

Abstract
The overall goal of this research was to separate out the effects of butyrate from its fiber source and determine in vivo if it upregulates colonic histone acetylation, p21Waf1/Cip1 expression (p21) and apoptosis and if this sequela of events is protective against aberrant crypt foci (ACF) formation. Eighty Sprague-Dawley rats were provided defined diets with either corn oil or fish oil as the lipid source, ± butyrate-containing capsules targeted for release in the colon and ± azoxymethane (AOM) (10 rats per group). Diets were provided for 11 weeks and at termination colonocyte nuclear histone H4 and p21 expression were determined by immunohistochemistry, apoptosis was measured by the terminal deoxynucleotide transferase biotin-dUTP nick end labeling assay and aberrant crypt numbers and multiplicity were enumerated. Luminal butyrate levels were also quantified. AOM injection repressed p21 expression, which was reversed by butyrate supplementation. Although butyrate enhanced p21 expression with both dietary lipid sources, the increase in p21 resulted in an increase in apoptosis and decrease in ACF with fish oil, but had no effect on apoptosis and increased ACF with corn oil. This significant interaction between fat, butyrate (fiber) and p21 expression with one combination being protective and the other promotive of colon carcinogenesis reinforces the importance of considering diet as a key factor in chemoprevention.
Introduction
We have shown previously that a diet containing pectin as the fiber source, and fish oil as the lipid component, is protective against experimentally induced colon cancer as compared with a cellulose–corn oil diet (1). We have also documented that the chemoprotective effects of the fish–pectin diet are due to upregulation of the apoptotic removal of DNA-damaged colon cells in response to an initiating event, which continues throughout cancer development, rather than a downregulation of colonic cell proliferation (2), and that the bioactive components from fish oil are the fatty acids eicosapentaenoic acid (20:5n-3) and docosahexaenoic acid (22:6n-3) rather than a contaminant or unknown protective factor in fish oil itself (3). A current interest of the laboratory and focus of this report is to identify, in vivo, the bioactive component from pectin and to determine how the combination of that bioactive compound with n-3 fatty acids from fish oil initiates the apoptotic response.
We hypothesized that butyrate (a fermentation product of pectin) is the ‘active’ agent for the fiber component. There are considerable in vitro data in support of this hypothesis that show the induction of apoptosis by butyrate in colon cancer cell lines (4) and a number of studies in vivo showing upregulation of colonic apoptosis with pectin (5,6). However, data are lacking that specifically document the effect of butyrate administration on colonic apoptosis in vivo. To determine whether the effects of fiber (pectin) are derived from its fermentation product butyrate, we designed diets with identical amounts and types of a non-fermentable fiber (cellulose) and provided (or not) pellets encapsulating butyrate for targeted release in the colon.
We further hypothesized that exposure of colonocytes to butyrate may initiate apoptosis by upregulating p21Waf1/Cip1 (p21) gene expression via targeted histone hyperacetylation. Butyrate is a well-established non-competitive, reversible inhibitor of histone deacetylase both in vitro and in vivo (7–9). In general, butyrate-targeted histone deacetylase inhibition leads to the upregulation of particular sequences within the genome, as evidenced by the well-documented induction of the cyclin-dependent kinase inhibitor, p21 (10). The relationship between an upregulation of p21, cell cycle arrest and apoptosis is less clear and the subject of active investigation (11). Paradoxically, p21 has been shown both to promote apoptosis and to protect tumor cells from apoptosis (12,13). It is therefore feasible that the role p21 plays in regulating apoptosis depends on whether cells are normal or are transformed into tumor cells. Whether or not butyrate upregulates p21 in vivo and enhances apoptosis has not been reported, but a few studies suggest that it may. In one report, mice with a mutant Apc allele and either heterozygous or homozygous for loss of p21 had increased tumor formation and decreased intestinal cell apoptosis (14). In another study, p21-deficient mice injected with azoxymethane (AOM) developed increased numbers of aberrant crypt foci (ACF) and reduced apoptosis of colon epithelial cells (15). These studies suggest that if p21 was to be upregulated because of butyrate administration, colonic apoptosis should be enhanced. Thus, the overall goal of this research was to separate out the effects of butyrate from its fiber source and determine in vivo if it upregulates colonic histone acetylation, p21 expression and apoptosis and if this sequela of events is protective against aberrant crypt formation (an intermediate marker for colon tumorigenesis in both rats and humans) (16,17).
Materials and methods
Animals and study design
The animal use protocol was approved by the University Laboratory Animal Care Committee of Texas A&M University and conformed to National Institutes of Health guidelines. Eighty male weanling Sprague-Dawley rats (Harlan Sprague-Dawley, Houston, TX) were individually housed in polycarbonate cages with raised double grid floors to help prevent coprophagy and maintained in a temperature and humidity-controlled animal facility with a 12 h light–dark cycle. Experimental diet per treatment groups were constructed using a 2 × 2 × 2 factorial design with 10 rats per experimental group: two dietary lipid sources (corn oil or fish oil), two sodium butyrate pellet supplementation groups (± sodium butyrate pellets) and two injection protocols (saline or 15 mg/kg body wt AOM). The rats were acclimated for 1 week, stratified by body weight and assigned to one of the defined diets. After 3 weeks of receiving the experimental diets, rats were injected (subcutaneously) with AOM or an equivalent volume of saline, followed by a second injection 1 week later. All rats were terminated after 11 weeks of receiving the experimental diets (7 weeks post second AOM injection).
Diets
The two defined diets (Table I) differed only in the type of fat (corn oil or fish oil). Fish oil contains significantly higher amounts of n-3 polyunsaturated fatty acids, eicosapentaenoic acid (20:5n-3) and docosahexaenoic acid (22:6n-3), whereas corn oil is rich in linoleic acid (18:2n-6). The fish oil diet contained 3.5 g corn oil/100 g diet to ensure that essential fatty acid requirements were met. Both diets contained 26 mg α-tocopherol, 14 mg γ-tocopherol and 2 mg tertiary butylhydroquinone/100 g diet as antioxidants. Sodium butyrate pellets (S.A.Valpharma, Serravalle, Italy) were added to both corn oil + butyrate and fish oil + butyrate diets at 5% of the diet by weight. The composition of these pellets is shown in a footnote to Table I. The coating is designed to dissolve at a pH of 7.0 or higher and thus remains relatively intact during transit through the stomach and begins to dissolve in the terminal ileum and cecum (18,19). In preliminary experiments, we provided rats with the butyrate + diets and assayed for intact pellets throughout the gastrointestinal tract. Intact pellets were observed throughout the small intestine with a lesser amount still extant in the cecum. No intact pellets were observed in the distal colon and we report our results from the distal colon. All diets were stored at −80°C to prevent lipid peroxidation and rats were provided with fresh diet every day. Rats had free access to food and water at all times. Forty-eight hour food intakes were measured prior to AOM injection, post second AOM injection and at termination. Body weights were recorded weekly.
Table I.
Experimental diet composition
| Ingredient | Grams/100 g total dieta |
| Dextrose | 51.06 |
| Casein | 22.35 |
| Methionine | 0.34 |
| Mineral mix, AIN-76 | 3.91 |
| Vitamin mix, AIN-76 | 1.12 |
| Choline bitartrate | 0.22 |
| Cellulose | 6.00 |
| Corn oil | 15.00 |
| Fish oil/corn oil | 11.50/3.50 |
A basal diet mixture without the oils was purchased from Harlan Teklad (Madison, WI). The oils, containing tertiary butylhydroquinone and α-, γ-tocopherols to equalize antioxidants across diets, were added later. Tertiary butylhydroquinone, (0.25 g/kg total oil) supplied by Tenox 20-A (Gilco, Vista, CA), was used to prevent oxidative damage to the oils. Synthetic α-, γ-tocopherols (0.698 g/kg fish oil) were supplied by MTS-70 (Archer Daniels Midland, Decatur, IL). For the diets containing the butyrate pellets, the pellets were added to the diet at 5% by weight. Pellets contained 595.0 mg/g sodium butyrate and the following non-active components (mg/g): povidone 20.0; methacrylic acid copolymer (type B) 31.2; triacetin 13.7; talc 67.4; hydroxypropyl methylcellulose phthalate 196.1; diethylphthalate 39.2 (total = 1000 mg/g).
Tissue collection
Rats were euthanized by CO2 overdose, followed by cervical dislocation. The colon was harvested and, after removal of the rectum, 1 cm of the distal end of the colon was fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). The remaining colon was divided longitudinally, and half was prepared for determination of ACF. The other half was scraped and the mucosal contents were stored at −80°C.
Short chain fatty acid analyses
Colonic luminal short chain fatty acids (SCFAs) were quantified as described previously (20). At termination, feces from the distal half of the colon were collected and flash frozen. Frozen samples were powdered, extracted and analyzed with appropriate standards by gas chromatography.
Histone H4 acetylation analysis
Acetylated histone H4 was visualized in situ via immunohistochemistry following an optimized procedure based on the protocol of Warrell et al. (21). Anti-acetyl-Histone H4 rabbit polyclonal antibody was diluted 1:750 (Upstate, Lake Placid, NY) followed by incubation with Biotin-SP-conjugated AffiniPure Goat Anti-Rabbit secondary antibody diluted 1:200 (Jackson ImmunoResearch Laboratories, West Grove, PA). Antibodies were diluted in 1% Normal Goat Serum (Jackson ImmunoResearch Laboratories). Subsequently, slides were incubated using an ABC kit (Vector Laboratory, Burlingame, CA). The entire complex was visualized with diaminobenzidine tetrahydrochloride. Omission of primary antibody was used as a negative control. Images were captured under a light microscope using National Institutes of Health Image Software 4.0. The staining intensity of each nucleus within one side of 15 crypt columns was determined (eight per treatment group). Both the average stain intensity per nucleus and the cell position were recorded. Background staining was detected by determining the average stain intensity of a 25 by 25 pixel square captured at the luminal surface of the crypt column and this value was subtracted from the average stain intensity of each nucleus. The resulting stain intensities were averaged and the mean acetylated histone H4 stain intensity was used for statistical analysis.
p21 analysis
The cyclin-dependent kinase inhibitor p21 was visualized in situ via immunohistochemistry on 4% paraformaldehyde-fixed, paraffin-embedded tissues according to the protocol described by Viale et al. (22). Slides were incubated overnight with primary monoclonal mouse anti-p21 antibody (BD Pharmingen, San Diego, CA) at a 1:10 dilution in 1% normal rabbit serum (Jackson ImmunoResearch). Negative control sections were incubated with 1% normal rabbit serum without primary antibody. Tissues were incubated for 2 h with a biotinylated rabbit anti-mouse secondary antibody (DakoCytomation, Carpinteria, CA) at a 1:250 dilution in 1% normal rabbit serum. Subsequently, slides were incubated using an ABC kit (Vector Laboratory). The entire complex was visualized with diaminobenzidine tetrahydrochloride. Images were captured under a light microscope using National Institutes of Health Image Software 4.0. The staining intensity of p21 was assessed by microscopic examination, image acquisition and image analysis as described above for the histone H4 assay.
Determination of apoptotic cells in the colonic mucosa
Apoptosis was assessed in situ by the terminal deoxynucleotide transferase biotin-dUTP nick end labeling assay using ApopTag kits (Serologicals Corporation, Norcross, GA) on non-serial 4 μm thick paraffin sections of 4% paraformaldehyde-fixed colon tissue. A positive control was generated by exposing selected tissue sections to DNase I (Ambion, Austin, TX); negative controls were DNase I-treated sections without addition of the terminal deoxynucleotidyl transferase enzyme. Fifty well-oriented crypt columns per rat were scored and both the total number of cells per column and the specific position of each apoptotic cell were recorded. A cell was considered apoptotic if it had positive staining and morphological features characteristic of apoptosis (2). The apoptotic index was calculated as the number of apoptotic cells per column divided by the total number of cells per column × 100.
Determination of aberrant crypts
Examination of aberrant crypts was conducted as described previously (23). Briefly, the total number of aberrant crypts was counted in each colon as well as the number of ACF and high multiplicity aberrant crypt foci (HM-ACF). A HM-ACF was considered to contain four or more aberrant crypts per focus (17).
Statistical analyses
Body weights and food intake data were analyzed using a two-way analysis of variance. SCFA quantification, ACF, apoptosis, p21 and histone acetylation were analyzed by three-way analysis of variance and by a split plot designed mixed model procedure (PROC MIXED) in SAS. The relationship between apoptosis and total ACF, as well as p21 and HM-ACF were determined by regression analysis in SAS. Differences were considered significant at P < 0.05.
Results
Food intake and weight gain
There were no differences in food intake among the eight groups at any of the intake assessment time points (data not shown). Although the AOM-injected rats fed corn oil (+ and − butyrate) had significantly less weight gain compared with those fed fish oil, there was no effect of body weight on any of the outcome measurements when the data were analyzed with weight as a covariate.
SCFA and butyrate levels in the distal colon
Butyrate supplementation increased the total concentration of SCFAs (acetic, propionic, butyric, isobutyric, valeric and isovaleric acids) in the distal colon [22.03 ± 0.79 versus 16.45 ± 0.47 μmol/g wet weight feces compared with that of unsupplemented groups, independent of dietary lipid source (P < 0.05)]. Dietary lipid source affected distal luminal butyrate concentrations in that the concentration of butyrate was higher in the fish oil + butyrate group than the corn oil + butyrate group (P = 0.017; Figure 1), but this effect was not seen in those rats injected with AOM (Figure 1).
Fig. 1.

Fish oil and butyrate (FOB) feeding increased fecal butyrate in the distal colon relative to corn oil and butyrate (COB)-fed rats, but only when the rats were injected with saline (SAL). FOB-fed rats injected with SAL had higher distal fecal butyrate levels than all other groups. Data are means ± SEMs for 9–10 rats per diet per treatment group. Bars with different letters are significantly different (P < 0.02).
Enumeration of aberrant crypts
Whereas AOM-treated rats developed aberrant crypts, their saline-treated counterparts did not. Therefore, all aberrant crypt results represent AOM-injected groups only. Total ACF and HM-ACF were higher (P < 0.0001; data not shown) in the distal colon compared with the proximal colon, independent of the diet. The corn oil + butyrate treatment resulted in a greater number of total ACF (P < 0.005; Figure 2A) and HM-ACF (P < 0.04; Figure 2B) compared with other treatment groups. In addition, the fish oil + butyrate-fed rats displayed the lowest incidence of HM-ACF compared with all other groups (P = 0.038; Figure 2B).
Fig. 2.

(A) Corn oil and butyrate combination increased total ACF. All rats were injected with AOM. Bars with different letters are significantly different (P < 0.005). (B) Effect of diet on HM-ACF. Data are means ± SEMs for 9–10 rats per treatment. Bars with different letters are significantly different (P < 0.04). Refer to Figure 1 for legend details.
In situ assessment of the apoptotic index and its relationship to ACF
Butyrate supplementation enhanced apoptosis in AOM-injected rats fed a diet containing fish oil (P = 0.014; Figure 3A), but not corn oil. This enhancement of apoptosis by fish oil + butyrate was not seen in control rats injected with saline (data not shown). Further, within the fish oil + butyrate group, there was an inverse relationship between the apoptotic index and the number of total ACF per rat (P < 0.05; Figure 3B). Specifically, as the apoptotic index increased, the number of total ACF decreased. This relationship was seen only in the fish oil + butyrate-fed animals, not in the corn oil + butyrate group.
Fig. 3.
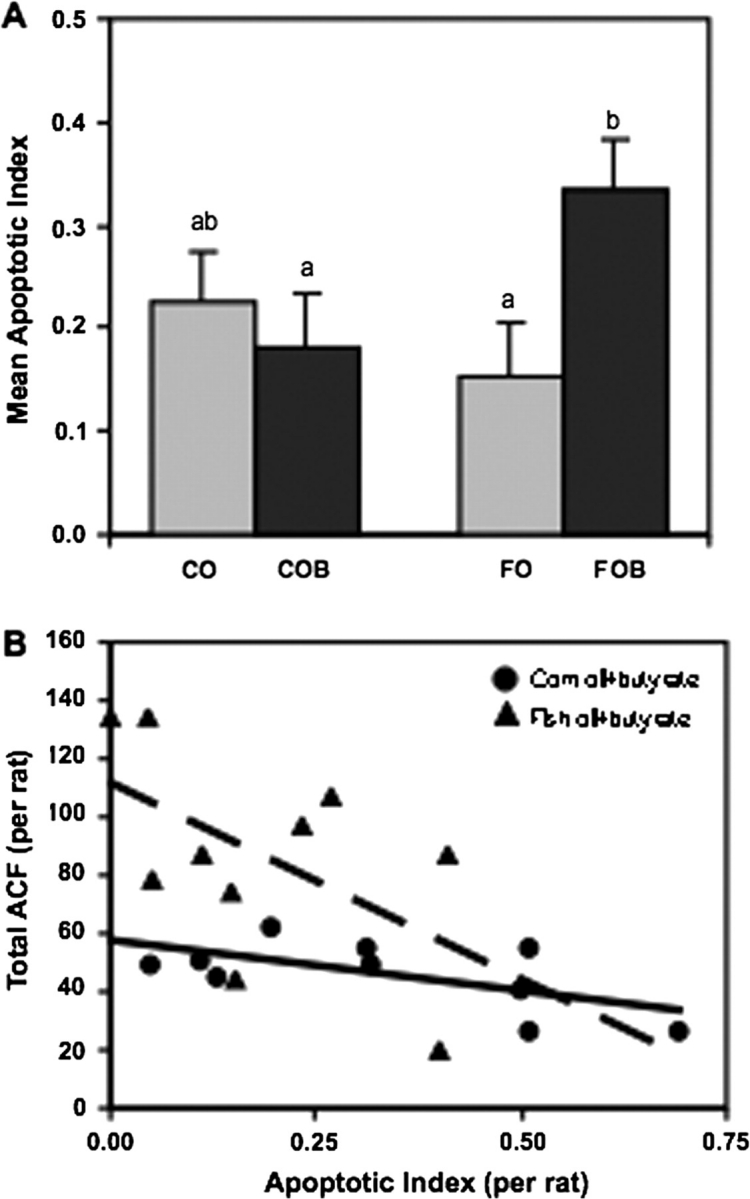
(A) Butyrate-supplemented diets enhanced apoptosis only in rats consuming fish oil. All animals were injected with AOM. Apoptosis was measured in the distal colon only. The apoptotic index represents number of apoptotic cells/total number of cells per crypt column × 100. Data are means ± SEMs for 10 rats per diet per treatment group. For each rat, 50 well-oriented crypt columns were assessed. Bars with different letters are significantly different (P < 0.02). (B) Inverse relationship between the apoptotic index and the number of ACF per animal (r = −0.63228; P < 0.05). No relationship between AOM-injected rats fed corn oil + butyrate diets (r = −0.2873; P > 0.05) was observed. Refer to Figure 1 for legend details.
In situ assessment of p21 expression and its relationship to ACF
In the absence of butyrate supplementation, treatment with AOM decreased protein expression of p21 (P = 0.03; Figure 4A). However, addition of butyrate to the diet restored p21 protein to control (saline injected) levels (P = 0.04; Figure 4A). While restoration of p21 levels was seen in both fish oil + butyrate and corn oil + butyrate animals, increased p21 levels were positively correlated with HM-ACF in rats fed corn oil + butyrate (P < 0.05; Figure 4B). However, this was not the case for fish oil + butyrate-fed rats (Figure 4B).
Fig. 4.

(A) There was a significant interaction between carcinogen treatment and butyrate administration on p21 expression (P = 0.03) in that AOM-injected rats not consuming butyrate exhibited decreased p21 levels. Data are means ± SEMs for 16 rats per butyrate per treatment group. For each rat, 15 well-oriented crypt columns were selected and each nucleus circled to determine the mean p21 stain intensity per rat. Bars with different letters are significantly different (P < 0.05). (B) Rats fed corn oil and butyrate diets (and injected with AOM) exhibited a positive relationship between the level of p21 expression and the number of HM-ACF (r = 0.62759, P < 0.05). In contrast, no relationship among rats fed fish oil and butyrate diets (r = 0.1338, P > 0.05) was observed. Data are means ± SEMs for eight rats.
In situ assessment of histone H4 acetylation status
Butyrate supplementation had no effect on histone H4 acetylation if corn oil was the lipid source in the diet. This was true whether or not the rats were injected with the colon-specific carcinogen (AOM) or saline (Figure 5A). In contrast, when fish oil was the lipid source, butyrate supplementation enhanced histone acetylation (P = 0.0331; Figure 5B) in saline-injected rats, but not in rats receiving AOM.
Fig. 5.
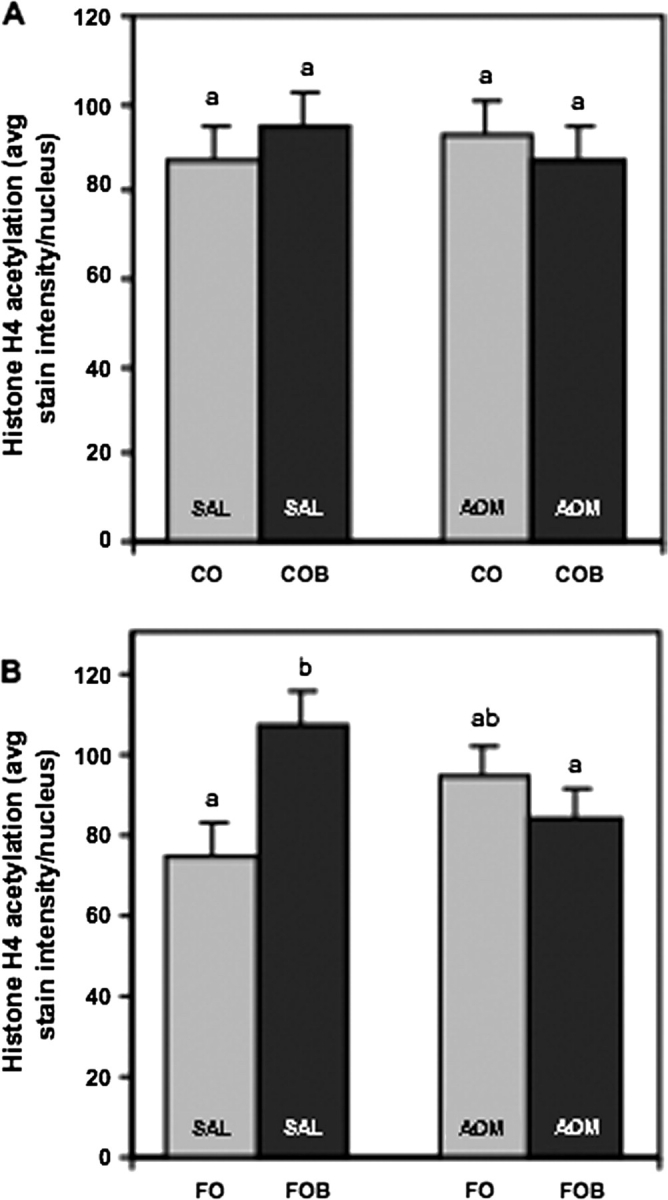
(A) Lack of treatment effects on histone H4 acetylation in the distal colon of corn oil-fed rats. Data are means ± SEMs for eight rats per diet per treatment group (P > 0.05). For each rat, 15 well-oriented crypt columns were selected and each nucleus circled to determine the mean histone H4 acetylation stain intensity per rat. (B) Dietary butyrate increased the level of histone H4 acetylation in the distal colon of rats fed fish oil and injected with saline. In contrast, when rats were injected with AOM, fish oil and butyrate treatment decreased the level of histone acetylation as compared with fish oil and butyrate rats injected with saline. Data are means ± SEMs for eight rats per diet per treatment group. Bars with different letters are significantly different (P < 0.04).
Discussion
There were two major findings from this study: (i) supplementation of butyrate, independent of fiber, explains our previously documented chemoprotective effects of a fish oil–pectin diet and (ii) upregulation of p21 expression as a consequence of butyrate supplementation can promote or protect against colon carcinogenesis depending on the lipid composition of the diet.
Butyrate is the bioactive compound
By supplementing diets with butyrate in the form of gastro-resistant sodium butyrate pellets targeted for release in the colon, we produced a colonic luminal environment similar to that found with a diet rich in a highly fermentable fiber such as pectin (24). Interestingly, dietary lipid source significantly affected the proportion of butyrate in the distal colon of rats not injected with AOM in that butyrate mole percentage was higher with fish oil supplementation compared with corn oil supplementation. Although we can only speculate as to the mechanism for this dietary lipid effect on colonic luminal butyrate, we have shown previously that diets containing fish oil versus corn oil can result in different fecal microbial profiles (25).
The results from this study support our hypothesis that butyrate is the active compound that protects against the promotion of colon carcinogenesis when supplied within a fish oil-rich diet. It should be noted, however, that the observed effects of butyrate administration are not necessarily direct effects of butyrate per se, but rather they may be indirect effects. For example, release of butyrate in the colon could lower colonic pH and many microbial enzymatic reactions such as the production of secondary bile acids from primary bile acids are highly pH sensitive (26). Alternatively, supplying the end product of fiber fermentation (butyrate) has the potential to alter the microbial population since certain bacteria (such as Syntrophomonas spp. and Syntrophotermus spp.) use butyrate for their growth (27).
The addition of butyrate to a fish oil-based diet resulted in a lower total number of aberrant crypts and a lower number of high multiplicity aberrant crypts compared with the corn oil + butyrate group. When rats were injected with AOM, butyrate addition increased the proportion of apoptotic cells in the fish oil, but not the corn oil-supplemented group. In addition, within the fish + butyrate group (but not the corn + butyrate group), there was a significant negative relationship between apoptosis and aberrant crypt formation. Although we have previously fed corn oil- versus fish oil-based diets and incubated colonocytes from those rats in short-term culture with or without butyrate and documented the induction of apoptosis with butyrate exposure in the fish oil-fed rats (3), this is our first report on providing butyrate per se in the diet, rather than producing butyrate by feeding a fermentable fiber. We used a similar approach to butyrate administration as did Caderni et al. (18,19) who found that the addition of butyrate neither protected against AOM-induced colon cancer nor increased the level of colonocyte apoptosis. The key difference between our study and those of Caderni et al. is that they provided butyrate in only a corn oil-based diet. If we only consider the results of the corn oil + butyrate-supplemented diet from the present study, our results are identical to those of Caderni et al. (18,19) that is corn oil + butyrate did not increase apoptosis and was not protective against aberrant crypt formation. In contrast, Wong et al. (28) used a more direct approach to deliver butyrate to the distal colon by infusing butyrate through a polyethylene tube passed through the cecum into the distal colon. Interestingly, both a saline infusion or a twice-daily infusion of 80 mM butyrate were equally protective against numbers of ACF when compared with no infusion, suggesting that the infusion itself was protective.
In contrast, when the amount of butyrate was raised to five times daily of 80 mM butyrate, the butyrate treatment was slightly more protective than the saline infusion, with both infusions being better than no infusion. It is difficult to make a direct comparison between our study and that of Wong et al. since the effect of the infusion itself on aberrant crypt formation was greater than the effect of butyrate, and we did not use an infusion system. However, since they only found a specific effect of butyrate at the higher doses, it is possible that our butyrate dose was lower than theirs in the distal colon since release of the butyrate from ours was not entirely in the distal colon. They used chow diets and defined diets with soybean oil as the lipid source. We have always found a synergistic effect of the combination of fish oil and a butyrate-producing fiber that may account for the protective effect of the fish oil–butyrate against aberrant crypt development and the lack of a protective effect of the corn oil–butyrate combination.
Upregulation of p21 expression by butyrate administration can be chemoprotective or chemopromotive depending on the lipid composition of the diet
Expression levels of p21 were reduced by AOM administration but restored to saline control values by butyrate administration in colons of rats receiving either fish oil or corn oil as the lipid component of their diets. However, the upregulation of p21 as a function of butyrate was associated with chemoprotective effects in fish oil-supplemented rats (enhanced apoptosis, which was inversely correlated with aberrant crypt formation), but not in corn oil-supplemented rats. When corn oil was the lipid component of the diet, butyrate did not enhance apoptosis, and p21 levels were positively correlated with high multiplicity aberrant crypts. These data suggest that upregulation of p21 by butyrate can have protective or promotive effects for colon carcinogenesis depending on the source of dietary fat.
The finding that AOM administration independently decreased expression of p21 is consistent with reports that p21 expression is downregulated during colon tumor development (29,30). The butyrate-induced upregulation of p21 expression in AOM-injected rats to levels observed in saline controls was not unexpected but has not been previously reported. The finding that butyrate-induced upregulation of p21 had dichotomous effects on the colon carcinogenic process depending on the lipid source of the diet was unexpected. This finding parallels the results of our previous studies in which we provided the butyrate-producing fiber pectin and showed that fish oil + pectin feeding resulted in the highest colonic apoptotic indices whereas corn oil + pectin resulted in the lowest (1,2). At this time, we can only speculate as to why p21 upregulation produces an anticarcinogenic effect with fish oil as the dietary lipid and a procarcinogenic effect when corn oil is the lipid source, but the data are consistent with recent findings that p21 may enhance or depress apoptosis (12,13). Previous studies from our laboratory have shown that the fatty acids from fish oil incorporate into mitochondrial membranes and, in the presence of butyrate, initiate apoptosis by way of a reactive oxygen species-initiated mechanism, translocation of cytochrome C to the cytosol and activation of caspase-3 (3,5,6). This apoptotic process does not occur when corn oil is the lipid source in the diet. Zhang et al. (31) found that caspase-3-mediated cleavage of p21 was necessary to convert human lung carcinoma cells from growth arrest to active apoptosis, and Chai et al. (32) reported a unique 15 kD protein fragment from caspase-3 cleavage of p21 produced during apoptosis in LIM 1215 colorectal cancer cells.
In conclusion, we found that administration of AOM repressed p21 expression in rat colonocytes that was restored to saline control levels with concomitant administration of butyrate. In rats provided fish oil-supplemented diets, enhanced p21 expression was associated with an increase in apoptosis and a decrease in aberrant crypt formation. In contrast, in rats provided corn oil-supplemented diets, enhanced p21 expression did not increase apoptosis and led to higher levels of aberrant crypt formation. Such important but often overlooked interactions between diet and gene expression patterns may help to explain why butyrate (or fiber) is sometimes found to be protective against colon cancer and other times promotive. These critical interactions may contribute to modifications in the cellular milieu, which determines if the p21 protein is pro- or antiapoptotic.
Funding
National Institutes of Health (CA 61750, CA 59034, CA 82907); National Institute of Environmental Health Sciences (P30-ES-09106); National Space Biomedical Research Institute (NASA NCC 9-58).
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ACF
aberrant crypt foci
- AOM
azoxymethane
- HM-ACF
high multiplicity aberrant crypt foci
- p21
p21Waf1/Cip1
- SCFA
short chain fatty acids
References
- 1.Chang WL, et al. Fish oil blocks azoxymethane-induced rat colon tumorigenesis by increasing cell differentiation and apoptosis rather than decreasing cell proliferation. J. Nutr. 1998;128:491–497. doi: 10.1093/jn/128.3.491. [DOI] [PubMed] [Google Scholar]
- 2.Chang WC, et al. Predictive value of proliferation, differentiation and apoptosis as intermediate markers for colon tumorigenesis. Carcinogenesis. 1997;18:721–730. doi: 10.1093/carcin/18.4.721. [DOI] [PubMed] [Google Scholar]
- 3.Hong MY, et al. Fish oil increases mitochondrial phospholipid unsaturation, upregulating reactive oxygen species and apoptosis in rat colonocytes. Carcinogenesis. 2002;23:1919–1925. doi: 10.1093/carcin/23.11.1919. [DOI] [PubMed] [Google Scholar]
- 4.McBain JA, et al. Apoptotic death in adenocarcinoma cell lines induced by butyrate and other histone deacetylase inhibitors. Biochem. Pharmacol. 1997;53:1357–1368. doi: 10.1016/s0006-2952(96)00904-5. [DOI] [PubMed] [Google Scholar]
- 5.Hong MY, et al. Fish oil enhances targeted apoptosis during colon tumor initiation in part by downregulating Bcl-2. Nutr. Cancer. 2003;46:44–51. doi: 10.1207/S15327914NC4601_06. [DOI] [PubMed] [Google Scholar]
- 6.Hong MY, et al. Dietary fish oil reduces O6-methylguanine DNA adduct levels in rat colon in part by increasing apoptosis during tumor initiation. Cancer Epidemiol. Biomarkers Prev. 2000;9:819–826. [PubMed] [Google Scholar]
- 7.Boffa LC, et al. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J. Biol. Chem. 1978;253:3364–3366. [PubMed] [Google Scholar]
- 8.Candido EP, et al. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 9.Sealy L, et al. The effect of sodium butyrate on histone modification. Cell. 1978;14:115–121. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- 10.Archer SY, et al. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl Acad. Sci. USA. 1998;95:6791–6796. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janicke R, et al. The multiple battles fought by anti-apoptotic p21. Cell Cycle. 2007;6:403–413. doi: 10.4161/cc.6.4.3855. [DOI] [PubMed] [Google Scholar]
- 12.Gartel AL, et al. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 13.Le H, et al. Cyclin-dependent kinase inhibitors uncouple cell cycle progression from mitochondrial apoptotic functions in DNA-damaged cancer cells. J. Biol. Chem. 2005;280:32018–32025. doi: 10.1074/jbc.M504689200. [DOI] [PubMed] [Google Scholar]
- 14.Yang WC, et al. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res. 2001;61:565–569. [PubMed] [Google Scholar]
- 15.Poole A, et al. Tumor suppressor functions for the Cdk inhibitor p21 in the mouse colon. Oncogene. 2004;23:8128–8134. doi: 10.1038/sj.onc.1207994. [DOI] [PubMed] [Google Scholar]
- 16.Pretlow TP, et al. Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res. 1991;51:1564–1567. [PubMed] [Google Scholar]
- 17.Bird RP, et al. The significance of aberrant crypt foci in understanding the pathogenesis of colon cancer. Toxicol. Lett. 2000;112–113:395–402. doi: 10.1016/s0378-4274(99)00261-1. [DOI] [PubMed] [Google Scholar]
- 18.Caderni G, et al. Slow-release pellets of sodium butyrate do not modify azoxymethane (AOM)-induced intestinal carcinogenesis in F344 rats. Carcinogenesis. 2001;22:525–527. doi: 10.1093/carcin/22.3.525. [DOI] [PubMed] [Google Scholar]
- 19.Caderni G, et al. Slow-release pellets of sodium butyrate increase apoptosis in the colon of rats treated with azoxymethane, without affecting aberrant crypt foci and colonic proliferation. Nutr. Cancer. 1998;30:175–181. doi: 10.1080/01635589809514660. [DOI] [PubMed] [Google Scholar]
- 20.Zoran DL, et al. Diet and carcinogen alter luminal butyrate concentration and intracellular pH in isolated rat colonocytes. Nutr. Cancer. 1997;27:222–230. doi: 10.1080/01635589709514530. [DOI] [PubMed] [Google Scholar]
- 21.Warrell RP, Jr., et al. Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J. Natl Cancer Inst. 1998;90:1621–1625. doi: 10.1093/jnci/90.21.1621. [DOI] [PubMed] [Google Scholar]
- 22.Viale G, et al. p21WAF1/CIP1 expression in colorectal carcinoma correlates with advanced disease stage and p53 mutations. J. Pathol. 1999;187:302–307. doi: 10.1002/(SICI)1096-9896(199902)187:3<302::AID-PATH243>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 23.Vanamala J, et al. Suppression of colon carcinogenesis by bioactive compounds in grapefruit. Carcinogenesis. 2006;27:1257–1265. doi: 10.1093/carcin/bgi318. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, et al. Dietary fibers stimulate colonic cell proliferation by different mechanisms at different sites. Nutr. Cancer. 1994;22:267–276. doi: 10.1080/01635589409514352. [DOI] [PubMed] [Google Scholar]
- 25.Maciorowski KG, et al. Diet and carcinogen alter the fecal microbial populations of rats. J. Nutr. 1997;127:449–457. doi: 10.1093/jn/127.3.449. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs LR, et al. Dietary wheat bran lowers colonic pH in rats. J. Nutr. 1982;112:592–594. doi: 10.1093/jn/112.3.592. [DOI] [PubMed] [Google Scholar]
- 27.Hatamoto M, et al. Identification and cultivation of anaerobic, syntrophic long-chain fatty acid-degrading microbes from mesophilic and thermophilic methanogenic sludges. Appl. Environ. Microbiol. 2007;73:1332–1340. doi: 10.1128/AEM.02053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong CSM, et al. The influence of specific luminal factors on the colonic epithelium: high-dose butyrate and physical changes suppress early carcinogenic events in rats. Dis. Colon Rectum. 2005;48:549–559. doi: 10.1007/s10350-004-0810-x. [DOI] [PubMed] [Google Scholar]
- 29.El-Deiry W, et al. Topological control of p21waf1/cip1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–2919. [PubMed] [Google Scholar]
- 30.Polyak K, et al. Early alteration of cell-cycle regulated gene expression in colorectal neoplasia. Am. J. Pathol. 1996;149:381–387. [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, et al. Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene. 1999;18:1131–1138. doi: 10.1038/sj.onc.1202426. [DOI] [PubMed] [Google Scholar]
- 32.Chai F, et al. Involvement of p21(Waf1/Cip1) and its cleavage by DEVD-caspase during apoptosis of colorectal cancer cells induced by butyrate. Carcinogenesis. 2000;21:7–14. doi: 10.1093/carcin/21.1.7. [DOI] [PubMed] [Google Scholar]