

Abstract
Interleukin-13 (IL-13), a Th2 cytokine, plays a pivotal role in pathogenesis of bronchial asthma via IL-13 receptor α1 (IL-13Rα1) and IL-4 receptor α (IL-4Rα). Recent studies show that a decoy receptor for IL-13, namely IL-13Rα2, mitigates IL-13 signaling and function. This study provides evidence for regulation of IL-13Rα2 production and release and IL-13-dependent signaling by lysophosphatidic acid (LPA) in primary cultures of human bronchial epithelial cells (HBEpCs). LPA treatment of HBEpCs in a time-dependent fashion increased IL-13Rα2 gene expression without altering the mRNA levels of IL-13Rα1 and IL-4Rα. Pretreatment with pertussis toxin (100 ng/ml, 4 h) or transfection of c-Jun small interference RNA or an inhibitor of JNK attenuated LPA-induced IL-13Rα2 gene expression and secretion of soluble IL-13Rα2. Overexpression of catalytically inactive mutants of phospholipase D (PLD) 1 or 2 attenuated LPA-induced IL-13Rα2 gene expression and protein secretion as well as phosphorylation of JNK. Pretreatment of HBEpCs with 1 μM LPA for 6 h attenuated IL-13- but not IL-4-induced phosphorylation of STAT6. Transfection of HBEpCs with IL-13Rα2 small interference RNA blocked the effect of LPA on IL-13-induced phosphorylation of STAT6. Furthermore, pretreatment with LPA (1 μM, 6 h) attenuated IL-13-induced eotaxin-1 and SOCS-1 gene expression. These results demonstrate that LPA induces IL-13Rα2 expression and release via PLD and JNK/AP-1 signal transduction and that pretreatment with LPA down-regulates IL-13 signaling in HBEpCs. Our data suggest a novel mechanism of regulation of IL-13Rα2 and IL-13 signaling that may be of physiological relevance to airway inflammation and remodeling.
The Th2-type cytokine, interleukin-13 (IL-13),2 has been shown to play a critical role in the pathogenesis of bronchial asthma (1-4). IL-13 is produced by Th2 cells and shares many of same biological activities of IL-4 (5, 6), including phosphorylation of signal transducer and activator of transcription 6 (STAT6). STAT6 activates the transcription of many proinflammatory genes in epithelial cells (7, 8). IL-13 levels are increased in bronchoalveolar lavage of asthma patients (9, 10) and ovalbumin-challenged mice (11). Experiments using IL-13 gene knock-out mice have shown that IL-13 regulates airway hyper-responsiveness and mucus hypersecretion (12). The biological activities of IL-13 are mediated through the IL-13 receptor α1(IL-13Rα1)·IL-4Rα heterodimer complex expressed on the surface of target cells (13-16). IL-13Rα1 binds to IL-13 at low affinity, whereas heterodimerization of the IL-13Rα1·IL-4Rα generates high-affinity receptor to IL-13, mediating IL-13-induced activation of Janus kinases and STAT6 (14-16).
The human IL-13 decoy receptor, termed IL-13Rα2, has been cloned and shares ∼33% homology and ∼21% identity with human IL-13Rα1 (17). IL-13Rα2 binds to IL-13 with much higher affinity (Kd = 0.25–1.2 nM), compared with IL-13Rα1 (Kd = 2–10 nM) (17-19). Overexpression of IL-13Rα2 in non-expressing heterologous cells inhibited IL-4- and IL-13-induced signal transduction in glioblastoma cells (20), whereas knock-out of IL-13Rα2 increased IL-13 function in vivo (21). The effect of IL-13 inhibition by IL-13Rα2 has been attributed to: 1) its high affinity for IL-13 and preventing IL-13 from binding the IL-13Rα1/IL-4Rα complex on cell surface; and 2) its short cytoplasmic tail, which has no signaling motif (17, 18, 20). In addition to the role of IL-13Rα2 in adaptive immunity, the role of IL-13Rα2 in cancer therapy has recently been demonstrated (22-24). Overexpression of IL-13Rα2 increased the sensitivity and antitumor activity of IL-13-PE38QQR toxin in cancer cells (22-24). The IL-13Rα2 promoter has been characterized, and several putative transcriptional factor binding sites for nuclear factor of activated T cells-1, AP-1 (c-Jun and c-Fos), AP2, GABP, OCT1, GATA3, PRE, and C-ETS1 were predicted in the promoter region (25). Elevated IL-13Rα2 mRNA expression has been detected in Schistosoma egg-induced liver fibrosis (26). In addition, interferon-γ or IL-13 treatment induced IL-13Rα2 mRNA expression in U937 (27) and bronchial epithelial cells (28, 29). However, the molecular mechanisms that regulate IL-13Rα2 gene expression and production are poorly understood.
Lysophosphatidic acid (LPA), a bioactive phospholipid, induces variety of biological activities on different cell types through G protein-coupled receptors (LPA1–3) (30-35). Recently, LPA receptors 4 (previously named GPR23) and 5 (previously named GPR92), which are related to the purinergic receptor family, have been cloned (36, 37). LPA exists in various biological fluids, including serum and bronchoalveolar lavage fluid (38, 39). Extracellular LPA is produced by the action of both secretory phospholipase A2 (40) and lysophospholipase D (also named autotoxin) (41) by using phosphatidic acid or lysophosphatidylcholine as substrate, respectively. The role of LPA in regulating expression of Th1 and Th2 type cytokines has been studied in bronchial epithelial cells and T cells. We previously demonstrated that treatment of HBEpCs with LPA induces IL-8 expression and secretion via LPA receptors regulated by changes in intracellular calcium and protein kinase C δ, p38 MAPK-dependent transcriptional activation of NF-κB, and JNK-dependent activation of AP-1 (42-44). In addition, LPA can activate cells via epidermal growth factor receptor (EGF-R) transactivation in HBEpCs (45). IL-8 is a potent chemoattractant for neutrophils and plays a pivotal role in innate immunity (46-48). In T-cells, LPA enhanced IL-13 gene expression under conditions of submaximal activation (49). Taken together, these studies suggest that LPA plays a critical role in airway inflammation by regulating immunomodulation of bronchial and alveolar epithelial cells However, mechanisms of LPA-dependent modulation of Th2 type cytokine-induced signaling are unclear.
To further determine the role of LPA in airway inflammation, we report here that LPA induces IL-13Rα2 mRNA expression through phospholipase D (PLD)-dependent JNK/AP-1 signaling in HBEpCs. This is the first report on LPA induced IL-13Rα2 gene expression and production in HBEpCs. Further, we found that elevated IL-13Rα2 expression attenuated IL-13-mediated phosphorylation of STAT6 and eotaxin and SOCS-1 gene expression in HBEpCs. These results demonstrate a novel role for LPA in attenuating Th2 cytokine signaling in airway inflammation.
EXPERIMENTAL PROCEDURES
Materials
1-Oleoyl (18:1) LPA, 1-butanol, 3-butanol, and dimethyl sulfoxide were purchased from Sigma. Antibodies to phospho-STAT6, STAT6, RelA, c-Jun, phospho-JNK, and JNK1 were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibodies to human phosphatidylcholine-specific PLD1 and mouse PLD2 were purchased from BIOSOURCE International (Camarillo, CA). Antibodies to IL-13Rα2 and recombinant IL-13 were procured from Cell Signaling Technology (Beverly, MA). Scrambled and IL-13Rα2 small interfering RNA (siRNA) were from Dharmacon Inc. (Lafayette, CO). Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies were purchased from Molecular Probes (Eugene, OR). An ECL kit for detection of proteins by Western blotting was obtained from Amersham Biosciences. Real-time PCR reagents were from Bio-Rad Laboratories. Transfection reagents were from Qiagen (Valencia, CA). Bronchial epithelial cell basal medium and a supplement kit were purchased from Cambrex Bio Science Inc. (Walkersville, MD). A Millipore™ 10 kit was purchased from Millipore (Bedford, MA). All other reagents were of analytical grade.
Cell Culture
HBEpCs were isolated from normal human lung obtained from lung transplant donors following previously described procedures (50, 51). The isolated passage 0 (P0) HBEpCs were cultured in serum-free basal essential growth medium supplemented with growth factors. Cells were incubated at 37 °C in 5% CO2 and 95% air to ∼80% confluence and subsequently were propagated in 35-mm or 6-well collagen-coated dishes. All experiments were carried out between passages 1 and 4.
Preparation of Cell Lysates, Media, and Western Blotting
After the indicated treatments, media were collected and centrifuged at 5,000 × g for 10 min, and supernatants were concentrated by Millipore™ 10 kit according to manufacturer’s instruction. Cells were rinsed twice with ice-cold phosphate-buffered saline and lysed in 200 μl of buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EGTA, 5 mM β-glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Cell lysates were incubated at 4 °C for 10 min, sonicated on ice for 10 s, and centrifuged at 5000 × g for 5 min at 4 °C in a microcentrifuge. Protein concentrations were determined with a BCA protein assay kit (Pierce Chemical Co.) using bovine serum albumin as standard. Equal amounts of protein (20 μg) or concentrated media (20 μl) were subjected to 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes, blocked with 5% (w/v) bovine serum albumin in TBST (25 mM Tris-HCl, pH 7.4, 137 mM NaCl, and 0.1% Tween 20) for 1 h, and incubated with primary antibodies in 5% (w/v) bovine serum albumin in TBST for 1–2 h at room temperature. The membranes were washed at least three times with TBST at 15-min intervals and then incubated with mouse, rabbit, or goat horseradish peroxidase-conjugated secondary antibody (1: 3,000) for 1 h at room temperature. The membranes were developed with an enhanced chemiluminescence detection system according to the manufacturer’s instructions.
RNA Isolation
Total RNA was isolated from cultured HBEpCs using TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions. RNA was quantified spectrophotometrically, and samples with an absorbance of ≥1.8 measured at 260/280 nm were analyzed by real-time RT-PCR.
Real-time RT-PCR and Reverse Transcription-PCR
1 μg of RNA was reverse-transcribed using a cDNA synthesis kit (Bio-Rad), and real-time PCR and regular PCR were performed to assess expression of the IL-13Rα1, IL-13Rα2, IL-4Rα, eotaxin-1, and SOCS-1 genes using primers designed on the basis of human mRNA sequences (Table 1). For real-time RT-PCR, amplicon expression in each sample was normalized to its 18 S RNA content. The relative abundance of target mRNA in each sample was calculated as 2 raised to the negative of its threshold cycle value times 106 after being normalized to the abundance of its corresponding 18 S, e.g. 2-(IL-13Rα2 threshold cycle)/2-(18 S threshold cycle) × 106.
TABLE 1.
Primers for RT-PCR and real-time RT-PCR
| Primers for RT-PCR | |
| IL-13Rα1 | Forward, 5′-GGAGAATACATCTTGTTTCATGG-3′ |
| Reverse, 5′-GCGCTTCTTACCTATACTCATTTCTTGG-3′ | |
| IL-13Rα2 | Forward, 5′-AATGGCTTTCGTTTGCTTGG-3′ |
| Reverse, 5′-ACGCAATCCATATCCTGAAC-3′ | |
| IL-4Rα | Forward, 5′-GACCTGGAGCAACCCGTATC-3′ |
| Reverse, 5′-CATAGCACAACAGGCAGACG-3′ | |
| GAPDHa | Forward, 5′-GGAGCCAAAAGGGTCATCATCT-3′ |
| Reverse, 5′-AGTGGGTGTCGCTGTTGAAGT-3′ | |
| Primers for real-time RT-PCR | |
| IL-13Rα2 | Forward, 5′-ACTGGTATGAGGGCTTGGAT-3′ |
| Reverse, 5′-TCTGATGCCTCCAAATAGGG-3′ | |
| SOCS-1 | Forward, 5′-TTGGAGGGAGCGGATGGGTGTAG-3′ |
| Reverse, 5′-AGAGGTAGGAGGTGCGAGTTCAGGTC-3′ | |
| Eotaxin-1 | Forward, 5′-AACCACCTGCTGCTTTAACC-3′ |
| Reverse, 5′-CACAGCTTTCTGGGGACATT-3′ | |
| 18S | Forward, 5′-GTAACCCGTTGAACCCCATT-3′ |
| Reverse, 5′-CCATCCAATCGGTAGTAGCG-3′ | |
Glyceraldehyde-3-phosphate dehydrogenase.
Infection with Adenoviral Constructs
Infection of HBEpCs (∼60% confluence) with purified adenoviral vectors of catalytically inactive mutants of human PLD1 (hPLD1) and mouse PLD2 (mPLD2) were carried out in 6-well plates as described previously (51). Cells were infected with different multiplicities of infection in 1 ml of serum-free basal essential growth medium for 48 h.
Transfection of siRNA for IL-13Rα2
HBEpCs (P1) cultured onto 6-well plates (40–50% confluence) were transiently transfected with SMARTpool RNA duplexes corresponding to human IL-13Rα2 and scrambled siRNA using Transmessenger transfection reagent. Briefly, IL-13Rα2 siRNA or scrambled siRNA (100 nM) was condensed with enhancer R and formulated with Transmessenger reagent according to the manufacturer’s instructions. The transfection complex was diluted into 900 μl of bronchial epithelial cell basal medium and added directly to the cells. The medium was replaced with bronchial epithelial cell basal medium after 3 h, and cells were used for experiments at 72 h after transfection.
Statistical Analyses
All results were subjected to statistical analysis using one-way analysis of variance and, whenever appropriate, were analyzed using the Student-Newman-Keuls test. Data are expressed as means ± S.D. of triplicate samples from at least three independent experiments, and the level of significance was taken to p < 0.05.
RESULTS
LPA Induces IL-13Rα2 Gene Expression and Secretion through Gαi in HBEpCs
Using a DNA microarray, we found that LPA treatment of HBEpCs induced IL-13Rα2 gene expression after 24 h.3 To confirm this result, we examined the changes in IL-13Rα1, IL-13Rα2, and IL-4Rα gene levels with or without LPA treatment by RT-PCR and related real-time RT-PCR. As shown in Fig. 1, A and B, compared with IL-13Rα1 and IL-4Rα gene levels, the IL-13Rα2 mRNA level is very low in HBEpCs. The increase of IL-13Rα2 gene expression was detected at 6 h (∼3-fold) after LPA (1 μM) treatment, reaching ∼7-fold after 24 h (Fig. 1B), whereas LPA (1 μM) treatment had no effect on IL-13Rα1 and IL-4Rα gene expression (Fig. 1A). These results show that only IL-13Rα2, not IL-13Rα1 or IL-4Rα, is inducible by LPA treatment in HBEpCs. Similar to LPA treatment, IL-13 (10 ng/ml, 6 h) and IL-4 (10 ng/ml, 6 h) also increased IL-13Rα2 gene expression (Fig. 1B).
FIGURE 1. LPA-induced IL-13Rα2 gene expression and protein release in HBEpCs.

HBEpCs grown to ∼70–80% confluence were challenged with LPA (1 μM), IL-13 (10 ng/ml), and IL-4 (10 ng/ml). Total RNA was extracted at different times, and mRNA levels were determined by RT-PCR (RT— indicates absence of reverse transcriptase) with primers for IL-13Rα1, IL-13Rα2, IL-4Rα, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (A) and real-time RT-PCR (B) with primers for IL-13Rα2. Data are the mean ± S.D. of three values. *, p < 0.05 versus 0 h cells. C, HBEpCs were treated as described in A, and then media were collected at different times, subjected to 10% SDS-PAGE, and analyzed with specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus 0 h cells. D, HBEpCs grown to ∼70–80% confluence were challenged with LPA at different concentration for 6 h, and then media were collected, subjected to 10% SDS-PAGE, and analyzed with specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus control cells. E, HBEpCs grown to ∼70–80% confluence were treated with pertussis toxin (PTx; 100 ng/ml) for 4 h. Cells were challenged with LPA (1 μM) for an additional 6 h. Total RNA was extracted, and IL-13Rα2 mRNA levels were determined by real-time RT-PCR with IL-13Rα2 primers. Data are the mean ± S.D. of three values. *, p < 0.05 versus vehicle (Veh) cells; **, p < 0.05 versus LPA treatment. F, HBEpCs were treated as described in E; media were collected, subjected to 10% SDS-PAGE, and analyzed with specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus vehicle cells; **, p < 0.05 versus LPA treatment.
To further confirm up-regulation of IL-13Rα2 by LPA, we next investigated IL-13Rα2 secretion after LPA treatment in HBEpCs. Using an anti-IL-13Rα2 extracellular domain antibody, we monitored soluble IL-13Rα2 protein levels in media after LPA (1 μM) treatment and observed a peak at 6 h (Fig. 1C). A 6-h incubation with LPA induced IL-13Rα2 secretion in a dose-dependent fashion (Fig. 1D). Pretreatment of HBEpCs with pertussis toxin (100 ng/ml) for 4 h attenuated LPA-induced IL-13Rα2 gene expression and soluble IL-13Rα2 secretion (Fig. 1, E and F). These results suggest that LPA-induced IL-13Rα2 gene expression and secretion is through Gi-coupled LPA receptors on the cell surface in HBEpCs.
Role of JNK/AP-1 Signaling in LPA-induced IL-13Rα2 Gene Expression and Secretion
Our previous studies (41-43) have shown that NF-κB and AP-1 transcriptional factors were involved in LPA-induced IL-8 secretion in HBEpCs. To determine the mechanisms of regulation of IL-13Rα2 gene expression and secretion by LPA, we inhibited NF-κB and AP-1 activities by transfection of siRNA or pharmacological inhibitors prior to LPA challenge. Transfection of NF-κB subunit RelA siRNA or pretreatment with the I-κB kinase inhibitor Bay11-7082 (5 and 10 μM) did not affect LPA-induced IL-13Rα2 gene expression and protein release (Fig. 2, A and C). In contrast, down-regulation of c-Jun protein expression (Fig. 2B) by transfection of c-Jun siRNA attenuated LPA-induced IL-13Rα2 secretion in HBEpCs (Fig. 2A). Further, pretreatment with the JNK inhibitor JNKiII for 1 h significantly attenuated LPA-mediated IL-13Rα2 mRNA expression (Fig. 2D). Fig. 2B shows that the transfection of RelA siRNA or c-Jun siRNA down-regulated expression of the respective transcription factors in HBEpCs as determined by Western blotting with specific antibodies to RelA or c-Jun. These results suggest that regulation of IL-13Rα2 gene expression and secretion by LPA is at least partly dependent on JNK/AP-1 signal transduction.
FIGURE 2. The JNK/AP-1, but not the NF-κB, pathway was involved in LPA-induced IL-13Rα2 gene expression and protein secretion in HBEpCs.

A, cells were transfected with NF-κB subunit RelA siRNA or c-Jun siRNA followed by challenge with LPA (1 μM) for 6 h. Media were collected, subjected to 10% SDS-PAGE, and analyzed with specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus scrambled (Scramb) siRNA-transfected cells; **, p < 0.05 versus LPA treatment. B, HBEpCs were treated transfected with scrambled, RelA, or c-Jun siRNAs for 72 h. Cell lysates were subjected to 10% SDS-PAGE and analyzed with specific antibody to RelA or c-Jun. C, HBEpCs grown to ∼70–80% confluence were treated with Bay11-7082 (5.0 and 10.0 μM) for 1 h. Cells were challenged with LPA (1 μM) for an additional 6 h, and media were collected, subjected to 10% SDS-PAGE, and analyzed with specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus control cells. D, HBEpCs grown to ∼70–80% confluence were treated with JNKiII (5.0, 10.0, and 20.0 μM) for 1 h, and then total RNA was extracted and IL-13Rα2 mRNA levels determined by real-time RT-PCR with IL-13Rα2 primers. Data are the mean ± S.D. of three values. *, p < 0.05 versus vehicle cells; **, p < 0.05 versus LPA treatment.
Role of PLD in LPA-induced IL-13Rα2 Gene Expression and Secretion
Our previous studies have shown that LPA stimulates PLD1 and -2 in HBEpCs (50). Furthermore, LPA-induced PLD activation could be blocked by overexpression of hPLD1 or mPLD2 catalytically inactive mutants,or by pretreatment with 1-butanol but not 3-butanol (50). Therefore, we next investigated the role of PLD activation in LPA-induced IL-13Rα2 release. LPA-induced IL-13Rα2 release was attenuated by pretreatment with 1-butanol (0.5%, 15 min) not 3-butanol (0.5%, 15 min; Fig. 3A), suggesting the involvement of PLD. To confirm these data, inhibition of PLD activity by overexpression of hPLD1 or mPLD2 mutants was performed by transfection of adenoviral vectors encoding hPLD1 or mPLD2 mutants (50). Consistent with the data using 1-butanol, both hPLD1 and mPLD2 mutants attenuated LPA-induced IL-13Rα2 gene expression and secretion (Fig. 3, B and D). Fig. 3C confirms overexpression of hPLD1 and mPLD2 mutants by Western blotting with specific antibodies to PLD1 or PLD2.
FIGURE 3. LPA-induced IL-13Rα2 gene expression and protein release was dependent on PLD in HBEpCs.

A, HBEpCs grown to ∼70–80% confluence were treated with 1-butanol (0.5%) and 3-butanol (0.5%) for 15 min. Cells were challenged with LPA (1 μM) for an additional 6 h, and then media were collected, subjected to 10% SDS-PAGE, and analyzed to a specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus control cells; **, p < 0.05 versus LPA treatment. B, HBEpCs grown to ∼50–60% confluence were infected with adenoviral vector containing cDNA of empty Ad5-vector, hPLD1 mutant, and mPLD2 mutant for 24 h. Cells were challenged with LPA (1 μM) for an additional 6 h, and then media were collected, subjected to 10% SDS-PAGE, and analyzed to a specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus adenoviral empty vector Infected cells; **, p < 0.05 versus LPA treatment. The expression of hPLD1 and mPLD2 were determined by immunoblotting with specific antibodies to PLD1 and PLD2 C and D, cells were treated as described in B. Total RNA was extracted, and IL-13Rα2 mRNA levels were determined by real-time RT-PCR with IL-13Rα2 primers. Data are the mean ± S.D. of three values. *,p < 0.05 versus adenoviral empty vector-infected cells; **, p < 0.05 versus LPA treatment. MOI, multiplicity of infection.
As JNK/AP-1 regulated LPA-mediated IL-13Rα2 expression, we further determined whether LPA-induced JNK phosphorylation is regulated by PLD1 or -2 in HBEpCs. Fig. 4 shows that LPA (1 μM, 15 min) induced phosphorylation of JNK1 and -2, whereas overexpression of hPLD1 or mPLD2 mutants attenuated LPA-induced phosphorylation of JNK1 and -2. Total JNK1 was detected by anti-JNK1 antibody, showing equal protein loading in Western blotting. These results support the data shown in Fig. 2 and suggest that LPA-induced IL-13Rα2 gene expression and secretion are dependent on PLD-mediated JNK/AP-1 activation in HBEpCs.
FIGURE 4. PLD1 and -2 were involved in LPA-induced phosphorylation of JNK in HBEpCs.
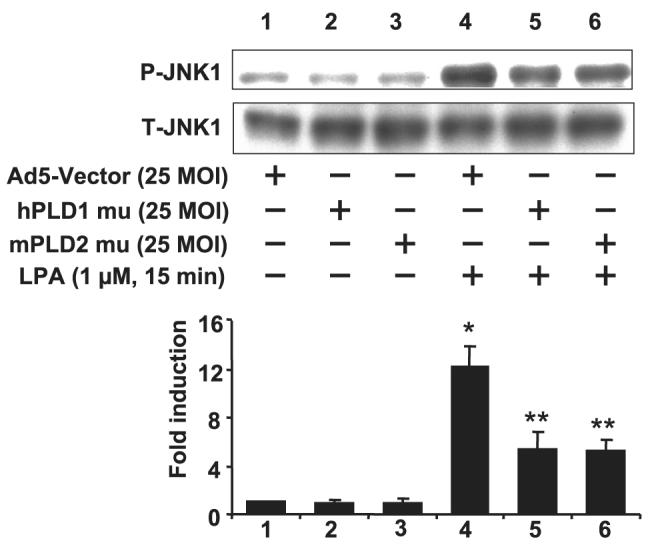
HBEpCs grown to ∼50–60% confluence were infected with adenoviral vector containing cDNA of empty Ad5-vector, hPLD1 mutant, and mPLD2 mutant for 24 h. Cells were challenged with LPA (1 μM) for an additional 15 min. Cell lysates were subjected to 10% SDS-PAGE and analyzed to specific antibodies to phospho-JNK1 or total JNK1. This is a quantitative analysis from three independent experiments (mean ± S.D.). *,p < 0.05 versus adenoviral empty vector-infected cells; **, p < 0.05 versus LPA treatment. MOI, multiplicity of infection.
Elevated IL-13Rα2 Attenuates IL-13-mediated Phosphorylation of STAT6 and Gene Expression of Eotaxin-1 and SOCS-1
STAT6 plays a critical role in IL-13-mediated gene expression and biological functions (7, 8, 14-16). To determine the role of elevated expression of IL-13Rα2 induced by LPA, the level of phosphorylation of STAT6 by IL-13 was examined with or without LPA pretreatment. As shown in Fig. 5A, IL-13 treatment induced phosphorylation of STAT6 in HBEpCs in a dose-dependent fashion. Pretreatment with LPA (1 μM, 6 h) attenuated IL-13-induced phosphorylation of STAT6, whereas, importantly, IL-4-induced phosphorylation of STAT6 was not affected. Western blotting with specific antibody to STAT6 showed that LPA treatment did not change the total STAT6 level (Fig. 5A). Thus LPA pretreatment specifically targets IL-13 but not IL-4 signaling, consistent with a role for IL-13Rα2. To further confirm the role of elevated IL-13Rα2 in the attenuation of IL-13-induced signaling, we next knocked down IL-13Rα2 expression using IL-13Rα2 siRNA and reasoned that the inhibitory effects of LPA on STAT6 phosphorylation would be attenuated. As shown in Fig. 5B, the ability of LPA pretreatment to inhibit STAT6 phosphorylation was indeed blocked by knockdown of IL-13Rα2 (Fig. 5B). The down-regulation of IL-13Rα2 by IL-13Rα2 siRNA was confirmed by Western blotting with anti-IL-13Rα2 antibody (Fig. 5C).
FIGURE 5. LPA attenuated IL-13-induced STAT6 phosphorylation in HBEpCS.
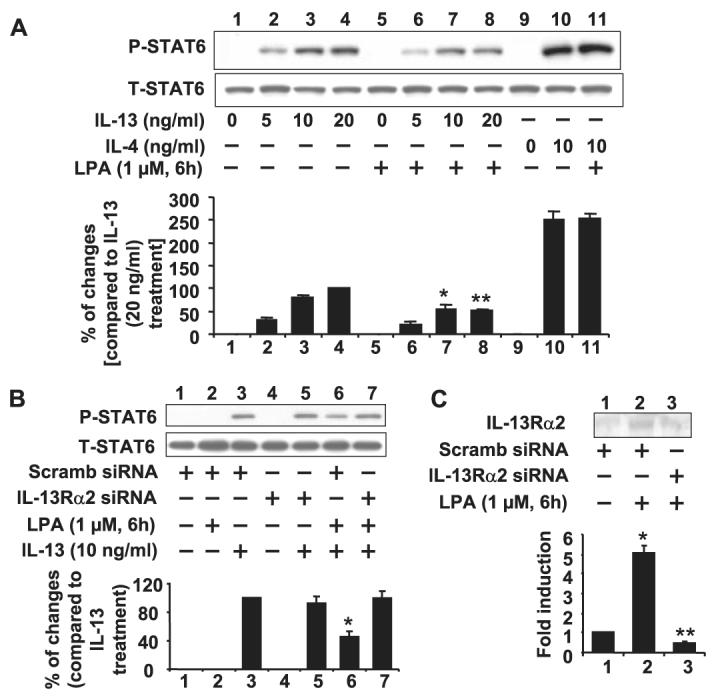
A, HBEpCs grown to ∼70–80% confluence were treated with LPA (1 μM) for 6 h. Cells were challenged with IL-13 (5, 10, and 20 ng/ml) and IL-4 (10 ng/ml) for 15 min. Cell lysates were subjected to 10% SDS-PAGE and analyzed to specific antibodies to phospho-STAT6 or total STAT6. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus IL-13 (10 ng/ml)-treated cells; **, p < 0.05 versus IL-13 (20 ng/ml)-treated cells. B, HBEpCs grown to ∼40–50% confluence were transfected with scrambled (Scramb) or IL-13Rα2 siRNAs for 72 h. Cells were pretreated with LPA (1 μM) for 6 h and then challenged with IL-13 (10 ng/ml) for 15 min. Cell lysates were subjected to 10% SDS-PAGE and analyzed to specific antibodies to phospho-STAT6 or total STAT6. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus IL-13 (10 ng/ml)-treated cells. C, cells were treated as described in B. Media were collected, subjected to 10% SDS-PAGE, and analyzed with specific antibody to IL-13Rα2. This is a quantitative analysis from three independent experiments (mean ± S.D.). *, p < 0.05 versus scrambled siRNA transfected cells; **, p < 0.05 versus scrambled siRNA + LPA treatment.
To confirm the physiological significance of attenuated IL-13 signaling, we measured the effect of LPA treatment on the induction of two well established IL-13/STAT6 target genes in HBEpCs, namely eotaxin-1 and SOCS-1. IL-13 (10 ng/ml, 3 h) increased eotaxin-1 and SOCS-1 mRNA expression, whereas pretreatment of HBEpCs with LPA (1 μM, 6 h) attenuated IL-13-mediated eotaxin-1 and SOCS-1 mRNA expression (Fig. 6). Taken together, these results suggest that elevated IL-13Rα2 by LPA treatment down-regulates IL-13-induced signaling and gene expression in HBEpCs.
FIGURE 6. LPA attenuated IL-13-induced gene expression of eotaxin-1 and SOCS-1 in HBEpCs.

HBEpCs grown to ∼70–80% confluence were treated with LPA (1 μM) for 6 h. Cells were challenged with IL-13 (10 ng/ml) for 3 h. Total RNAs were extracted, and eotaxin-1 and SOCS-1 mRNA levels were determined by real-time RT-PCR with eotaxin-1 primers (A)or SOCS-1 primers (B). Data are the mean ± S.D. of three values. *, p < 0.05 versus vehicle cells; **, p < 0.05 versus IL-13 treatment. Veh, vehicle.
DISCUSSION
We have previously demonstrated that LPA-induced IL-8 expression and secretion in HBEpCs is dependent on p38 MAPK, NF-κB, JNK/AP-1, and EGF-R transactivation (42-45). In this report we show that LPA exerts a negative regulation effect on Th2-type cytokine IL-13-induced signaling by up-regulating IL-13Rα2 in airway epithelium. Our observations indicate that LPA has a novel role in protecting airway epithelium by reducing Th2-type cytokine function.
IL-13 is known to regulate airway hyperresponsiveness and allergic inflammation by binding to heterodimers of IL-13Rα1 and IL-4Rα (9-16). Another IL-13 receptor, IL-13Rα2, binds IL-13 with high affinity and, in contrast to IL-13Rα1, is a decoy receptor and a negative regulator of IL-13-induced biological functions (17-24, 27-29). In HBEpCs, we found that LPA treatment increased IL-13Rα2 gene expression and protein production without affecting IL-13Rα1 and IL-4Rα (Fig. 1). These results are similar to studies by two other research groups, David et al. (60) and Yasunaga et al. (29), who found that the Th2 cytokines IL-4 and IL-13 induce IL-13Rα2 gene expression without affecting IL-13Rα1 and IL-4Rα. Ours is the first report showing that a lysophospholipid can regulate IL-13Rα2 expression. Our observation that in HBEpCs, LPA-induced IL-13Rα2 gene expression and protein production is dependent on Gi-coupled LPA receptors (Fig. 1) is in keeping with our prior studies of LPA signaling in HBEpCs (42-45). We have shown previously that LPA-mediated transactivation of EGF-R via LPA receptors regulates IL-8 secretion (45); however, increased IL-13Rα2 expression by LPA is not dependent on EGF-R transactivation.4 In future studies, the isoforms of LPA receptor(s) involved in LPA-induced IL-13Rα2 expression will be investigated.
The promoter region of the human IL-13Rα2 gene has been cloned and analyzed. The IL-13Rα2 promoter contains several putative transcriptional factor binding sites for NFAT1, AP-1, AP-2, GABP, OCT1, GATA3, PRE, and C-ETS1 (25). Also, STAT6 has been shown to regulate IL-13Rα2 gene expression in response to IL-13 treatment in bronchial epithelial cells (29). In the present studies, LPA treatment alone had no effect on the phosphorylation of STAT6, indicating that STAT6 is not involved in LPA-induced IL-13Rα2 expression. Our previously studies showed that NF-κB and AP-1 pathways play critical roles in LPA-induced IL-8 gene expression in HBEpCs (42-44). Here, we found that the JNK/AP-1, but not the NF-κB pathway, regulated IL-13Rα2 gene expression by LPA treatment in HBEpCs (Fig. 2). These results are consistent with the studies from Wu and Low (25) who observed that the JNK inhibitor SP600125 decreases IL-13Rα2 gene expression from 34 to 7.23% in the U87MG cell line. Therefore, these data suggest that IL-13Rα2 gene expression is enhanced by JNK/AP-1 activation in response to LPA. IL-13 or IL-4 treatment had no effect on phosphorylation of JNK in HBEpCs (data not shown), suggesting the involvement of different signal pathways in IL-13Rα2 expression by LPA and IL-13/IL-4.
To further determine the regulation of the JNK/AP-1 pathway in HBEpCs, we focused on the role of PLD in LPA-induced JNK activation and IL-13Rα2 expression. The PLD isoenzymes PLD1 and PLD2 catalyze the hydrolysis of the phosphodiester bond of glycerophospholipids to generate phosphatidic acid and a free head group and are activated by hormones, growth factors, neurotransmitters, cytokines, and reactive oxygen species (52, 53). PLD is a critical enzyme in intracellular signal transduction. Phosphatidic acid, considered as a second messenger, can be subsequently converted to LPA or diacylglycerol by phospholipase A1/A2 (40). Our previous studies demonstrated that LPA treatment induces PLD activation and that PLD2 is involved in LPA-induced platelet-derived growth factor (PDGF) receptor transactivation in HBEpCs (51). Interestingly, we found that PLD1 and -2 functioned upstream of JNK in HBEpCs. Down-regulation of PLD isoforms attenuated LPA-induced JNK phosphorylation as well as IL-13Rα2 gene expression and protein production in HBEpCs (Figs. 3 and 4). The mechanisms of regulation of JNK activation by PLD are still not clear. In B-cells, JNK activation is regulated by both PLD-dependent and -independent mechanisms (55). Kogut et al. (56) show that complement receptor-induced degranulation in chicken heterophiles is dependent on PLD-mediated JNK activation. Nozawa and colleagues (57) have demonstrated that in primary cultured rat hepatocytes, PLD inhibitor suppresses hepatocyte growth factor-induced expression of c-jun and c-fos mRNAs. In future studies, the mechanism(s) by which AP-1 proteins mediates LPA-induced IL-13Rα2 gene expression and the mechanisms of regulation of JNK by PLD will need to be determined.
IL-13Rα2 has been known to act as a decoy receptor and to down-regulate IL-13 effects on airway epithelial cells. In BEAS-2B cells overexpressing IL-13Rα2, STAT6 phosphorylation by IL-13 is completely attenuated, whereas IL-4 signaling is not changed (29). In glioblastoma cells, IL-13Rα2 acts as an inhibitor of IL-4-dependent STAT6 phosphorylation (20). Consistent with the specificity of IL-13Rα2 for IL-13, we found that elevated IL-13Rα2 by LPA treatment attenuated IL-13-dependent STAT6 phosphorylation but had no effect on IL-4-depedent signaling (Fig. 5). Further, we found LPA pretreatment attenuated IL-13-mediated eotaxin-1 and SOCS-1 gene expression (Fig. 6). Taken together, our data suggest that pretreatment of HBEpCs by LPA attenuates IL-13-mediated signaling and biological functions in an IL-13Rα2-dependent manner. Our previous studies also showed that LPA treatment enhances IL-13 gene expression and secretion in T cells (49), suggesting that the effects of LPA on different types of immune cells results in different immune responses. In contrast to the effect of IL-13α2 as decoy receptor, Fichtner-Feigl et al. (59) recently showed that in some situations IL-13Rα2 appears to have a signaling function in macrophages. However, none of our data are consistent with a signaling function for IL-13Rα2 in HBEpCs. LPA is constitutively present in serum, and its concentration is increased in bronchoalveolar lavage fluids following allergen challenge in human subjects (38). Therefore, the data presented in this study suggest that LPA may play a previously underappreciated role during resolution of Th2-dependent airway inflammation.
Compared with its low expression in normal airway epithelial cells and other normal cell types, IL-13Rα2 has been detected in many human cancer cell lines and has been demonstrated to serve as a tumor diagnostic marker of human head and neck cancer (54). In addition to its function in a Th2-dominant immune response, IL-13Rα2 is being investigated as a therapeutic target for many human cancers, including prostate tumor, pediatric brain tumor, and breast and pancreatic tumors (22-24). Although plasma LPA levels are elevated in ovarian and breast cancer (58), no causal relationship has been proposed between LPA and IL-13Rα2 in cancer.
In summary, we have shown that LPA negatively regulates IL-13-induced signaling by elevating IL-13Rα2 gene expression and protein production, which is dependent on PLD-dependent JNK activation in HBEpCs (Fig. 7). Our results have demonstrated that in addition to its role in producing epithelial chemokines, LPA may also protect airway epithelium during Th2-dominant immune responses. Future studies using gene-targeted mice and/or specific LPA receptor antagonists should help to refine our understanding of the role of LPA and its receptors during lung inflammation.
FIGURE 7. Mechanisms of LPA signaling in IL-13Rα2 expression and IL-13-induced STAT6 phosphorylation.
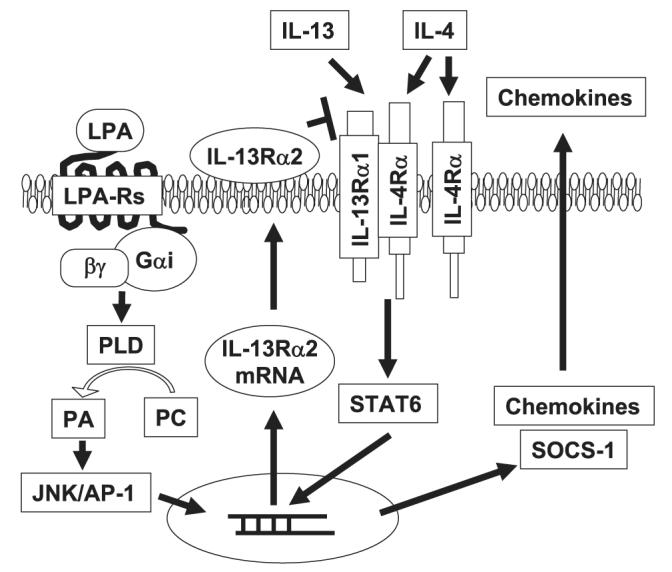
Ligation of LPA to G protein-coupled LPA receptors results in IL-13Rα2 expression through activation of PLD and JNK/AP-1. Elevated IL-13Rα2 expression attenuated the IL-13- but not the IL-4-mediated STAT6 pathway. PA, phosphatidic acid; PC, phosphatidylcholine.
Acknowledgment
We thank Dr. Anne J. Sperling for helpful discussion.
Footnotes
This work was supported by National Institutes of Health Grant HL71152 (to V. N.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- IL
- interleukin
- IL-13R
- IL-13 receptor
- LPA
- lysophosphatidic acid
- HBEpCs
- human bronchial epithelial primary cells
- STAT6
- signal transducer and activator of transcription 6
- PLD
- phospholipase D
- hPLD1
- human phospholipase D1
- mPLD2
- mouse phospholipase D2
- siRNA
- small interference RNA
- MAPK
- mitogen-activated protein kinase
- JNK
- c-Jun N-terminal kinase
- AP-1
- activator protein 1
- EGF-R
- epidermal growth factor receptor
- RT
- reverse transcription
L. Wang and V. Natarajan, unpublished data.
Y. Zhao and V. Natarajan, unpublished data.
REFERENCES
- 1.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 2.Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksiey RM, Corry DB. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. J. Clin. Investig. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wills-Karp M. Immunol. Rev. 2004;202:1750190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 5.Hilton DJ, Zhang JG, Metcalf D, Alexander WS, Nicola NA, Willson TA. Proc. Natl. Acad. Sci. U. S. A. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murata T, Obiri NI, Puri RK. Int. J. Mol. Med. 1998;1:551–557. doi: 10.3892/ijmm.1.3.551. [DOI] [PubMed] [Google Scholar]
- 7.Taube C, Duez C, Cui Z-H, Katsuyuki T, Rha Y-H, Park J-W, Balhorn A, Donaldson DD, Dakhama A, Gelfand EW. J. Immunol. 2002;169:6482–6489. doi: 10.4049/jimmunol.169.11.6482. [DOI] [PubMed] [Google Scholar]
- 8.Thomas AW. Annu. Rev. Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 9.Huang SK, Xiao HQ, Klein-Tebbe J, Paciotti G, Marsh DG, Lichtenstein LM, Liu MC. J. Immunol. 1995;55:2688–2694. [PubMed] [Google Scholar]
- 10.Bodey KJ, Semper AE, Redington AE, Madden J, Teran LM, Holgate ST, Frew AJ. Allergy (Cph.) 1999;54:1083–1093. doi: 10.1034/j.1398-9995.1999.00889.x. [DOI] [PubMed] [Google Scholar]
- 11.Henderson WR, Jr., Tang LO, Chu SJ, Tsao S-M, Chiang GK, Jones F, Jonas M, Pae C, Wang H, Chi EY. Am. J. Respir. Crit. Care Med. 2002;165:108–116. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- 12.Webb DC, McKenzie AN, Koskinen AM, Yang M, Mattes J, Foster PS. J. Immunol. 2000;165:108–113. doi: 10.4049/jimmunol.165.1.108. [DOI] [PubMed] [Google Scholar]
- 13.Malabarba MG, Rui H, Deutsch HH, Chung J, Kalthoff FS, Farrar WL, Kirken RA. Biochem. J. 1996;319:865–872. doi: 10.1042/bj3190865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zurawski SM, Chomarat P, Djossou O, Bidaud C, McKenzie AN, Miossec P, Banchereau J, Zurawski G. J. Biol. Chem. 1995;270:13869–13878. doi: 10.1074/jbc.270.23.13869. [DOI] [PubMed] [Google Scholar]
- 15.Kotowicz K, Callard RE, Friedrich K, Matthews DJ, Klein N. Int. Immunol. 1996;8:1915–1925. doi: 10.1093/intimm/8.12.1915. [DOI] [PubMed] [Google Scholar]
- 16.Miloux B, Laurent P, Bonnin O, Lupker J, Caput D, Vita N, Perrara P. FEBS Lett. 1997;401:163–166. doi: 10.1016/s0014-5793(96)01462-7. [DOI] [PubMed] [Google Scholar]
- 17.Zhang JG, Hilton DJ, Willson TA, McFarlane C, Roberts BA, Mortiz RL, Simpson RJ, Alexander WS, Metcalf D, Nicola NA. J. Biol. Chem. 1997;272:9474–9480. doi: 10.1074/jbc.272.14.9474. [DOI] [PubMed] [Google Scholar]
- 18.Donaldson DD, Whitters MJ, Fitz LJ, Neben TY, Finnerty H, Henderson SL, O’Hara RM, Jr., Beier DR, Turner KJ, Wood CR, Collins M. J. Immunol. 1998;161:2317–2324. [PubMed] [Google Scholar]
- 19.Kawakami K, Taguchi J, Murata T, Puri RK. Blood. 2001;97:2673–2679. doi: 10.1182/blood.v97.9.2673. [DOI] [PubMed] [Google Scholar]
- 20.Rahaman SO, Sharma P, Harbor PC, Aman MJ, Vogelbaum MA, Haque SJ. Cancer Res. 2002;62:1103–1109. [PubMed] [Google Scholar]
- 21.Wood N, Whitters MJ, Jacobson BA, Witek J, Sypek JP, Kasaian M, Eppihimer MJ, Unger M, Tanaka T, Goldman SJ, Collins M, Donaldson DD, Grusby MJ. J. Exp. Med. 2003;197:703–709. doi: 10.1084/jem.20020906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawakami K, Husain SR, Bright RK, Puri RK. Cancer Gene Ther. 2001;8:861–868. doi: 10.1038/sj.cgt.7700373. [DOI] [PubMed] [Google Scholar]
- 23.Kawakami K, Kawakami M, Snoy PJ, Husain SR, Puri RK. J. Exp. Med. 2001;194:1743–1754. doi: 10.1084/jem.194.12.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawakami K, Terabe M, Kawakami M, Berzofsky JA, Puri RK. Cancer Res. 2006;66:4434–4442. doi: 10.1158/0008-5472.CAN-05-1265. [DOI] [PubMed] [Google Scholar]
- 25.Wu AH, Low WC. Neuro-Oncology. 2003;5:179–187. doi: 10.1215/S1152-8517-02-00051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiaramonte MG, Mentink-Kane M, Jacobson BA, Cheever AW, Whitters MJ, Goad ME, Wong A, Collins M, Donaldson DD, Grusby MJ, Wynn TA. J. Exp. Med. 2003;197:687–701. doi: 10.1084/jem.20020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daines MO, Hershey GK. J. Biol. Chem. 2002;277:10387–10393. doi: 10.1074/jbc.M108109200. [DOI] [PubMed] [Google Scholar]
- 28.Heller NM, Matsukura S, Georas SN, Boothby MR, Rothman PB, Stellato C, Schleimer RP. Am. J. Respir. Cell Mol. Biol. 2004;31:573–582. doi: 10.1165/rcmb.2004-0195OC. [DOI] [PubMed] [Google Scholar]
- 29.Yasunaga S, Yuyama N, Arima K, Tanaka H, Toda S, Maeda M, Matsi K, Goda C, Yang Q, Sugita Y, Nagai H, Izuhara K. Cytokine. 2003;24:293–303. doi: 10.1016/j.cyto.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Chun J, Contos JJ, Munroe D. Cell Biochem. Biophys. 1999;30:213–242. doi: 10.1007/BF02738068. [DOI] [PubMed] [Google Scholar]
- 31.Fukushima N, Chun J. Prostaglandins. 2001;64:21–32. doi: 10.1016/s0090-6980(01)00105-8. [DOI] [PubMed] [Google Scholar]
- 32.Casas-Gonzalez P, Ruiz-Martinez A, Garcia-Sainz JA. Biochim. Biophys. Acta. 2003;1633:75–83. [PubMed] [Google Scholar]
- 33.Chou CH, Wei LH, Kuo ML, Huang YJ, Lai KP, Chen CA, Hsieh CY. Carcinogenesis. 2005;26:45–52. doi: 10.1093/carcin/bgh301. [DOI] [PubMed] [Google Scholar]
- 34.Xu YJ, Saini HK, Cheema SK, Dhalla NS. Cell Calcium. 2005;38:569–579. doi: 10.1016/j.ceca.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Chun J, Rosen H. Curr. Pharm. Des. 2006;12:161–171. doi: 10.2174/138161206775193109. [DOI] [PubMed] [Google Scholar]
- 36.Noguchi K, Ishii S, Shimizu T. J. Biol. Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 37.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. J. Biol. Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 38.Georas SN, Berdyshev E, Hubbard W, Gorshkova IA, Usatyuk PV, Saatian B, Myers AC, Williams MA, Xiao HQ, Liu M, Natarajan V. Clin. Exp. Allergy. 2007 doi: 10.1111/j.1365-2222.2006.02626.x. in press. [DOI] [PubMed] [Google Scholar]
- 39.Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, Bittman R, Tigyi G, Aepfelbacher M. Proc. Natl. Acad. Sci. U. S. A. 1999;96:6931–6936. doi: 10.1073/pnas.96.12.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Budnik LT, Mukhopadhyay AK. Biol. Reprod. 2002;66:859–865. doi: 10.1095/biolreprod66.4.859. [DOI] [PubMed] [Google Scholar]
- 41.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. J. Cell Biol. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JG, Watkins T, He D, Saatian B, Natarajan V. J. Biol. Chem. 2004;279:41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Usatyuk PV, Cummings R, Saatian B, He D, Watkins T, Morris A, Spannhake EW, Brindley DN, Natarajan V. Biochem. J. 2005;385:493–502. doi: 10.1042/BJ20041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saatian B, Zhao Y, He D, Georas SN, Watkins T, Spannhake EW, Natarajan V. Biochem. J. 2006;393:657–668. doi: 10.1042/BJ20050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. J. Biol. Chem. 2006;281:19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koch AE, Polverini PJ, Kunkel EJ, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Science. 1992;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 47.Car BD, Meloni F, Luisetti M, Semenzato G, Gialdroni-Grassi G, Walz A. Am. J. Respir. Crit. Care Med. 1994;149:655–659. doi: 10.1164/ajrccm.149.3.8118632. [DOI] [PubMed] [Google Scholar]
- 48.Folkard SG, Westwick J, Millar AB. Eur. Respir. J. 1997;10:2097–2104. doi: 10.1183/09031936.97.10092097. [DOI] [PubMed] [Google Scholar]
- 49.Rubenfeld J, Guo J, Sookrung N, Chen R, Chaicumpa W, Casolaro V, Zhao Y, Natarajan V, Georas S. Am. J. Physiol. 2006;290:L66–L74. doi: 10.1152/ajplung.00473.2004. [DOI] [PubMed] [Google Scholar]
- 50.Bernacki SH, Nelson AL, Abdullah L, Sheehan JK, Harris A, Davis CW, Randell SH. Am. J. Respir. Cell Mol. Biol. 1999;20:595–604. doi: 10.1165/ajrcmb.20.4.3442. [DOI] [PubMed] [Google Scholar]
- 51.Wang L, Cummings R, Zhao Y, Kazlauskas A, Sham JK, Morris A, Georas S, Brindley DN, Natarajan V. J. Biol. Chem. 2003;278:39931–39940. doi: 10.1074/jbc.M302896200. [DOI] [PubMed] [Google Scholar]
- 52.Jenkins GM, Frohman MA. Cell. Mol. Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cazzolli R, Shemon AN, Fang MO, Hughes WE. IUBMB Life. 2006;58:457–461. doi: 10.1080/15216540600871142. [DOI] [PubMed] [Google Scholar]
- 54.Kawakami M, Kawakami K, Kasperbauer JL, Hinkley LL, Tsukuda M, Strome SE, Puri RK. Clin. Cancer Res. 2003;9:381–6388. [PubMed] [Google Scholar]
- 55.Snyder MD, Pierce SK. Traffic. 2006;7:993–1006. doi: 10.1111/j.1600-0854.2006.00450.x. [DOI] [PubMed] [Google Scholar]
- 56.Kogut MH, Lowry VK, Farnell M. Int. Immunopharmacol. 2003;3:693–706. doi: 10.1016/S1567-5769(03)00057-2. [DOI] [PubMed] [Google Scholar]
- 57.Adachi T, Nakashima S, Saji S, Nakamura T, Nozawa Y. Hepatology. 1996;24:1274–1281. doi: 10.1053/jhep.1996.v24.pm0008903410. [DOI] [PubMed] [Google Scholar]
- 58.Xu Y, Shen Z, Wiper DW, Wu M, Morton RE, Elson P, Kennedy AW, Belinson J, Markman M, Casey G. J. Am. Med. Assoc. 1998;280:719–723. doi: 10.1001/jama.280.8.719. [DOI] [PubMed] [Google Scholar]
- 59.Fichtner-Feigl S, Warren S, Kawakami K, Puri RK, Kitani A. Nat. Med. 2005;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 60.David M, Ford D, Bertoglio J, Maizel AL, Pierre J. Oncogene. 2001;20:6660–6668. doi: 10.1038/sj.onc.1204629. [DOI] [PubMed] [Google Scholar]