;247:464–467. doi: 10.1126/science.2405488. [DOI] [PubMed] [Google Scholar]
- 41.Chan JR, Hyduk SJ, Cybulsky MI. {alpha}4{beta}1 Integrin/VCAM-1 Interaction Activates {alpha}L{beta}2 Integrin-Mediated Adhesion to ICAM-1 in Human T Cells. J Immunol. 2000;164:746–753. doi: 10.4049/jimmunol.164.2.746. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J-L, Huang Y, Qiu L-Y, Nickel J, Sebald W. von Willebrand Factor Type C Domain-containing Proteins Regulate Bone Morphogenetic Protein Signaling through Different Recognition Mechanisms. J. Biol. Chem. 2007;282:20002–20014. doi: 10.1074/jbc.M700456200. [DOI] [PubMed] [Google Scholar]
- 43.Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, Vulsma T, Ris-Stalpers C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. The New England journal of medicine. 2002;347:95–102. doi: 10.1056/NEJMoa012752. [DOI] [PubMed] [Google Scholar]
- 44.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 45.Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal. 2005;7:308–317. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 46.Krijnen PA, Meischl C, Hack CE, Meijer CJ, Visser CA, Roos D, Niessen HW. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol. 2003;56:194–199. doi: 10.1136/jcp.56.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, Steger K, Krause K-H, Jaconi ME. The NADPH Oxidase NOX4 Drives Cardiac Differentiation: Role in Regulating Cardiac Transcription Factors and MAP Kinase Activation. Mol. Biol. Cell. 2006 doi: 10.1091/mbc.E05-06-0532. E05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. 1S. Specificity of anti-VPO1 antibody. Western blotting was carried out to determine the specificity of the anti-VPO1 antibody. Blotting was also carried out using commercially available antibody to MPO (Anti-MPO) and LPO (Anti-LPO). Lanes contain the following proteins or cell lysates: 1. control HEK293 cell lysates; 2. GJ-4 (VPO1-expressing HEK293 cells) cell lysates; 3. Purified human MPO (59kDa); 4. Purified bovine LPO (79 kDa); 5. Lysates of HEK293 cells expressing human EPO (34kDa); 6. Lysates of HEK293 cells expressing human TPO (105kDa). Molecular size scale is shown at the left. Bars on the right indicate the respective proteins recognized by the antibodies in the panels. Peroxidase activity was confirmed in cell lysates of lane 2, 5 and 6. Anti-LPO antibody also recognizes MPO (dot in the right of the blot). The lowest panel uses anti-tubulin antibody as loading control.