

Abstract
HTLV-1 and HTLV-2 are highly related complex retroviruses that have been studied intensely for nearly three decades because of their association with neoplasia, neuropathology, and/or their capacity to transform primary human T lymphocytes. The study of HTLV also represents an attractive model that has allowed investigators to dissect the mechanism of various cellular processes, several of which may be critical steps in HTLV-mediated pathogenesis. Both HTLV-1 and HTLV-2 can efficiently immortalize and transform T lymphocytes in cell culture and persist in infected individuals or experimental animals. However, the clinical manifestations of these two viruses differ significantly. HTLV-1 is associated with adult T-cell leukemia (ATL) and a variety of immune-mediated disorders including the chronic neurological disease termed HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). In contrast, HTLV-2 is much less pathogenic with reports of only a few cases of variant hairy cell leukemia and neurological disease associated with infection. The limited number of individuals shown to harbor HTLV-2 in association with specific diseases has, to date, precluded convincing epidemiological demonstration of a definitive etiologic role of HTLV-2 in human disease. Therefore, it has become clear that comparative studies designed to elucidate the mechanisms by which HTLV-1 and HTLV-2 determine distinct outcomes are likely to provide fundamental insights into the initiation of multistep leukemogenesis.
Keywords: HTLV-1, HTLV-2, tax, cellular transformation, immortalization, leukemia
Introduction
Human T-cell lymphotropic viruses type 1 (HTLV-1) and type 2 (HTLV-2) are closely related human complex deltaretroviruses. Infection with HTLV-1 has been linked to the development of adult T-cell leukemia/lymphoma (ATL/ATLL), a clonal aggressive malignancy of CD4+ T lymphocytes. HTLV-1 infection is endemic in Japan, Africa, South America, and the Caribbean basin. Intriguingly, HTLV-1 also is the causative agent of a distinct neurological disorder termed HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) (for review see Green and Chen, 2001). HTLV-2 was first identified in a T cell line established from a patient with hairy-cell leukemia (Saxon et al., 1978). In addition, HTLV-2 infection has been associated with sporadic cases of a myelopathy resembling HAM/TSP (Hjelle et al., 1992; Dooneief et al., 1996; Murphy et al., 1997), but has not been clearly linked with the development of lymphoproliferative disorders. HTLV-2 infection is highly concentrated in Central and West Africa (Gessain et al., 1993; Goubau et al., 1993), native Amerindian populations in North, Central, and South America (Hjelle et al., 1990; Lairmore et al., 1990; Heneine et al., 1991; Levine et al., 1993), and among cohorts of intravenous drug users (IVDUs) in the United States and Europe (Tedder et al., 1984; Robert-Guroff et al., 1986; Lee et al., 1989; Khabbaz et al., 1991; Toro et al., 2005).
HTLV-1 and HTLV-2 have a similar genome structure and share approximately 70% nucleotide sequence homology. In addition to the essential viral genes gag, pol, and env, HTLV-1 and HTLV-2 encode regulatory and accessory genes from pX region open reading frames (ORFs) found in the 3′ portion of the viral genome (Figure 1). ORFs IV and III encode the Tax and Rex regulatory proteins, respectively. Tax acts in trans to activate transcription initiating from the viral promoter in the U3 region of the long terminal repeat (Cann et al., 1985; Chen et al., 1985; Felber et al., 1985; Fujisawa et al., 1985; Seiki et al., 1986; Ross et al., 1997) and Rex regulates viral gene expression post-transcriptionally by facilitating the cytoplasmic expression of the incompletely spliced viral mRNAs (Younis and Green, 2005). HTLV-1 ORFs I, and II encode the accessory proteins p12/p27 and p13/p30, respectively, whereas the HTLV-2 ORFs I, II, and V encode the p10, p28, and p11 accessory gene products, respectively. The functional roles of these proteins in HTLV biology are not clearly understood. However, studies have indicated that they are dispensable for infection and transformation of activated T cells in vitro (Green et al., 1995; Derse et al., 1997), but are important for the ability of the virus to infect, spread, and persist in vivo (Cockerell et al., 1996; Collins et al., 1998; Bartoe et al., 2000; Silverman et al., 2004). This review will discuss important aspects of HTLV biology highlighting differences between HTLV-1 and HTLV-2. It will emphasize studies investigating the transforming potential of HTLV-2 and the Tax-2 oncoprotein with appropriate comparisons to HTLV-1.
Figure 1.
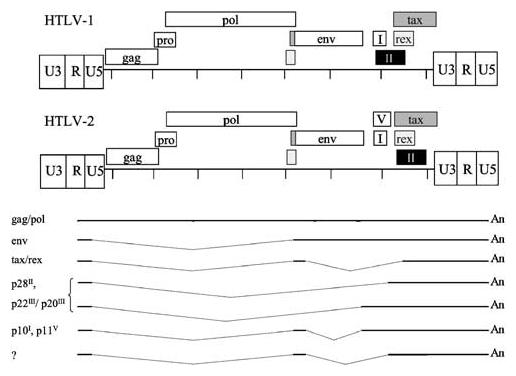
Genome organization of HTLV-1 and HTLV-2. HTLV-1 and HTLV-2 proviral genomes and the open reading frames are shown. Highlights the HTLV-2 mRNA species, including at least seven mRNAs. The genomic unspliced mRNA encodes the Gag, Pol, and Pro proteins. In addition to the unspliced species, three singly spliced mRNAs contain exon 1 (nt 1–134) linked to splice acceptor sites at 4729 (Env), 6629 (p28, p22/p20), or 6899 (p28, p22/p20). The major doubly spliced mRNA encodes Tax/Rex and contains exons 1 and 2 (nt 4729–4868) linked to a splice acceptor at position 6899. The other doubly spliced mRNAs contain exons 1 and 2 linked to a splice acceptor at 6629 or 6491; their protein products are less characterized
HTLV experimental system
Since their discovery, experimental assay systems for the study of HTLV have been complicated by the poor replication of these viruses in culture as compared to the avian and murine retroviruses, as well as HIV-1. Although a reliable infection-based animal model of disease for studies of HTLV pathogenesis is lacking, transformation of primary T lymphocytes represents a widely accepted experimental system for exploring the early events associated with malignancy. HTLV has been a difficult virus to work with for a number of reasons. First, cell-free infection by HTLV is very inefficient and efficient infection of cells requires cocultivation of peripheral blood mononuclear cells (PBMC) with irradiated HTLV-producer cells. Secondly, although HTLV has the capacity to infect a number of cell types including T cells, B cells, endothelial cells, glial cells, and monocytes of both human and nonhuman origin (Ho et al., 1984; Hoffman et al., 1984; Akagi et al., 1992; Koyanagi et al., 1993), the only cells susceptible to HTLV transformation are primary T lymphocytes. For the context of this article transformation is defined as continuous growth in the absence of exogenous IL-2; immortalization is defined as continuous IL-2-dependent growth. Transformed cells typically are evident microscopically as refractile cell clusters within 7–10 weeks of cocultivation. However, establishment of hearty IL-2-independent transformed cell lines usually requires months in culture. Lastly, the isolation, amplification, and manipulation of HTLV infectious DNA clones are not trivial due to problems of viable plasmid growth and amplification in bacteria.
Over the past 25 years, experimental studies have focused on dissecting functions of the HTLV gene products. Initial HTLV studies were restricted to examination of infected patients, overexpression of individual viral genes using reporter assays in cell lines, or characterization of infected cell lines or animals using viral isolates obtained directly from patients. Although these types of studies have been very informative, the understanding of HTLV biology and pathogenesis has benefited further from the isolation and manipulation of proviral clones capable of generating infectious virus, and the development and refinement of methodologies for characterization of these clones in primary human T lymphocytes and relevant animal models. It is important to note that HTLV gene structure function studies in the context of a replicating virus were first performed in HTLV-2 due to the availability of an HTLV-2 infectious molecular clone (Chen et al., 1983) over a decade prior to HTLV-1 (Kimata et al., 1994; Derse et al., 1995).
HTLV cellular transformation and pathogenesis
Although HTLV-1 and HTLV-2 display distinct pathogenic properties in vivo, both viruses infect and transform primary human T cells in cell culture. The basis for HTLV-mediated cellular transformation is not completely understood, but clearly involves the viral transactivator protein Tax. Tax displays oncogenic potential in several experimental systems including the morphological transformation of rodent fibroblasts, induction of tumors in transgenic animals, and Herpes samiri vector immortalization of human T cells (Nerenberg et al., 1987; Tanaka et al., 1990; Grassmann et al., 1992; Yamaoka et al., 1992; Grossman et al., 1995). More recently, studies using infectious molecular clones showed directly that a functional Tax is essential for HTLV-1- and HTLV-2-mediated cellular transformation of primary human T cells (Ross et al., 1996; Robek and Ratner, 1999). The precise mechanism by which Tax initiates the malignant process is unclear, but is proposed to involve several points of transcriptional and post-transcriptional dysregulation in the infected T cell. Tax activities implicated in the malignant process, with emphasis on those supported by studies on both Tax-1 and Tax-2 will be discussed in more detail below.
Investigation of HTLV cell tropism in infected patients has indicated that HTLV-1 has a preferential tropism for CD4+ T cells in both asymptomatic patients and those with neurological disease (Richardson et al., 1990). More recently, studies demonstrated that CD8+ T cells are an additional viral reservoir in vivo for HTLV-1 in HAM/TSP patients (Nagai et al., 2001). In vitro, it has been shown that Tax-mediated transcription of HTLV-1 is significantly increased in purified CD4+ versus CD8+ T cell subsets (Newbound et al., 1996). This is consistent with the hypothesis that this enhanced rate of transcription ultimately is responsible for the cell tropism and the leukemogenic potential of HTLV-1.
HTLV-2 in vivo tropism appears to be less clear. One in vivo study indicated that HTLV-2 has a preferential tropism for CD8+ T cells (Ijichi et al., 1992), whereas others have detected HTLV-2 in both CD4+ and CD8+ T cell subsets with a greater proviral burden in CD8+ T cells (Miyamoto et al., 1991; Lal et al., 1993a). In contrast to HTLV-1, both purified CD4+ and CD8+ T cells are equally susceptible to HTLV-2 infection and subsequent viral gene expression (Wang et al., 2000). However, coculture of irradiated HTLV-2 producer cells with PBMCs or purified T cell subsets resulted in preferential transformation of CD8+ T cells (Wang et al., 2000; Ye et al., 2003). The first viral recombination studies exchanging the tax/rex genes of HTLV-1 and HTLV-2 infectious molecular clones recently were performed in an attempt to identify the genetic basis for this distinct transformation tropism. The results showed that the gene exchanges were well tolerated by the viruses. However, the Tax and overlapping Rex sequences did not confer the distinct transformation tropisms of HTLV-1 and HTLV-2 (Ye et al., 2003) suggesting the contribution of other viral genes or sequences.
Another difference in HTLV-1 and HTLV-2 cellular transformation involves the Jak/STAT activation pathway, which has been shown to be constitutively activated in several hematopoietic malignancies, including HTLV-1-associated adult T cell leukemia (Takemoto et al., 1997). T cells transformed by HTLV-1 exhibited constitutive Jak/STAT activation and this activation correlated with the transition from IL-2-dependent to IL-2-independent phase of growth. Interestingly, examination of the activation status of T cells transformed by HTLV-2 and STLV-2pan-p revealed that the Jak/STAT signaling pathway is not activated in any of the HTLV-2/STLV-2-transformed T cell lines examined, but this pathway could be induced upon IL-2 treatment of the cells (Mulloy et al., 1998). These results suggest that HTLV-2 and the cognate virus STLV-2 from Pan paniscus transforms T cells in vitro by mechanisms at least partially different from those used by HTLV-1.
Role of tax in HTLV pathogenesis
It remains unclear as to why infection with HTLV-1 is more pathogenic in comparison to infection with HTLV-2. It has been hypothesized that this differential pathogenic potential might be attributable to differences in the respective Tax activities. The HTLV-1-encoded transactivator Tax (Tax-1) and the HTLV-2 Tax (Tax-2) have ∼78% homology at the amino acid level and display many properties characteristic of viral oncoproteins. Furthermore, strong evidence supports the premise that Tax plays an important role in HTLV pathogenesis. HTLV Tax is expressed early after infection and is a transactivator of viral gene expression. Tax increases transcription of the viral promoter by exploiting the cyclic-AMP-response element (CRE) and activating transcription factor (ATF) binding proteins (CREB/ATF) family of transcription factors (Zhao and Giam, 1992; Wagner and Green, 1993; Adya et al., 1994; Anderson and Dynan, 1994; Bantignies et al, 1996; Yin et al., 1995b). Tax facilitates the binding of these proteins to the nonpalindromic consensus cAMP response element (CRE) sequences contained within the 21 bp repeats in the HTLV promoter (Adya et al., 1994; Yin et al., 1995a). Formation of the Tax/CREB/promoter bound ternary complex appears to be critical for the recruitment of the multifunctional CBP/p300 coactivators and their associated cellular coactivator PCAF, which physically interacts with Tax, ultimately resulting in efficient viral transcription. Tax also modulates the expression of cellular genes, and many cellular transcription factors including serum response factor (SRF), NFκB, and bHLH proteins have been implicated in this interaction. Of the factors targeted by Tax, NFκB clearly plays a prominent role in deregulation of cellular gene expression. Tax induces the nuclear expression of members of the NFκB family of transcription factors, which leads to expression of many gene promoters containing NFκB motifs (Lacoste et al., 1991; Lindholm et al., 1991; Li et al., 1993; Kanno et al., 1994; Brockman et al., 1995; Maggirwar et al., 1995; McKinsey et al., 1996). These genes include those encoding interleukin-2 (IL-2), IL-2 receptor α (IL-2Rα), IL-3, GM-CSF, as well as HIV-1 (Siekevitz et al., 1987; Ballard et al., 1988; Leung and Nabel, 1988; Wano et al., 1988; Böhnlein et al., 1989; Nimer, 1991). The ability to transcriptionally activate a variety of cellular genes, including proto-oncogenes, is a key mechanism by which Tax is thought to modulate cellular transformation. Four subtypes of HTLV-2 (A to D) have been described that encode slightly variable Tax proteins. Tax-2B, 2C, and 2D are similar, but not identical, in length (356, 356, 344 amino acids, respectively), whereas Tax-2A is shorter (331 amino acids). Tax-1 (353 amino acids) and Tax-2 isolates have shown a remarkable similarity with respect to transcriptional transactivation via CREB/ATF and NFκB pathways (Ross et al., 1997; Lewis et al., 2002). Mutational analysis and amino-acid sequence alignment have indicated that the domains important for transactivation through CREB/ATF or NFκB signaling pathways are similar, but not identical, in Tax-1 and Tax-2 (Figure 2) (Smith and Greene, 1990; Semmes and Jeang, 1992; Ross et al., 1997).
Figure 2.

Structural and functional domains of Tax-1 and Tax-2A. Specific domains, mutations and the transcriptional activation pathways interrupted are indicated
Role of NFκB and CREB/ATF in HTLV Tax-mediated transformation
A number of mutations in Tax-1 and Tax-2 have been described that selectively abrogate the ability of Tax to activate transcription through defined molecular pathways. These mutants have been invaluable for dissecting cell signaling pathways and for determining the interplay between Tax and cellular transformation. Mutants of Tax-1 that have been used substantially and have been the most informative regarding function are termed Tax M47 and Tax M22 (Smith and Greene, 1990). Tax M47 does not activate CREB/ATF responsive promoters, but maintains the capacity to activate NFκB promoters; Tax M22 activates CREB/ATF promoters, but fails to activate NFκB. Extensive mutational analysis of Tax-2 mapped and characterized identical mutations (Ross et al., 1997). Several approaches have been used to evaluate domains of Tax-1 required to immortalize/transform rodent cell lines or primary human T cells. These analyses have yielded conflicting results as to whether Tax-1 activation of CREB/ATF or NFκB pathway is critical for cellular transformation (Smith and Greene, 1991; Akagi et al., 1997; Rosin et al., 1998; Iwanaga et al., 1999; Robek and Ratner, 1999). More recently, a unique HTLV-2 provirus (HTLVc-enh), which replicates by a Tax-independent mechanism due to replacement of the TRE with the cytomegalovirus immediate-early promoter enhancer (Ross et al., 1996), was utilized to identify the respective roles of the CREB/ATF or NFκB signaling pathway in HTLV-2-mediated transformation of primary human T lymphocytes. The advantage of using the novel HTLVc-enh for these studies is that viral gene expression and replication is not significantly disrupted by mutations in Tax-2. Results from these studies demonstrated that Tax-2 activation of both NFκB and CREB/ATF is required to induce IL-2 independent T lymphocyte transformation. These data are consistent with the conclusion that the activation of NFκB provides a critical proliferative signal early in the cellular transformation process. HTLVc-enh containing a mutant Tax that fails to activate NFκB is indistinguishable in phenotype from a Tax knockout virus. It appears that CREB/ATF activation by Tax is required to promote sustained cell growth and IL-2-independent cellular transformation (Ross et al., 2000).
Tax and cell cycle dysregulation
T lymphocyte cell division and proliferation generally is a tightly regulated process. Perturbation of the cell cycle is a common feature in the transformation of cells by viral oncoproteins. Defects in the regulation of cell cycle progression and induction of DNA damage are one of the common features of transformed cells, including HTLV-transformed cells. Eukaryotic cell cycle progression is regulated by sequential activation and the subsequent inactivation of a series of cyclin-dependent kinases (CDKs) at different stages of the cell cycle. D-type cyclins (D1, D2, D3) and cyclin E are involved in regulating G1 progression and entry into S phase. Tax-1 has been shown to compel cells to egress from G1 into S phase (Suzuki et al., 1996; Low et al., 1997; Neuveut et al., 1998; Schmitt et al., 1998; Haoudi and Semmes, 2003). Infection with HTLV-1 has been shown to be capable of influencing G1/S transition as well (Schmitt et al., 1998; Lemoine and Marriott, 2001; Haoudi and Semmes, 2003). Tax induces T-cell proliferation in the absence of appropriate signals and dysregulation of cellular gene expression important for cell growth and cell cycle progression has been hypothesized to be the molecular basis for cellular transformation. Tax-1 modifies the cell cycle, by directly binding CDK-4 and CDK-6 and their inhibitors such as p16INK4a. Several cellular genes are also transcriptionally repressed by Tax-1 including β-polymerase, lck, p18INK4c gene, bax, cyclin 1A, cyclin D3, p53 and myb (Jeang et al., 1990; Uittenbogaard et al., 1995; Brauweiler et al., 1997; Lemasson et al., 1997; Suzuki et al., 1999a; Riou et al., 2000; Kibler and Jeang, 2001). Experimental evidence has shown that the mechanisms of repression include modulation of bHLH factors, NFκB, or direct competition between Tax and cellular transcription factors for limiting intracellular CBP/p300. Thus, Tax-1 affects G1/S transition by overriding cell cycle control regulation and releasing cells from growth arrest.
Tax-1 also has been documented to induce cell cycle arrest (Lemoine and Marriott, 2001; Van et al., 2001; Liang et al., 2002; Haoudi et al., 2003; Haoudi and Semmes, 2003). HTLV-1-infected T-cell lines have a characteristic expression pattern of cell cycle regulatory genes, distinct from that of uninfected T cells. HTLV-1-infected T cells preferentially express cyclin D1 and D2, as well as high levels of the CDK inhibitors p16INK4 and p21cip1/waf1 (Akagi et al., 1996; Mori et al., 2002). p21cip1/waf1 is a CDK inhibitor, forms part of the cyclin D1/CDK2/PCNA complex and has been shown to be transcriptionally upregulated by Tax-1 (Yamada et al., 1994; de La Fuente et al., 2000; Sieburg et al., 2004). Overexpression of p21cip1/waf1 inhibits two critical checkpoints in the cell cycle, namely G1 and G2, via p53-independent and -dependent pathways (Macleod et al., 1995). Tax-2 also activates expression of the p21cip1/waf1 promoter, although at much more modest levels (∼25 to 35%) as compared to Tax-1 (de La Fuente et al., 2000; Sieburg et al., 2004). Tax-1 also can induce arrest of cells at G2/M (Liang et al., 2002; Haoudi and Semmes, 2003). De novo expression of Tax-1 using lentivirus vectors recently has been shown to arrest cells at G2/M by interaction with Chk2 tumor suppressor protein (Haoudi et al., 2003). Although induction of cell cycle, arrest by an oncoprotein may seem counter-intuitive, Tax clearly dysregulates the cell cycle although the precise end point of perturbation is cell type dependent. The observation that HIV-1 Vpr also is capable of G2 arrest, suggests HTLV is not unique in its ability to dysregulate cell cycle and that control of cell cycle progression may be an advantage to virus replication.
Cell lines stably transduced to express Tax-1 or Tax-2 have recently been shown to be a useful model system to directly compare the roles of constitutive Tax-1 and Tax-2 expression on cellular proliferation kinetics. Studies using Jurkat cells that constitutively express Tax-1 or Tax-2 demonstrated reduced cellular proliferation kinetics, although Tax-1 displayed a more robust effect on reducing cell proliferation kinetics in comparison to cells expressing Tax-2. In addition, formation of binucleated and multinucleated lymphoid cells was detected in Tax-1-expressing Jurkat cell lines (Liang et al., 2002; Sieburg et al., 2004). Tax-2-expressing cells also demonstrated multinucleated cells, but to a lesser degree than Tax-1 cells. The abnormal cellular morphology suggests that Tax-1 and Tax-2 may block progression or completion of mitosis or cytokinesis and bolsters the tenet that Tax-1 and Tax-2 disrupt cell cycle checkpoints resulting in unscheduled S phase entry and accumulation of DNA damage (Lemoine and Marriott, 2001; Marriott et al., 2002).
Although investigations of Tax-1 and Tax-2 function in transformed cell lines have been useful and convenient, it is more appropriate and critical to ascribe the functions of these oncoproteins in cell types most likely to be encountered by the virus, particularly with respect to the characterization of dysregulation of the cell cycle. Recently, the role of Tax-1 and Tax-2 in cell cycle progression has been documented in primary human hematopoietic CD34+ progenitor cells (HPCs), a heterogeneous cell population that includes hematopoietic stem cells (HSCs). Hematopoiesis is a tightly regulated process involving self-renewal, expansion of lineage-committed progenitors and maturation into terminally differentiated cells. HSCs generally are quiescent, which is critical to prevent exhaustion under conditions of stress. HSCs are subject to strict regulation of cell division and differentiation during normal development and, thus, are exquisitely sensitive to perturbation of the cell cycle by oncoproteins. Lentivirus-mediated transduction of Tax-1 in HPCs suppressed hematopoiesis in vitro and led to increased cell cycle arrest in G0/G1. Tax-1 transduced HPCs displayed concomitant activation of endogenous p21cip1/waf1 and p27kip1 expression (Tripp et al., 2003) (Tripp and Feuer, unpublished observations). Interestingly, Tax-2 failed to arrest HPCs in G0/G1, failed to suppress hematopoiesis and did not detectably modulate endogenous p21cip1/waf1 and p27kip1 gene expression. The dramatically different phenotypes displayed by Tax-1 and Tax-2 in primary hematopoietic cells with respect to cell cycle progression prompts speculation that HTLV-1 may be more effective at facilitating viral latency in vivo following infection of HPCs and HSCs and these infection events may contribute to the distinct pathogenesis of HTLV-1 and HTLV-2.
Differential modulation of apoptosis by Tax-1 and Tax-2
Apoptosis is an active physiological process that plays an essential role in the elimination of virus-infected or cancerous cells. Previous evidence suggested that imbalances between cellular death-inducing and proliferation pathways significantly contribute to oncogenesis. Defects in the mechanisms controlling apoptosis play a major role in the pathogenesis of cancer and neurode-generative and immunological diseases. Therefore, it is not entirely surprising that Tax-1 and Tax-2 have been reported to prevent induction of apoptosis or programed cell death (PCD). Although Tax-1 plays an essential role during the immortalization of CD4+ T lymphocytes and is invariably integrated in ATL cells, numerous studies have shown that persistent Tax expression is toxic to cells and is associated with apoptosis in nonlymphoid as well as lymphoid-derived cell lines (Yamada et al., 1994; Chlichlia et al., 1995; Nicot et al., 1997; Los et al., 1998; Nicot and Harrod, 2000). Tax-1 has been shown to induce apoptosis in Jurkat cells (Chlichlia et al., 1995; Los et al., 1998; Rivera-Walsh et al., 2001), Rat-1 (Tanaka et al., 1990), human endothelial cells (Nicot et al., 1997; Nicot and Harrod, 2000) and sensitizes cells to apoptosis following DNA damage by UV irradiation (Kao et al., 2000). Jurkat cell lines that constitutively express Tax-1 or Tax-2 had significantly down-modulated Tax expression during cell expansion in vitro, suggesting that elevated levels of Tax are detrimental for cellular replication and survival (Sieburg et al., 2004). Previous difficulties in the generation of stably transduced Tax cell lines may reflect the elevated sensitivity to apoptosis demonstrated by clonal cells that express high levels of Tax during drug selection coupled with decreased cellular proliferation kinetics displayed by Tax-transduced cells. In animal models, tumor cells derived from Tax-1 transgenic mice had enhanced levels of apoptosis (Hall et al., 1998) and neuronal cells showed enhanced apoptosis in HTLV-1-infected WKAH rats that displayed myeloneuropathy (Ohya et al., 1997).
Expression of Tax-1 and Tax-2 has been shown to protect cells from PCD. Specifically, Tax-1 protects cells from apoptosis following serum starvation (Liang et al., 2002; Sieburg et al., 2004) by upregulation of Bcl-2 and repression of Bax (Brauweiler et al., 1997; Tsukahara et al., 1999; Nicot et al., 2000). Both Tax-1 and Tax-2 inhibit apoptosis in Jurkat cells and in murine fibro-blasts following serum deprivation (Saggioro et al., 2001; Sieburg et al., 2004). Tax-2 has been shown to protect cells from Fas-mediated apoptosis by upregulation of Bcl-XL expression (Zehender et al., 2001). In contrast, Tax-1 uniquely confers resistance to apoptosis following treatment of cells with the topoisomerase inhibitors camptothecin or etoposide (Sieburg et al., 2004). The ability of Tax-1 to protect cells from a broad range of PCD stimuli and from apoptosis due to DNA damage may be a critical distinguishing component of the association of HTLV-1 with leukemogenesis (Haoudi and Semmes, 2003), particularly since Tax-1 has been associated with increased DNA damage and the accumulation of mutations (Gatza et al., 2003). The more robust ability of Tax-1 to protect cells from apoptosis could reflect the ability of HTLV-1 infected lymphocytes to persist in vivo and undergo further mutagenic events leading to full transformation. The role of Tax-1 in modulation of apoptosis clearly is cell-type dependent and ultimately could provide an environment conducive to viral replication.
Distinct Tax-1 and Tax-2 activities
To date, infection with HTLV-2 has not been conclusively associated with the development of ATL-like malignancies as those seen with HTLV-1 infection. This is intriguing from the perspective that HTLV-1 and HTLV-2 share a high degree of sequence homology, particularly in the Tax region and that both viruses have the capability of immortalizing T cells in vitro. Distinct phenotypic differences between Tax-1 and Tax-2 have been documented in certain cell culture model systems. These differences have been the focus of speculation as to why HTLV-1 and not HTLV-2 is associated with disease. Tax-1 and Tax-2 display a differential ability to induce cellular gene transcription (Tanaka et al., 1990; Ejima et al., 1993; Lal et al., 1993b; Mori and Prager, 1996; Ye et al., 2003). For example, Tax-1 has been shown to have a higher intrinsic transactivation activity for the viral LTR than Tax-2 (Ye et al., 2003). This would not only increase viral gene expression and infectivity but also would enhance expression of cellular genes responsive to Tax. Tax-1 has been shown to induce micronuclei formation, a marker for genetic instability. Genetic instability correlates well with biological findings of DNA damage in ATLL cells (Semmes et al., 1996). Micronuclei induction capacity maps to the carboxyl amino acids of Tax-1. Interestingly, Tax-2 isolates that are missing the carboxy terminus of Tax-1 (Tax-2A) or contain unrelated amino acids (Tax-2C or -2D) display a significantly less capacity to induce micronuclei. More recently, Tax-2 was shown to be impaired for the inhibition of p53 functions in comparison to Tax-1 (Mahieux et al., 2000). Tax-2 also was shown to be less efficient at transforming rat embryo fibroblasts in vitro, in comparison to Tax-1 (Endo et al., 2002). HTLV-2-transformed lymphoid cell lines (JLB-II) demonstrated lower tumorigenic potential in comparison to HTLV-1 transformed cells (SLB-1) when inoculated into SCID mice (Feuer et al., 1995). Results in a recent report demonstrated that while Tax-1 is localized to the cytoplasm and the nucleus, a 10 amino acid domain unique to Tax-2 is responsible for the predominant cytoplasmic localization of Tax-2 (Meertens et al., 2004). Collectively, the phenotypes displayed by Tax-2 in these models are consistent with Tax-2 retaining a less robust transformation potential in comparison to Tax-1. Furthermore, the ability of Tax-1 to induce G0/G1 cell cycle arrest in HPCs and suppress hematopoiesis, in distinct contrast to Tax-2, allows speculation that the fate of infected HPCs following HTLV-1 and HTLV-2 infection may be fundamentally different in vivo (Tripp et al., 2003; Feuer, unpublished observations).
Role of Tax-1 C-terminus in transformation
The C-terminus of Tax-1 recently has received much attention concerning domains that differentiate it from Tax-2. The C-terminal 53 amino acids of Tax-1 were shown to be responsible for increased transformation efficiency in rodent fibroblasts and increased micronuclei induction; this region is noticeably absent in Tax-2A and significantly different in the other Tax-2 isolates. A PDZ binding motif (PBM) has been identified in this C-terminal fragment of Tax-1 and is absent in Tax-2 (see also Hall and Fujii, this issue). The PDZ domain was named after the first identified PDZ-containing proteins, postsynaptic density protein (PSD-95), Drosophila discs large protein (DLG) and epithelial tight junction protein (Zonula Occludens-1). It is one of the protein–protein interaction modules commonly used in eukaryotic cells. PDZ domain-containing proteins play a key role in recruiting and organizing the appropriate proteins to sites of cellular signaling, as well as polar sites of cell–cell communication (Fanning and Anderson, 1999; Harris and Lim, 2001; Sheng and Sala, 2001). Tax-1 has been shown to interact with the human homolog of Drosophila neoplasia related protein, hDLG (Rousset et al., 1998). Tax-1 competes with the binding domain of hDLG and APC tumor suppressor protein and rescues cells from cell cycle arrest induced by hDLG (Suzuki et al., 1999b). Additional studies have implicated other cellular PDZ domain-containing proteins as Tax-1 targets, such as precursor of interleukin-16 (pro-IL-16) and a membrane-associated guanylate kinase (MAGUK) with inverted orientation (MAGI)-3. Pro-IL-16 is an abundant protein constitutively expressed in human peripheral blood T cells that can induce cell growth arrest. MAGI-3 belongs to the same MAGUK family as hDlg and has been implicated in several cellular signaling pathways involved in cell survival as well as cell polarity (Wu et al., 2000; Adamsky et al., 2003; Wilson et al., 2003; Ohashi et al., 2004; Yao et al., 2004).
A chimeric Tax-2 encoding the last 53 amino acids of Tax-1 (Tax221), which contains the PBM, demonstrated an increased transforming potential in rat fibroblast cells (Endo et al., 2002). It was demonstrated further that deletion of only the PBM from Tax-1 led to reduced transformation activity in rat fibroblasts (Hirata et al., 2004). Transduction of Tax221 into HPCs results in suppression of hematopoiesis in vitro and induction of p21cip1/waf1 and p27kip1 expression. This suggests that domains in the C terminus of Tax-1 contribute to suppression of hematopoiesis and transcriptional trans-activation of endogenous genes that regulate the entry of HPCs into the cell cycle (Feuer, unpublished results).
Recently, the contribution of the Tax-1 PBM to HTLV-induced proliferation and immortalization of primary T cells in vitro and viral survival in an infectious rabbit animal model was investigated. Coculture microtiter proliferation assays revealed that the Tax-1 PBM significantly increased both HTLV-1- and HTLV-2-induced primary T-cell proliferation. In addition, Tax-1 PBM was shown to be responsible for the micronuclei induction activity of Tax-1 relative to Tax-2. Viral infection and persistence were severely attenuated in rabbits inoculated with HTLV-1DPBM (Xie and Green, unpublished results). Together, these studies imply that the PBM of Tax-1 and its interacting partners, the cellular PDZ domain containing proteins, are a major determinant of the differences in pathogenicity between HTLV-1 and HTLV-2.
Conclusions
HTLV-1 and HTLV-2 can efficiently immortalize and transform T lymphocytes in vitro and persist in infected individuals or experimental animals. HTLV-1 infection leads to leukemia and HAM/TSP, whereas HTLV-2 is associated with only a few cases of disease. The differences between these two viruses, focusing primarily on the Tax oncoproteins, have been highlighted in this review. These differences likely provide the basis for the different clinical manifestations of these two viruses. Further comparative studies in the context of infectious molecular clones in primary human T cells in vitro and relevant animal models will be required to understand the multistep leukemogenic process associated with HTLV-1 infection.
Acknowledgments
Acknowledgements We thank Tim Vojt for preparation of the figures and Kate Hayes for editorial comments. This work is supported in part by National Institutes of Health Grants CA100730 (PLG) and the Hendricks Foundation (GF).
References
- Adamsky K, Arnold K, Sabanay H, Peles E. J. Cell Sci. 2003;116:1279–1289. doi: 10.1242/jcs.00302. [DOI] [PubMed] [Google Scholar]
- Adya N, Zhao L-J, Huang W, Boros I, Giam C-Z. Proc. Natl. Acad. Sci. USA. 1994;91:5642–5646. doi: 10.1073/pnas.91.12.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi T, Ono H, Nyunoya H, Shimotohno K. Oncogene. 1997;14:2071–2078. doi: 10.1038/sj.onc.1201045. [DOI] [PubMed] [Google Scholar]
- Akagi T, Ono H, Shimotohno K. Oncogene. 1996;12:1645–1652. [PubMed] [Google Scholar]
- Akagi T, Hoshida Y, Yoshino T, Teramoto N, Kondo E, Hayashi K, Takahashi K. Acta Neuropathol. 1992;84:147–152. doi: 10.1007/BF00311387. [DOI] [PubMed] [Google Scholar]
- Anderson MG, Dynan WS. Nucleic Acids Res. 1994;22:3194–3201. doi: 10.1093/nar/22.15.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard DW, Bohnlein E, Lowenthal JW, Wano Y, Franza BR, Greene WC. Science. 1988;241:1652–1655. doi: 10.1126/science.241.4873.1652. [DOI] [PubMed] [Google Scholar]
- Bantignies F, Rousset R, Desbois C, Jalinot P. Mol. Cell. Biol. 1996;16:2174–2182. doi: 10.1128/mcb.16.5.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoe JT, Albrecht B, Collins ND, Robek MD, Ratner L, Green PL, Lairmore MD. J. Virol. 2000;74:1094–1100. doi: 10.1128/jvi.74.3.1094-1100.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhnlein E, Siekevitz M, Ballard DW, Lowenthal JW, Rimsky L, Bogerd H, Hoffman J, Wano Y, Franza BR, Greene WC. J. Virol. 1989;63:1578–1586. doi: 10.1128/jvi.63.4.1578-1586.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauweiler A, Garrus JE, Reed JC, Nyborg JK. Virology. 1997;231:135–140. doi: 10.1006/viro.1997.8509. [DOI] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee W, Ballard DW. Mol. Cell. Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann AJ, Rosenblatt JD, Wachsman W, Shah NP, Chen ISY. Nature. 1985;318:571–574. doi: 10.1038/318571a0. [DOI] [PubMed] [Google Scholar]
- Chen ISY, Cann AJ, Shah NP, Gaynor RB. Science. 1985;230:570–573. doi: 10.1126/science.2996140. [DOI] [PubMed] [Google Scholar]
- Chen IY, McLaughlin J, Gasson JC, Clark SC, Golde DW. Nature. 1983;305:502–505. doi: 10.1038/305502a0. [DOI] [PubMed] [Google Scholar]
- Chlichlia K, Moldenhauer G, Daniel PT, Busslinger M, Gazzolo L, Schirrmacher V, Khazaie K. Oncogene. 1995;10:269–277. [PubMed] [Google Scholar]
- Cockerell GL, Rovank J, Green PL, Chen ISY. Blood. 1996;87:1030–1035. [PubMed] [Google Scholar]
- Collins ND, Newbound GC, Albrecht B, Beard J, Ratner L, Lairmore MD. Blood. 1998;91:4701–4707. [PubMed] [Google Scholar]
- de La Fuente C, Santiago F, Chong SY, Deng L, Mayhood T, Fu P, Stein D, Denny T, Coffman F, Azimi N, Mahieux R, Kashanchi F. J. Virol. 2000;74:7270–7283. doi: 10.1128/jvi.74.16.7270-7283.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derse D, Mikovitis J, Polianova M, Felber BK, Ruscetti F. J. Virol. 1995;69:1907–1912. doi: 10.1128/jvi.69.3.1907-1912.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derse D, Mikovits J, Ruscetti F. Virology. 1997;237:123–128. doi: 10.1006/viro.1997.8781. [DOI] [PubMed] [Google Scholar]
- Dooneief G, Marlink R, Bell K, Marder K, Renjifo B, Stern Y, Mayeux R. Neurology. 1996;46:1556–1560. doi: 10.1212/wnl.46.6.1556. [DOI] [PubMed] [Google Scholar]
- Ejima E, J.D R, Massari M, Quan E, Stephens D, Rosen CA, Prager D. Blood. 1993;81:1017–1024. [PubMed] [Google Scholar]
- Endo K, Hirata A, Iwai K, Sakurai M, Fukushi M, Oie M, Higuchi M, Hall WW, Gejyo F, Fujii M. J. Virol. 2002;76:2648–2653. doi: 10.1128/JVI.76.6.2648-2653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning AS, Anderson JM. Curr. Opin. Cell Biol. 1999;11:432–439. doi: 10.1016/S0955-0674(99)80062-3. [DOI] [PubMed] [Google Scholar]
- Felber BK, Paskalis H, Kleinman-Ewing C, Wong-Staal F, Pavlakis GN. Science. 1985;229:675–679. doi: 10.1126/science.2992082. [DOI] [PubMed] [Google Scholar]
- Feuer G, Stewart SA, Baird SM, Lee F, Feuer R, Chen IS. J. Virol. 1995;69:1328–1333. doi: 10.1128/jvi.69.2.1328-1333.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa J, Seiki M, Kiyokawa T, Yoshida M. Proc. Natl. Acad. Sci. USA. 1985;82:2277–2281. doi: 10.1073/pnas.82.8.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatza ML, Watt JC, Marriott S. Oncogene. 2003;22:5141–5149. doi: 10.1038/sj.onc.1206549. [DOI] [PubMed] [Google Scholar]
- Gessain A, Fretz C, Koulibaly M, Boudret ML, Bah A, Raphael M, de The G, Fournel JJ. J. AIDS Hum. Retro. 1993;6:324–325. [PubMed] [Google Scholar]
- Goubau P, Liu HF, De Lange GG, Vandamme AM, Desmyter J. AIDS Res. Hum. Retroviruses. 1993;9:709–713. doi: 10.1089/aid.1993.9.709. [DOI] [PubMed] [Google Scholar]
- Grassmann R, Berchtolds S, Radant I, Alt M, Fleckenstein B, Sodroski JG, Haseltine WA, Ramstedt U. J. Virol. 1992;66:4570–4575. doi: 10.1128/jvi.66.7.4570-4575.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PL, Chen ISY. In: Fields Virology. 4th edn Knipe Dm, Howley P, Griffin D, Lamb R, Martin M, Straus S., editors. Lippincott Williams & Wilkins; Philidelphia; 2001. pp. 1941–1969. [Google Scholar]
- Green PL, Ross TM, Chen ISY, Pettiford S. J. Virol. 1995;69:387–394. doi: 10.1128/jvi.69.1.387-394.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman WJ, Kimata JT, Wong FH, Zutter M, Ley TJ, Ratner L. Proc. Natl. Acad. Sci. USA. 1995;92:1057–1061. doi: 10.1073/pnas.92.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AP, Irvine J, Blyth K, Cameron ER, Onions DE, Campbell ME. J. Pathol. 1998;186:209–214. doi: 10.1002/(SICI)1096-9896(1998100)186:2<209::AID-PATH162>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Haoudi A, Daniels RC, Wong E, Kupfer G, Semmes OJ. J. Biol. Chem. 2003;278:37736–37744. doi: 10.1074/jbc.M301649200. [DOI] [PubMed] [Google Scholar]
- Haoudi A, Semmes OJ. Virology. 2003;305:229–239. doi: 10.1006/viro.2002.1642. [DOI] [PubMed] [Google Scholar]
- Harris BZ, Lim WA. J. Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Heneine W, Kaplan JE, Gracia F, Lal R, Levine PH, Reeves WC. N. Engl. J. Med. 1991;324:565. doi: 10.1056/NEJM199102213240815. [DOI] [PubMed] [Google Scholar]
- Hirata A, Higuchi M, Niinuma A, Ohashi M, Fukushi M, Oie M, Akiyama T, Tanaka Y, Gejyo F, Fujii M. Virology. 2004;318:327–336. doi: 10.1016/j.virol.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Hjelle B, Appenzeller O, Mills R, Alexander S, Torrez-Martinez N, Jahnke R, Ross G. Lancet. 1992;339:645–646. doi: 10.1016/0140-6736(92)90797-7. [DOI] [PubMed] [Google Scholar]
- Hjelle B, Scalf R, Swenson S. Blood. 1990;76:450–454. [PubMed] [Google Scholar]
- Ho DD, Rota TR, Hirsch MS. Proc. Natl. Acad. Sci. USA. 1984;81:7588–7590. doi: 10.1073/pnas.81.23.7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PM, Dhib-Jalbut S, Mikovits JA, Robbins DS, Wolf AL, Bergey GK, Lohrey NC, Weislow OS, Ruscetti FW. Proc. Natl. Acad. Sci. USA. 1984;89:11784–11788. doi: 10.1073/pnas.89.24.11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi S, Ramundo MB, Takahashi H, Hall WW. J. Exp. Med. 1992;176:293–296. doi: 10.1084/jem.176.1.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga Y, Tsukahara T, Ohashi T, Tanaka Y, Arai M, Nakamura M, Ohtani K, Koya Y, Kannagi M, Yamamoto N, Fujii M. J. Virol. 1999;73:1271–1277. doi: 10.1128/jvi.73.2.1271-1277.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeang KT, Widen SG, Semmes OJ, Wilson SH. Science. 1990;247:1082–1084. doi: 10.1126/science.2309119. [DOI] [PubMed] [Google Scholar]
- Kanno T, Borwn K, Franzoso G, Siebenlist U. Mol. Cell. Biol. 1994;14:6443–6451. doi: 10.1128/mcb.14.10.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SY, Lemoine FJ, Marriott SJ. J. Biol. Chem. 2000;275:35926–35931. doi: 10.1074/jbc.M004397200. [DOI] [PubMed] [Google Scholar]
- Khabbaz RF, Hartel D, Lairmore M, Horsburgh CR, Schoenbaum EE, Roberts B, Hartley TM, Friedland G. J. Infect. Dis. 1991;163:252–256. doi: 10.1093/infdis/163.2.252. [DOI] [PubMed] [Google Scholar]
- Kibler KV, Jeang KT. J. Virol. 2001;75:2161–2173. doi: 10.1128/JVI.75.5.2161-2173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata JT, Wong FH, Wang JJ, Ratner L. Virology. 1994;204:656–664. doi: 10.1006/viro.1994.1581. [DOI] [PubMed] [Google Scholar]
- Koyanagi Y, Itoyama Y, Nakamura N, Takamatsu K, Kira J, Iwamasa T, Goto I, Yamamoto N. Virology. 1993;196:25–33. doi: 10.1006/viro.1993.1451. [DOI] [PubMed] [Google Scholar]
- Lacoste J, Cohen L, Hiscott J. Virology. 1991;184:553–562. doi: 10.1016/0042-6822(91)90425-b. [DOI] [PubMed] [Google Scholar]
- Lairmore MD, Jacobson S, Gracia F, De BK, Castillo L, Larreategui M, Roberts BD, Levine PH, Blattner WA, Kaplan JE. Proc. Natl. Acad. Sci. USA. 1990;87:8840–8844. doi: 10.1073/pnas.87.22.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal RB, Rudolph D, Buckner C, Pardi D, Hooper WC. Blood. 1993a;81:1827–1832. [PubMed] [Google Scholar]
- Lal RB, Rudolph DL, Folks TM, Hooper WC. Leuk. Res. 1993b;17:31–35. doi: 10.1016/0145-2126(93)90138-b. [DOI] [PubMed] [Google Scholar]
- Lee H, Swanson P, Shorty VS, Zack JA, Rosenblatt JD, Chen ISY. Science. 1989;244:471–475. doi: 10.1126/science.2655084. [DOI] [PubMed] [Google Scholar]
- Lemasson I, Robert-Hebmann V, Hamaia S, Duc Dodon M, Gazzolo L, Devaux C. J. Virol. 1997;71:1975–1983. doi: 10.1128/jvi.71.3.1975-1983.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Marriott SJ. J. Biol. Chem. 2001;276:31851–31857. doi: 10.1074/jbc.M105195200. [DOI] [PubMed] [Google Scholar]
- Leung K, Nabel GJ. Nature. 1988;333:776–778. doi: 10.1038/333776a0. [DOI] [PubMed] [Google Scholar]
- Levine PH, Jacobson S, Elliott R, Cavallero A, Colclough G, Dorry C, Stephenson C, Knigge RM, Drummond J, Nishimura M, Taylor ME, Wiktor S, Shaw GM. AIDS Res. Hum. Retroviruses. 1993;9:123–127. doi: 10.1089/aid.1993.9.123. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Sheehy N, Salemi M, VanDamme AM, Hall WW. Virology. 2002;295:182–189. doi: 10.1006/viro.2002.1357. [DOI] [PubMed] [Google Scholar]
- Li C-CH, Ruscetti FW, Rice N, Chen E, Yang N-S, Mikovits J, Longo DL. J. Virol. 1993;67:4205–4213. doi: 10.1128/jvi.67.7.4205-4213.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang MH, Geisbert T, Yao Y, Hinrichs SH, Giam CZ. J. Virol. 2002;76:4022–4033. doi: 10.1128/JVI.76.8.4022-4033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm P, Marriott S, Gitlin S, Bohan C, Brady J. New Biol. 1991;2:1034–1043. [PubMed] [Google Scholar]
- Los M, Khazaie K, Schulze-Osthoff K, Baeuerle PA, Schirrmacher V, Chlichlia K. J. Immunol. 1998;161:3050–3055. [PubMed] [Google Scholar]
- Low KG, Dorner LF, Fernando DB, Grossman J, Jeang KT, Comb MJ. J. Virol. 1997;71:1956–1962. doi: 10.1128/jvi.71.3.1956-1962.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Harhaj E, Sun S-C. Oncogene. 1995;11:993–998. [PubMed] [Google Scholar]
- Mahieux R, Pise-Masison CA, Lambert PF, Nicot C, De Marchis L, Gessain A, Green P, Hall W, Brady JN. J. Virol. 2000;74:6866–6874. doi: 10.1128/jvi.74.15.6866-6874.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott SJ, Lemoine FJ, Jeang KT. J. Biomed. Sci. 2002;9:292–298. doi: 10.1007/BF02256583. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Brockman JA, Scherer DC, Al-Murrani SW, Green PL, Ballard DW. Mol. Cell. Biol. 1996;16:2083–2090. doi: 10.1128/mcb.16.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Chevalier S, Weil R, Gessain A, Mahieux R. J. Biol. Chem. 2004;279:43307–43320. doi: 10.1074/jbc.M400497200. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Kamiya T, Minowada J, Tomita N, Kitajima K. Jpn. J. Cancer Res. 1991;82:1178–1183. doi: 10.1111/j.1349-7006.1991.tb01775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, Prager D. Blood. 1996;87:3410–3417. [PubMed] [Google Scholar]
- Mori N, Fujii M, Hinz M, Nakayama K, Yamada Y, Ikeda S, Yamasaki Y, Kashanchi F, Tanaka Y, Tomonaga M, Yamamoto N. Int. J. Cancer. 2002;99:378–385. doi: 10.1002/ijc.10388. [DOI] [PubMed] [Google Scholar]
- Mulloy JC, Migione T-S, Ross TM, Ton N, Green PL, Leonard WJ, Franchini G. J. Virol. 1998;72:4408–4412. doi: 10.1128/jvi.72.5.4408-4412.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EL, Fridey J, Smith JW, Engstrom J, Sacher RA, Miller K, Gibble J, Stevens J, Thomson R, Hansma D, Kaplan J, Khabbaz R, Nemo G. Neurology. 1997;48:315–320. doi: 10.1212/wnl.48.2.315. [DOI] [PubMed] [Google Scholar]
- Nagai M, Yamano Y, Brennan MB, Mora CA, Jacobson S. Ann. Neurol. 2001;50:807–812. doi: 10.1002/ana.10065. [DOI] [PubMed] [Google Scholar]
- Nerenberg M, Hinrichs SM, Reynolds RK, Khoury G, Jay G. Science. 1987;237:1324–1329. doi: 10.1126/science.2888190. [DOI] [PubMed] [Google Scholar]
- Neuveut C, Low KG, Maldarelli F, Schmitt I, Majone F, Grassmann R, Jeang KT. Mol. Cell. Biol. 1998;18:3620–3632. doi: 10.1128/mcb.18.6.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbound GC, Andrews JM, O'Rourke JP, Brady JN, Larimore MD. J. Virol. 1996;70:2101–2106. doi: 10.1128/jvi.70.4.2101-2106.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot C, Astier-Gin T, Guillemain B. Virology. 1997;236:47–53. doi: 10.1006/viro.1997.8720. [DOI] [PubMed] [Google Scholar]
- Nicot C, Harrod R. Mol. Cell. Biol. 2000;20:8580–8589. doi: 10.1128/mcb.20.22.8580-8589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot C, Mahieux R, Takemoto S, Franchini G. Blood. 2000;96:275–281. [PubMed] [Google Scholar]
- Nimer SD. New Biol. 1991;3:997–1004. [PubMed] [Google Scholar]
- Ohashi M, Sakurai M, Higuchi M, Mori N, Fukushi M, Oie M, Coffey RJ, Yoshiura K, Tanaka Y, Uchiyama M, Hatanaka M, Fujii M. Virology. 2004;320:52–62. doi: 10.1016/j.virol.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Ohya O, Tomaru U, Yamashita I, Kasai T, Morita K, Ikeda H, Wakisaka A, Yoshiki T. Leukemia. 1997;11(Suppl 3):255–257. [PubMed] [Google Scholar]
- Richardson JH, Edwards AJ, Cruickshank JK, Rudge P, Dalgleish AG. J. Virol. 1990;64:5682–5687. doi: 10.1128/jvi.64.11.5682-5687.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou P, Bex F, Gazzolo L. J. Biol. Chem. 2000;275:10551–10560. doi: 10.1074/jbc.275.14.10551. [DOI] [PubMed] [Google Scholar]
- Rivera-Walsh I, Waterfield M, Xiao G, Fong A, Sun SC. J. Biol. Chem. 2001;276:40385–40388. doi: 10.1074/jbc.C100501200. [DOI] [PubMed] [Google Scholar]
- Robek MD, Ratner L. J. Virol. 1999;73:4856–4865. doi: 10.1128/jvi.73.6.4856-4865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Guroff M, Weiss SH, Giron JA, Jennings AM, Ginzburg HM, Margolis IB, Blattner WA, Gallo RC. JAMA. 1986;255:3133–3137. [PubMed] [Google Scholar]
- Rosin R, Koch C, Schmitt I, Semmes OJ, Jeang K-T, Grassmann R. J. Biol. Chem. 1998;273:6698–6703. doi: 10.1074/jbc.273.12.6698. [DOI] [PubMed] [Google Scholar]
- Ross TM, Minella AC, Fang ZY, Pettiford SM, Green PL. J. Virol. 1997;71:8912–8917. doi: 10.1128/jvi.71.11.8912-8917.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross TM, Narayan M, Fang ZY, Minella AC, Green PL. J. Virol. 2000;74:2655–2662. doi: 10.1128/jvi.74.6.2655-2662.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross TM, Pettiford SM, Green PL. J. Virol. 1996;70:5194–5202. doi: 10.1128/jvi.70.8.5194-5202.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset R, Fabre S, Desbois C, Bantignies F, Jalinot P. Oncogene. 1998;16:643–654. doi: 10.1038/sj.onc.1201567. [DOI] [PubMed] [Google Scholar]
- Saggioro D, Barp S, Chieco-Bianchi L. Exp. Cell Res. 2001;269:245–255. doi: 10.1006/excr.2001.5310. [DOI] [PubMed] [Google Scholar]
- Saxon A, Stevens RH, Quan SG, Golde DW. 1978;120:777–782. [PubMed] [Google Scholar]
- Schmitt I, Rosin O, Rohwer P, Gossen M, Grassmann R. J. Virol. 1998;72:633–640. doi: 10.1128/jvi.72.1.633-640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiki M, Inoue J, Takeda T, Yoshida M. EMBO J. 1986;5:561–565. doi: 10.1002/j.1460-2075.1986.tb04247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmes OJ, Jeang KT. J. Virol. 1992;66:7183–7192. doi: 10.1128/jvi.66.12.7183-7192.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmes OJ, Majone F, Cantemir C, Turchetto L, Hjelle B, Jeang KT. Virology. 1996;217:373–379. doi: 10.1006/viro.1996.0126. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C. Ann. Rev. Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- Sieburg M, Tripp A, Jung-Woo M, Feuer G. J. Virol. 2004;78:10399–10409. doi: 10.1128/JVI.78.19.10399-10409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekevitz M, Josephs SF, Dukovich M, Peffer N, Wong-Staal F, Greene WC. Science. 1987;238:1575–1578. doi: 10.1126/science.2825351. [DOI] [PubMed] [Google Scholar]
- Silverman LR, Phipps AJ, Montgomery A, Ratner L, Lairmore MD. J. Virol. 2004;78:3837–3845. doi: 10.1128/JVI.78.8.3837-3845.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Greene WC. Genes Dev. 1990;4:1875–1885. doi: 10.1101/gad.4.11.1875. [DOI] [PubMed] [Google Scholar]
- Smith MR, Greene WC. J. Clin. Invest. 1991;88:1038–1042. doi: 10.1172/JCI115364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Kitao S, Matsushime H, Yoshida M. EMBO J. 1996;15:1607–1614. [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Narita T, Uchida-Toita M, Yoshida M. Virology. 1999a;259:384–391. doi: 10.1006/viro.1999.9760. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ohsugi Y, Uchida-Toita M, Akiyama T, Yoshida M. Oncogene. 1999b;18:5967–5972. doi: 10.1038/sj.onc.1203008. [DOI] [PubMed] [Google Scholar]
- Takemoto S, Mulloy JC, Cereseto A, Migone TS, Patel BK, Matsuoka M, Yamaguchi K, Takatsuki K, Kamihira S, White JD, Leonard WJ, Waldmann T, Franchini G. Proc. Natl. Acad. Sci. USA. 1997;94:13897–13902. doi: 10.1073/pnas.94.25.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Takahashi C, Yamaoka S, Nosaka T, Maki M, Hatanaka M. Proc. Natl. Acad. Sci. USA. 1990;87:1071–1075. doi: 10.1073/pnas.87.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder RS, Shanson DC, Jeffries DJ, Cheingsong-Popov R, Dalgleish A, Clapham P, Nagy K, Weiss RA. Lancet. 1984;2:125–128. doi: 10.1016/s0140-6736(84)91046-8. [DOI] [PubMed] [Google Scholar]
- Toro C, Rodes B, Bassani S, Jimenez V, Tuset C, Brugal MT, Fuente Lde L, Soriano V. J. Clin. Virol. 2005;33:65–70. doi: 10.1016/j.jcv.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Tripp A, Liu Y, Sieburg M, Montalbano J, Wrzesinski S, Feuer G. J. Virol. 2003;77:12152–12164. doi: 10.1128/JVI.77.22.12152-12164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara T, Kannagi M, Ohashi T, Kato H, Arai M, Nunez G, Iwanaga Y, Yamamoto N, Ohtani K, Nakamura M, Fujii M. J. Virol. 1999;73:7981–7987. doi: 10.1128/jvi.73.10.7981-7987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uittenbogaard MN, Giebler HA, Reisman D, Nyborg JK. J. Biol. Chem. 1995;270:28503–28506. doi: 10.1074/jbc.270.48.28503. [DOI] [PubMed] [Google Scholar]
- Van PL, Yim KW, Jin DY, Dapolito G, Kurimasa A, Jeang KT. J. Virol. 2001;75:396–407. doi: 10.1128/JVI.75.1.396-407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Green MR. Science. 1993;262:395–399. doi: 10.1126/science.8211160. [DOI] [PubMed] [Google Scholar]
- Wang T-G, Ye J, Lairmore M, Green PL. AIDS Res. Hum. Retroviruses. 2000;16:1661–1668. doi: 10.1089/08892220050193119. [DOI] [PubMed] [Google Scholar]
- Wano Y, Feinberg M, Hosking JB, Bogerd H, Greene WC. Proc. Natl. Acad. Sci. USA. 1988;85:9733–9737. doi: 10.1073/pnas.85.24.9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KC, Center DM, Cruikshank WW, Zhang Y. Virology. 2003;306:60–67. doi: 10.1016/s0042-6822(02)00056-9. [DOI] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA. J. Biol. Chem. 2000;275:21477–21485. doi: 10.1074/jbc.M909741199. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yamaoka S, Goto T, Nakai M, Tsujimoto Y, Hatanaka M. J. Virol. 1994;68:3374–3379. doi: 10.1128/jvi.68.5.3374-3379.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka S, Tobe T, Hatanaka M. Oncogene. 1992;7:433–437. [PubMed] [Google Scholar]
- Yao R, Natsume Y, Noda T. Oncogene. 2004;23:6023–6030. doi: 10.1038/sj.onc.1207817. [DOI] [PubMed] [Google Scholar]
- Ye J, Xie L, Green PL. J. Virol. 2003;77:7728–7735. doi: 10.1128/JVI.77.14.7728-7735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M-J, Paulssen EJ, Seeler JS, Gaynor RB. J. Virol. 1995a;69:3420–3432. doi: 10.1128/jvi.69.6.3420-3432.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin MJ, Paulssen E, Seeler J, Gaynor RB. J. Virol. 1995b;69:6209–6218. doi: 10.1128/jvi.69.10.6209-6218.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis I, Green PL. Frontiers Biosci. 2005;10:431–445. doi: 10.2741/1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehender G, Varchetta S, De Maddalena C, Colasante C, Riva A, Meroni L, Moroni M, Galli M. Virology. 2001;281:43–50. doi: 10.1006/viro.2000.0765. [DOI] [PubMed] [Google Scholar]
- Zhao L-J, Giam C-Z. Proc. Natl. Acad. Sci. USA. 1992;89:7070–7074. doi: 10.1073/pnas.89.15.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]