

Abstract
We have demonstrated that LPA (lysophosphatidic acid)-induced IL (interleukin)-8 secretion was partly mediated via transactivation of EGFR [EGF (epidermal growth factor) receptor] in HBEpCs (human bronchial epithelial primary cells). The present study provides evidence that LPA-induced transactivation of EGFR regulates COX (cyclo-oxygenase)-2 expression and PGE2 [PG (prostaglandin) E2] release through the transcriptional factor, C/EBPβ (CCAAT/enhancer-binding protein β), in HBEpCs. Treatment with LPA (1 μM) stimulated COX-2 mRNA and protein expression and PGE2 release via Gαi-coupled LPARs (LPA receptors). Pretreatment with inhibitors of NF-κB (nuclear factor-κB), JNK (Jun N-terminal kinase), or down-regulation of c-Jun or C/EBPβ with specific siRNA (small interference RNA) attenuated LPA-induced COX-2 expression. Downregulation of EGFR by siRNA or pretreatment with the EGFR tyrosine kinase inhibitor, AG1478, partly attenuated LPA-induced COX-2 expression and phosphorylation of C/EBPβ; however, neither of these factors had an effect on the NF-κB and JNK pathways. Furthermore, LPA-induced EGFR transactivation, phosphorylation of C/EBPβ and COX-2 expression were attenuated by overexpression of a catalytically inactive mutant of PLD2 [PLD (phospholipase D) 2], PLD2-K758R, or by addition of myristoylated PKCζ [PKC (protein kinase C) ζ] peptide pseudosubstrate. Overexpression of the PLD2-K758R mutant also attenuated LPA-induced phosphorylation and activation of PKCζ. These results demonstrate that LPA induces COX-2 expression and PGE2 production through EGFR transactivation-independent activation of transcriptional factors NF-κB and c-Jun, and EGFR transactivation-dependent activation of C/EBPβ in HBEpCs. Since COX-2 and PGE2 have been shown to be anti-inflammatory in airway inflammation, the present data suggest a modulating and protective role of LPA in regulating innate immunity and remodelling of the airways.
Keywords: bioactive phospholipid, gene expression, receptor tyrosine kinase (RTK), signal transduction, transcription factor
INTRODUCTION
A classical role of human airway epithelium is to function as a passive barrier between inhaled gases/particles and intima of the airways [1-3]. Recent studies suggest that the airway epithelial cells participate in innate immunity through the release of cytokines, chemokines, lipid mediators and other inflammatory mediators in response to a variety of inhaled stimuli [4-6]. LPA (lysophosphatidic acid) is a bioactive lipid mediator that plays an important role in regulating intracellular mobilization of Ca2+, cytoskeletal reorganization, cell growth, differentiation, motility and survival [7-10]. LPA has been found in various biological fluids, including plasma and BAL (bronchoalveolar lavage) [11-13]. Recent studies have demonstrated that LPA also plays a critical role in airway inflammation [14-22], and we have shown that levels of LPA in BAL fluid are higher in a group of allergen-challenged asthmatic patients compared with a normal group [13]. Furthermore, treatment of HBEpCs (human bronchial epithelial primary cells) with LPA-induced IL (interleukin)-8 secretion [15-20], and intratracheal administration of LPA to mice induced infiltration and accumulation of neutrophils in BAL fluid [15]. These results suggested that LPA plays an important role in innate immunity through the secretion of Th-1 type cytokines. Additionally, LPA treatment of HBEpCs attenuated IL-13-induced STAT (signal transducer and activator of transcription)-6 phosphorylation via increased expression of the IL-13 decoy receptor, IL-13Rα2 (IL-13 receptor α2) [19]. These results suggest that LPA may play an anti-inflammatory role in airway adaptive immunity by reducing Th-2 cytokine function. Thus, LPA appears to induce pro-inflammatory signals in innate immunity and anti-inflammatory factors in adaptive immunity during airway inflammation.
Ligation of LPA to its GPCR (G-protein-coupled receptor) activates MAPKs (mitogen-activated protein kinases), PLD (phospholipase D) and transcriptional factors such as NF-κB (nuclear factor-κB) and JNK (Jun N-terminal kinase)/c-Jun pathways [14-19]. A growing body of literature shows that LPA induces intracellular signals through transactivation of RTKs (receptor tyrosine kinases) in various cell lines [14,18,20,23- 25]. Among the various RTKs, transactivation of EGFR [EGF (epidermal growth factor) receptor] [18,23,24], PDGFR (platelet-derived growth factor receptor) [14] and TrkA [25] have been well described. In addition to transactivation, transinactivation of RTKs by GPCRs has been reported [20,26]. LPA treatment induces serine phosphorylation of c-Met and downregulates HGF (hepatocyte growth factor)/c-Met signalling in HBEpCs [20]. In HBEpCs, LPA-stimulated tyrosine phosphorylation of PDGFRβ by a transactivation mechanism is regulated by PLD2, and also part of the LPA-induced threonine/tyrosine phosphorylation of ERK1/2 (extracellular-signal-regulated kinase 1/2) was linked to the transactivation of PDGFRβ [14]. Also, LPA-mediated tyrosine phosphorylation of EGFR through PKCδ [PKC (protein kinase C) δ]-mediated MMP2/9 (matrix metalloproteinase 2 and 9) activation, HB-EGF (heparin-binding EGF) shedding, and binding of HB-EGF to EGFR, results, in part, in IL-8 secretion from HBEpCs [18].
PGE2 [PG (prostaglandin) E2] is an autocrine mediator derived from AA (arachidonic acid) metabolism either through the constitutive active COX (cyclo-oxygenase)-1 and/or an inducible COX-2 [27,28]. A growing body of evidence suggests that upregulation of COX-2 expression and PGE2 release plays a protective role in the innate immunity response and tissue repair process in airway inflammation [29-31]. This is apparent from the findings that administration of COX-1/COX-2 inhibitors to allergen-sensitized mice increased expression of Th2 cytokines (IL-5 and IL-13), mRNA expression of CCR (CC chemokine receptor) 1/5 and airway hyper-responsiveness [30]. However, the regulation of COX-2 expression and the physiological effect of PGE2 on airway epithelium have not been well examined.
In this study, we demonstrated that LPA treatment induces COX-2 expression and PGE2 production through a PLD2 and PKCζ -mediated cross-link of LPA receptors and EGFR in HBEpCs. This is the first report which shows that the trans-activation of EGFR regulates the C/EBPβ (CCAAT/enhancer-binding protein β) pathway and leads to COX-2 expression and PGE2 release. These results demonstrate novel mechanisms for regulation of airway immunity by LPA.
EXPERIMENTAL
Materials
LPA, in the form 1-oleoyl (18:1)-LPA, was purchased from Sigma-Aldrich. PTx (pertussis toxin) was from Calbiochem. AG1478 was purchased from AG Scientific. MBP (myelin basic protein) and antibodies for PLD1, PLD2, phospho-IκB (inhibitory κB) (Ser32), phospho-JNK1/2, JNK1, NF-κB (RelA), phospho-C/EBPβ and C/EBPβ were from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Antibodies for EGFR and phospho-EGFR (Tyr1068), PKCζ and phospho-PKCζ (Thr410) were procured from Cell Signaling Technology (Beverly, MA, U.S.A.) and human recombinant EGF was from Upstate Biotechnology. Scrambled siRNA (small interference RNA) was from Dharmacon and EGFR siRNA, c-Jun siRNA, and C/EBPβ siRNA were from Santa Cruz Biotechnology. TransMessenger™ Transfection Reagent was from Qiagen. Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies were purchased from Molecular Probes (Eugene, OR, U.S.A.). ECL® (enhanced chemiluminescence) kit for detection of proteins by Western blotting was obtained from Amersham Pharmacia. EIA (enzyme immunoassay) kit for PGE2 measurement and antibodies for COX-1 and COX-2 were from Cayman Chemical (Ann Arbor, MI, U.S.A.). All other reagents were of analytical grade.
Cell culture
Primary human bronchial epithelial cells were isolated from normal human lung obtained from lung transplant donors, purchased from Clonetics/Lonza, following typical procedures as previously described [14,15]. The isolated P0 (passage zero) HBEpCs were then seeded, at a density of 1.5 × 104 cells/cm2, onto T-75 flasks in serum-free BEGM (basal essential growth medium; supplied by Clonetics/Lonza). Growth factors were supplemented according to Clonetics/Lonza’s instructions. Cells were incubated at 37 °C in 5% CO2/95% air to ∼80% confluence and subsequently propagated in 35-mm-diameter collagen-coated dishes or 6-well plates. All experiments were carried out between P1 and P3.
Preparation of cell lysates and Western blotting
After indicated treatments, HBEpCs were rinsed twice with ice-cold PBS and lysed in 200 μl of lysis buffer (20mM Tris/HCl, pH 7.4, 150 mM NaCl, 2 mM EGTA, 5 mM β-glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml aprotinin, 1 μg/ml leupeptin and 1 μg/ml pepstatin). Cell lysates were incubated at 4 °C for 15 min, sonicated on ice for 15 s, and centrifuged at 5000 g for 5 min at 4 °C. Protein concentration was determined with a BCA (bicinchoninic acid) protein assay kit (Pierce) using BSA as standard. Equal amounts of protein (20 μg) were subjected to SDS/PAGE (10% gels), transferred to PVDF membranes, blocked with 5% (w/v) BSA in TBST (25 mM Tris/HCl, pH 7.4, 137 mM NaCl and 0.1% Tween 20) for 1 h, and incubated with primary antibodies in 5% (w/v) BSA in TBST for 1 h at room temperature (25 °C). The membranes were washed at least three times with TBST at 15 min intervals and then incubated with either mouse or rabbit horseradish peroxidase-conjugated secondary antibody (1:3000 dilution) for 1 to 2 h at room temperature. The membranes were developed with an ECL® detection system according to the manufacturer’s instructions.
Transfection of EGFR, c-Jun, COX-2 or C/EBPβ siRNA
siRNAs (20 μM) were designed and synthesized by Santa Cruz Biotechnology. HBEpCs (P1 or P2) were cultured onto 6-well plates. At 50–60% confluence, transient transfection of siRNAs was carried out using TransMessenger™ Transfection Reagent. siRNA (2.5 μl) was diluted with kit contents Enhancer R(4 μl) in buffer EC-R (93.5 μl), and mixed by vortexing for 10 s. After 5 min of incubation at room temperature, 8 μl of TransMessenger™ Transfection Reagent was added to the RNA-Enhancer R mixture and mixed by vortexing for 10 s. After 10 min of incubation at room temperature, the mixture was added drop-wise onto the cells with 900 μl of BEBM (bronchial epithelial basal medium). The basal medium was replaced with complete BEGM medium after 3 h. Cells were lysed 72 h after transfection and analysed using Western blotting as above.
RNA extraction and real-time RT—PCR (reverse transcription—PCR)
Total RNA was extracted from cultured HBEpCs using TRIzol® reagent (Life Technology) according to the manufacturer’s instructions. RNA was quantified spectrophotometrically and samples with an absorbance of ≥1.8 measured at 260/280 nm were analysed by real-time RT—PCR. RNA (0.5 μg) was reverse transcribed using a cDNA synthesis kit (Bio-Rad), and real-time PCR and quantitative PCR were performed to assess expression of COX-1 and COX-2 using primers designed based on human mRNA sequences. COX-1 primers: forward, GAGTACTGGAAGCCGAGCAC and reverse, AGGGACAGGTCTTGGTGTTG; COX-2 primers: forward, ATCGATGCTGTGGAGCTGTA and reverse, AAGGAGAATGGTGCTCCAAC; 18S primers: forward, GTAACCCGTTGAACCCCATT and reverse, CCATCCAATCGGTAGTAGCG. Amplicon expression in each sample was normalized to its 18S RNA content. The relative abundance of target mRNA in each sample was calculated as 2 raised to the negative of its threshold cycle value times 106 after being normalized to the abundance of its corresponding 18S, {e.g. [2-(IL-13Rα2 threshold cycle)/2-(18S threshold cycle)] × 106}.
Transfection of adenoviral constructs
Infection of HBEpCs (∼60% confluence) with purified adenoviral vectors of catalytically inactive mutants of hPLD1b (human PLD1b)-K898R and mPLD2 (mouse PLD2)-K758R, or empty Ad5 vector were carried out in 6-well plates as described previously [14,19]. Following infection with different MOI (multiplicity of infection) in 1 ml of BEGM for 48 h, the virus-containing medium was replaced with complete BEBM prior to LPA challenge.
Measurement of PGE2 secretion
HBEpCs grown on 6-well plates were challenged with LPA or EGF for 3 h and media were collected and centrifuged at 5000 g for 10 min at 4°C. The supernatants were transferred to new eppendorf tubes and frozen at −80°C for later analysis of PGE2 with PGE2 ELISA kit according to the manufacturer’s instructions.
Immunocytochemistry
HBEpCs grown on coverslips to ∼80% confluence were challenged with LPA (1 μM) for 15 min. Coverslips were rinsed with PBS and treated with 3.7% (v/v) formaldehyde in PBS at room temperature for 20 min. After washing with PBS, coverslips were incubated in blocking buffer [1% (w/v) BSA in TBST] for 1 h, cells were subjected to immunostaining with NF-κB antibody (RelA) (1:200 dilution) for 1 h, and washed four times with TBST followed by staining with a secondary antibody with Alexa Fluor 488 (1:200 dilution in blocking buffer) for 1 h. After washing four times with TBST, the coverslips were mounted using commercial mounting medium for fluorescent microscopy (Kirkegaard and Perry Laboratories, Gaithersburg, MD, U.S.A.) and were examined using an immunofluorescent microscope with a Hamamatsu digital camera using a 60 × oil immersion objective and MetaVue software.
PKCζ activity assay
HBEpCs grown in 100-mm-diameter dishes were challenged with LPA for 15 min, cell lysates were centrifuged at 5000 g for 10 min at 4°C, and lysate containing 1 mg of protein was incubated with 10 μl of PKCζ antibody overnight at 4°C. The immune complexes were incubated with 50 μl of Protein A/G beads for 2 h at 4°C with gentle rotation. Immunoprecipitates were washed five times with lysis buffer, modified to contain 500 mM NaCl, and incubated with 2 μg of MBP and 10 μCi of [γ -32P]ATP for 30 min at 37°C in kinase buffer (35 mM Tris/HCl, pH 7.5, 10 mM MgCl2, 5mM EGTA,1 mM CaCl2 and 1 mM phenylphosphate) for PKCζ activity assays. Proteins were separated by SDS/PAGE (20% gels), gels were dried and MBP was detected as a 20 kDa band by autoradiography.
Statistical analyses
All results were subjected to statistical analysis using one-way ANOVA and, where appropriate, analysed by Student-Newman-Keuls test. Results are expressed as means ± S.D. of triplicate samples from at least three independent experiments and level of significance was taken to be P < 0.05.
RESULTS
LPA induces COX-2 expression and PGE2 release
We have demonstrated earlier that LPA treatment induces IL-8 [15-18] and IL-13Rα2 secretion in HBEpCs [19]. Here, we examined the effect of LPA treatment on COX-2 expression and PGE2 secretion. Analyses of total RNA by real-time RT—PCR showed that both COX-1 and COX-2 are expressed in HBEpCs and that the expression of COX-1 is ∼2-fold higher than COX-2 in unstimulated cells (Figure 1A). Exposure of HBEpCs to LPA (1 μM) induced COX-2 gene expression in a time-dependent fashion with a ∼4-fold increase at 1 h and a maximal level reached at 3 h (∼ 12.8-fold), whereas such exposure had no effect on COX-1 gene expression at up to 24 h (Figure 1B). As shown in Figure 1(C), LPA (1 μM) induced COX-2 protein expression after 3 h (5.2-fold), which continued up to 24 h (4.5-fold), without altering COX-1 protein expression.
Figure 1. LPA induces COX-2 expression in HBEpCs.
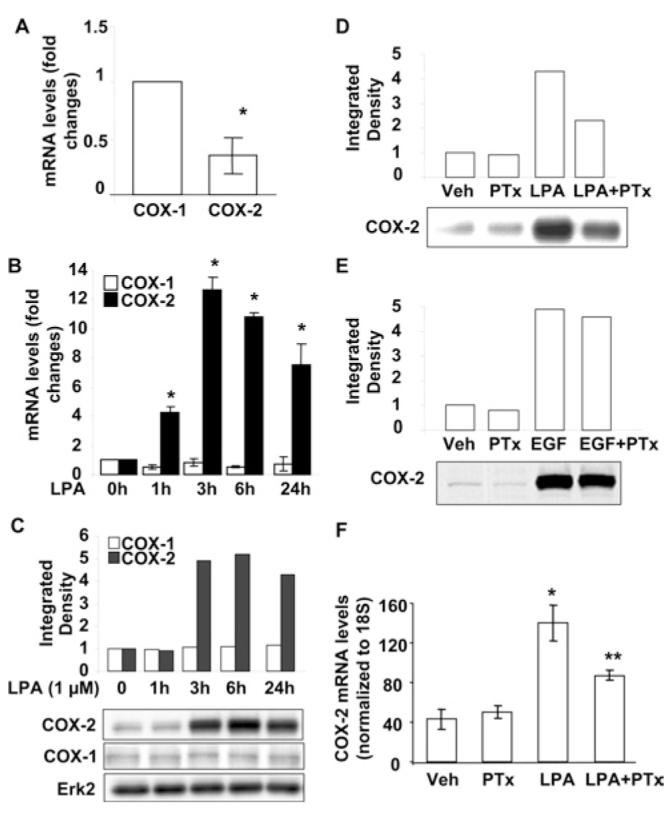
(A) Total RNA was extracted from HBEpCs, and COX-1 and COX-2 mRNA expression were examined by real-time RT—PCR with specific primers for human COX-1 and COX-2. Results are the means ± S.D. (n = 3). *P < 0.05 against COX-1. (B) HBEpCs grown to ∼80–90% confluence were treated with LPA (1 μM), then total RNA was extracted at the times indicated and COX-1 and COX-2 mRNA expression were determined by real-time RT—PCR with specific primers for human COX-1 and COX-2. Results are the means ± S.D. (n = 3). *P < 0.05 against cells treated for 0 h. (C) HBEpCs grown to ∼80–90% confluence were treated with LPA (1 μM) for 1–24 h, cell lysates were collected and equivalent amounts of protein were analysed by Western blotting with COX-1, COX-2 or Erk2 antibodies. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots (Erk2 expression is shown as Western blot only as control). (D and E) HBEpCs grown to ∼80–90% confluence were pretreated with PTx (100 ng/ml) for 4 h, then challenged with LPA (1 μM) (D) or EGF (20 ng/ml) (E) for 3 h. Cell lysates were analysed by Western blotting with COX-2 antibody. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot. (F) HBEpCs grown to ∼80–90% confluence were pretreated with PTx (100 ng/ml) for 4 h, then challenged with LPA (1 μM) for 3 h. Total RNA was extracted and COX-2 mRNA expression was by real-time RT—PCR with specific primers for human COX-2. Results are the means ± S.D. (n = 3). *P < 0.05 against vehicle (Veh) (medium alone); **P < 0.05 against LPA.
LPA induces various cellular responses through its ligation to GPCRs (LPA1–5) in mammalian cells [17,32-34]. We have shown that LPA-induced intracellular signalling transduction, IL-8 and IL-13Rα2 secretion were dependent on Gαi proteins [15-19]. To examine whether LPA-induced COX-2 expression was through Gαi-protein-coupled receptors, HBEpCs were pretreated with PTx (100 ng/ml for 4 h) prior to an LPA (1 μM for 3 h) challenge. As shown in Figures 1(D) and 1(F), PTx attenuated LPA-induced COX-2 mRNA and protein expression; however, PTx had no effect on EGF-induced COX-2 expression (Figure 1E), suggesting the involvement of Gαi-coupled LPARs (LPA receptors). Next, we measured secretion of PGE2 in media by cells after LPA challenge. HBEpCs were challenged with LPA (1 μM) for 3 h, media were collected, and PGE2 levels were measured by a PGE2-specific ELISA kit. As shown in Figure 2(A), media from control cells contained 196 ± 12 pg/ml of PGE2, and LPA treatment increased the PGE2 levels to 723 ± 45 pg/ml. To examine the role of inducible COX-2 in LPA-induced PGE2 release, COX-2 expression was downregulated by transfection of COX-2 siRNA (50 nM) for 72 h. COX-2 siRNA effectively down-regulated COX-2 protein expression (Figure 2B) and attenuated LPA-induced PGE2 release by ∼50% compared with scrambled siRNA cells exposed to LPA (723 ± 45 pg/ml and 375 ± 17 pg/ml respectively) (Figure 2A). These results show that LPA-induced PGE2 secretion was dependent on expression of COX-2 in HBEpCs.
Figure 2. LPA induces PGE2 release through inducible COX-2 expression.
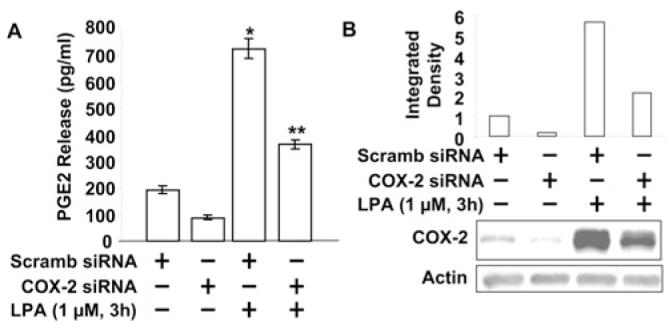
(A) HBEpCs grown to ∼50–60% confluence were transfected with scrambled (Scramb) siRNA and COX-2 siRNA for 72 h, then challenged with LPA (1 μM) for 3 h. Media were collected and analysed for PGE2 levels by the PGE2 ELISA kit. *P <0.05 against scrambled siRNA transfected cells; **P <0.05 against LPA treatment of scrambled siRNA transfected cells. (B) Cell lysates were analysed by Western blotting with COX-2 antibody. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot (actin was Western blotted as a control).
Role of transcription factors NF-κB, c-Jun and C/EBPβ in LPA-induced COX-2 expression
As the human COX-2 promoter region contains NF-κB, AP (activating protein)-1 and C/EBPβ binding sites [35], we examined the role of these transcriptional factors in the regulation of COX-2 expression by LPA. Treatment of HBEpCs with LPA (1 μM for 15 min) induced phosphorylation of IκB, JNK and C/EBPβ (Figure 3A), suggesting LPA activates these three transcription factors. To determine the role of the NF-κB pathway in LPA-induced COX-2 expression, HBEpCs were pretreated with varying concentrations of an IκB kinase-specific inhibitor, Bay11-7082 (1.0, 5.0 and 10.0 μM), for 1 h, then challenged with LPA (1 μM) for a further 3 h. As shown in Figure 2(B), Bay11-7082 (5 and 10 μM) attenuated LPA-induced COX-2 expression by 36% and 55% respectively. Next, we examined the role of the JNK/c-Jun pathway in LPA-induced COX-2 expression. HBEpCs were transfected with c-Jun siRNA (50 nM for 72 h) or pretreated with a JNK-specific inhibitor (JNKi II, at 10, 20 and 40 μM) prior to LPA challenge (1 μM for 3 h). Transfection of cells with c-Jun siRNA reduced c-Jun expression by ∼80% and attenuated LPA-induced COX-2 expression by ∼35% (Figure 3C). Similarly, pretreatment of HBEpCs with JNKi II attenuated LPA-induced COX-2 expression in a dose-dependent manner (Figure 3D). Furthermore, transfection of HBEpCs with C/EBPβ siRNA (50 nM for 72 h) reduced C/EBPβ expression by 70% and attenuated LPA-induced COX-2 expression by 48% (Figure 2E). These results suggest that LPA-induced COX-2 expression is dependent on activation of NF-κB, JNK/c-Jun and C/EBPβ in HBEpCs.
Figure 3. LPA induces COX-2 expression through NF-κB, JNK/c-Jun and C/EBPβ pathways.
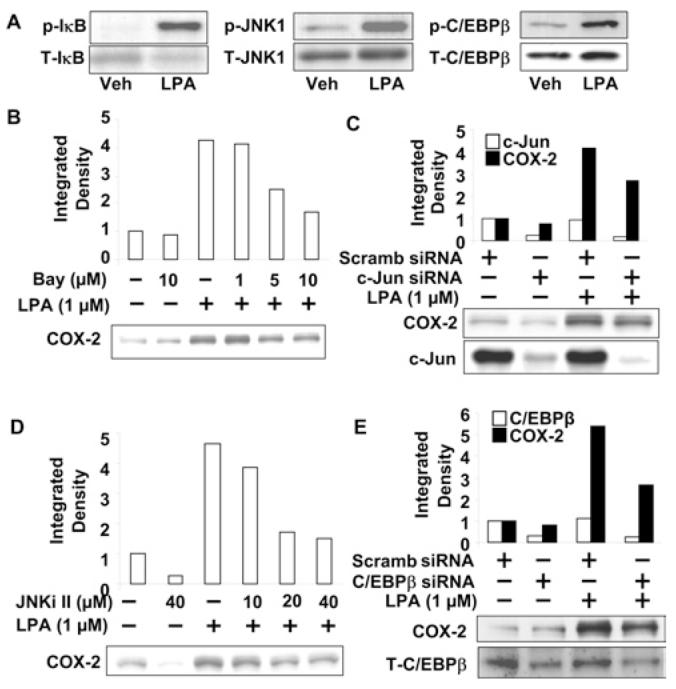
(A) HBEpCs (80–90% confluence on 6-well plates) were treated with LPA (1 μM) for 15 min. Cell lysates were analysed by Western blotting with antibodies to phospho-IκB (p-IκB), total IκB(T-IκB), phospho-JNK1 (p-JNK1), total JNK1 (T-JNK1) phospho-C/EBPβ (p-C/EBPβ) and total C/EBPβ (T-C/EBPβ). Representative blots of three independent experiments are shown. (B) HBEpCs (80% confluence on 6-well plates) were pretreated with Bay11-7082 (Bay; 1, 5 and 10 μM) for 1 h, then challenged with LPA (1 μM) for 3 h. Cell lysates were analysed by Western blotting with antibody to COX-2. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot of three independent experiments. (C and E) HBEpCs (50–60% confluence on 6-well plates) were transfected with scrambled (Scramb) siRNA, c-Jun siRNA (C)orC/EBPβ siRNA (E) for 72 h, then challenged with LPA (1 μM) for 3 h. Cell lysates were analysed by Western blotting with antibodies to COX-2, c-Jun (C) or C/EBPβ (E). Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots. (D) HBEpCs (80% confluence on 6-well plates) were pretreated with JNK inhibitor II (JNKi II; 10, 20 and 40 μM) for 1 h, then challenged with LPA (1 μM) for 3 h. Cell lysates were analysed by Western blotting with COX-2 antibody. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot.
Transactivation of EGFR by LPA regulates COX-2 expression and PGE2 release
We have demonstrated previously that transactivation of EGFR by LPA regulates LPA-induced IL-8 secretion in HBEpCs [18]. To determine further the molecular mechanisms of LPA-induced COX-2 expression and PGE2 release in HBEpCs, we first examined the effect of EGFR siRNA on EGF-induced COX-2 expression. HBEpCs were transfected with scrambled siRNA (50 nM) or EGFR siRNA (50 nM) for 72 h and then challenged with EGF (20 ng/ml) for 3 h. Analyses of total cell lysates by Western blotting showed that transfection of cells with EGFR siRNA almost completely blocked EGFR protein expression (>95%), and partly attenuated EGF-induced COX-2 expression (∼50%, compared with scrambled siRNA cells exposed to EGF) (Figure 4A). To examine the role of transactivation of EGFR on LPA-induced COX-2 expression, HBEpCs were transfected with EGFR siRNA (50 nm for 72 h) or pretreated with the EGFR tyrosine kinase inhibitor AG1478 (0.1 and 0.5 μM for 1 h). As shown in Figures 4(B) and 4(C), EGFR siRNA or AG1478 attenuated LPA-induced COX-2 expression. Furthermore, EGFR siRNA attenuated LPA- and EGF-induced PGE2 secretion in HBEpCs [Scrambled siRNA transfected cells: vehicle (medium alone), 225 ± 45 pg/ml; LPA, 798 ± 70 pg/ml; EGF, 375 ± 50 pg/ml. EGFR siRNA transfected cells: vehicle,85 ± 25 pg/ml; LPA, 360 ± 27 pg/ml; EGF, 250 ± 45 pg/ml] (Figure 4D). These results suggest that LPA-induced COX-2 expression is partly regulated by transactivation of EGFR in HBEpCs.
Figure 4. LPA induces COX-2 expression and PGE2 release through LPA-mediated EGFR transactivation.
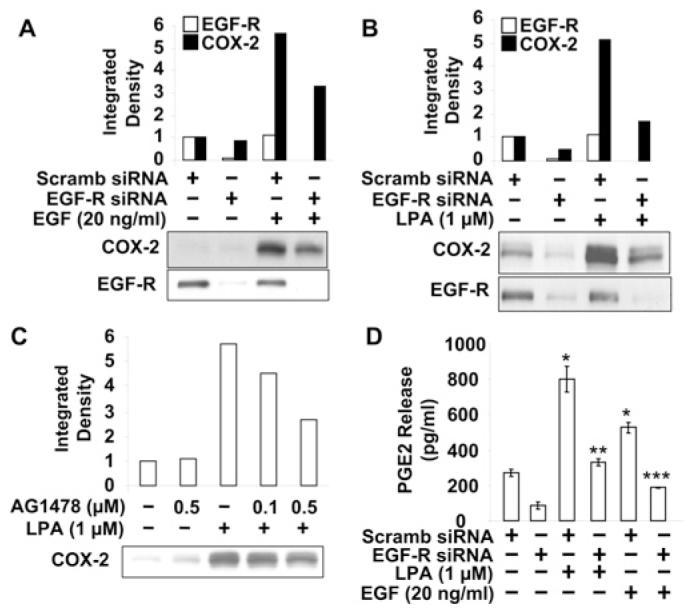
HBEpCs (50–60% confluence) were transfected with scrambled (Scramb) siRNA (50 nM) or EGFR siRNA (50 nM) for 72 h, then challenged with EGF (20 ng/ml) (A) or LPA (1 μM) (B) for 3 h. Cell lysates were analysed with antibodies to COX-2 and EGFR. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots. (C) HBEpCs (80–90% confluence) were pretreated with AG1478 (0.1 and 0.5 μM) for 1 h and then challenged with LPA (1 μM) for 3 h. Cell lysates were analysed with COX-2 antibody. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot. (D) The supernatant from EGF and LPA treatment (A and B) was collected and PGE2 levels were measured by the PGE2 ELISA kit. Results are the means ± S.D. (n = 3). *P < 0.05 against scrambled siRNA-transfected cells; **P < 0.05 against LPA treatment of scrambled siRNA transfected cells; **P < 0.05 against EGF treatment of scrambled siRNA transfected cells.
Role of PLD2 rather than PLD1 in LPA-induced COX-2 expression and EGFR transactivation
We have previously shown that LPA stimulated PLD1 and PLD2 expression, and that PLD2 regulated LPA-induced PDGFRβ transactivation in HBEpCs [14]. To investigate the role of PLD1 and PLD2 isoenzymes in regulating LPA-induced COX-2 expression, we overexpressed catalytically-inactive mutants of hPLD1 (10 MOI) and mPLD2 (10 MOI) for 24 h [14] and cell lysates were analysed for enhanced expression of PLD1 and PLD2 proteins by Western blotting with specific antibodies. As shown in Figures 5(A) and 5(B), infection of HBEpCs with adenoviral constructs increased the expression of PLD1 and PLD2 proteins. Furthermore, overexpression of the catalytically inactive mutant of mPLD2 (K758R), but not hPLD1 (K898R), attenuated LPA-induced COX-2 expression (Figures 5A and 5B). Next, we investigated if LPA-induced EGFR transactivation was regulated by PLD. HBEpCs were infected with adenoviral constructs of the catalytically inactive mutants of hPLD1 (K898R) and mPLD2 (K758R) (10 MOI) for 24 h prior to LPA stimulation. As shown in Figure 5(C), LPA stimulated tyrosine phosphorylation of EGFR at Tyr1068 (∼5.0-fold change); and overexpression of the mPLD2 mutant, but not of the hPLD1 mutant, attenuated LPA-induced tyrosine phosphorylation of EGFR. However, over-expression of both hPLD1 and mPLD2 mutants had no effect on EGF-induced EGFR tyrosine phosphorylation. These results establish the involvement of PLD2 in LPA-mediated COX-2 expression and EGFR transactivation in HBEpCs.
Figure 5. PLD2 regulates LPA-induced COX-2 expression and EGFR trans-activation.

HBEpCs (50–60% confluence) were infected with adenoviral vector containing mutant mPLD2 (mPLD 2 mut) (10 MOI) (A), hPLD1 mutant (hPLD 1b mut) (10 MOI) (B) or control empty vector (Ad-Vector) (10 MOI) (A and B) for 24 h, then challenged with LPA (1 μM) for 3 h. Cell lysates were analysed with antibodies to COX-2 (A and B), PLD2 (A) or PLD1 (B). Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots. (C) HBEpCs (50—60% confluence) were infected with adenoviral vector containing hPLD1 mutant or mPLD2 mutant (10 MOI) or control empty vector (10 MOI) for 24 h, then challenged with LPA (1 μM) or EGF (20 ng/ml) for 15 min. Cell lysates were analysed with antibodies to phospho-EGFR (P-EGFR) or total EGFR (T-EGFR). Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots.
Role of PKCζ in LPA-induced COX-2 expression and EGFR transactivation
We have previously shown that, among the various PKC isoforms, PKCδ regulates LPA-induced IL-8 secretion [14] and EGFR transactivation [18] in HBEpCs. To determine further the role of other PKC isoforms in LPA-induced COX-2 expression, HBEpCs were infected with adenoviral constructs of dn (dominant-negative) PKCα (K368R) δ (K376A) and ζ (K282R) (10 MOI) for 24 h prior to LPA challenge (1 μM for 3 h). Overexpression of dn-PKCδ and ζ , but not dn-PKCα, partially blocked LPA-induced COX-2 expression (results not shown). To study the role of PKCζ further, cells were pretreated with different concentrations of a myristoylated PKCζ peptide inhibitor for 1 h prior to LPA challenge. As shown in Figure 6(A), the myristoylated PKCζ peptide inhibitor attenuated LPA-induced COX-2 expression in a dose-dependent fashion. Furthermore, LPA-induced tyrosine phosphorylation of EGFR was attenuated by the myristoylated PKCζ peptide inhibitor (Figure 6B). These results demonstrate that PKCζ regulates LPA-induced COX-2 expression and transactivation of EGFR in HBEpCs.
Figure 6. PKCζ regulates LPA-induced COX-2 expression and EGFR transactivation.
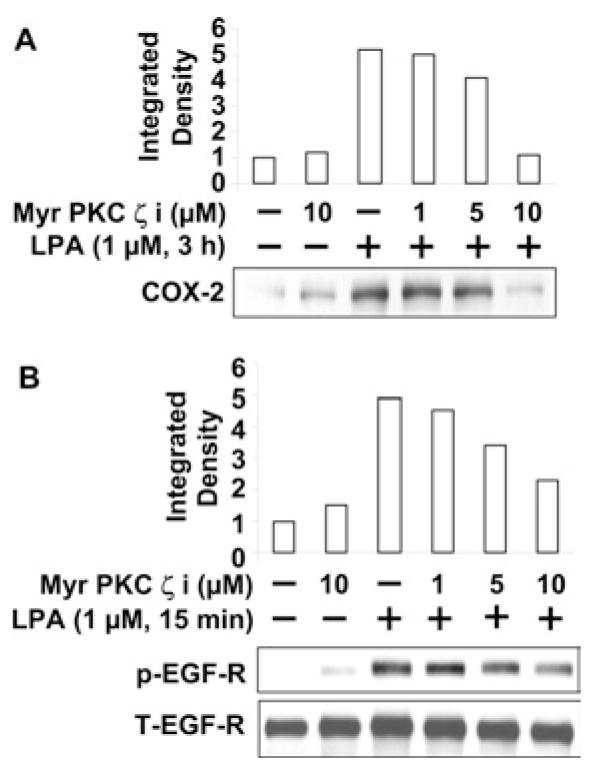
(A) HBEpCs were pretreated with myristoylated PKCζ peptide inhibitor (myr PKC ζ i, at 1, 5 and 10 μM) for 1 h, and then challenged with LPA (1 μM) for 3 h. Cell lysates were analysed with COX-2 antibody. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot. (B) HBEpCs were pretreated with myristoylated PKCζ peptide inhibitor (myr PKCζ i, at 1, 5, and 10 μM) for 1 h, and then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with phospho-EGFR (p-EGF-R) or total EGFR (T-EGF-R) antibodies. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots.
PLD2 regulates LPA-induced PKCζ activation
Since PLD2 and PKCζ regulate LPA-induced COX-2 expression and EGFR transactivation, we investigated whether PKCζ activation by LPA was downstream of PLD2. HBEpCs were infected with an adenoviral construct of the catalytically inactive mutant PLD2 (K758R) (10 MOI for 24 h), and then challenged with LPA for 15 min. As shown in Figure 7(A), LPA treatment induced phosphorylation of PKCζ , as determined by Western blotting with phospho-specific PKCζ antibody, and overexpression of the PLD2 mutant attenuated LPA-induced phosphorylation of PKCζ. The effect of overexpression of the catalytically inactive mutant of PLD2 on PKCζ was also determined in vitro using an MBP phosphorylation assay. HBEpCs, infected with adenoviral mPLD2 mutant (K758R) (10 MOI) for 24 h, were challenged with LPA (1 μM) for 15 min, cell lysates (of equal protein concentration) were subjected to immunoprecipitation with PKCζ, and immunoprecipitates were assayed for PKCζ activity using [γ -32P]ATP and MBP as a substrate. LPA increased phosphorylation of MBP (∼3.9 fold), compared with empty vector-treated cells, and over-expression of mPLD2 mutant blocked LPA-mediated phosphorylation of MBP (Figure 7B). These results show that LPA-induced stimulation of PKCζ is downstream of PLD2 activation in HBEpCs.
Figure 7. PLD2 regulates PKCζ phosphorylation and activation.
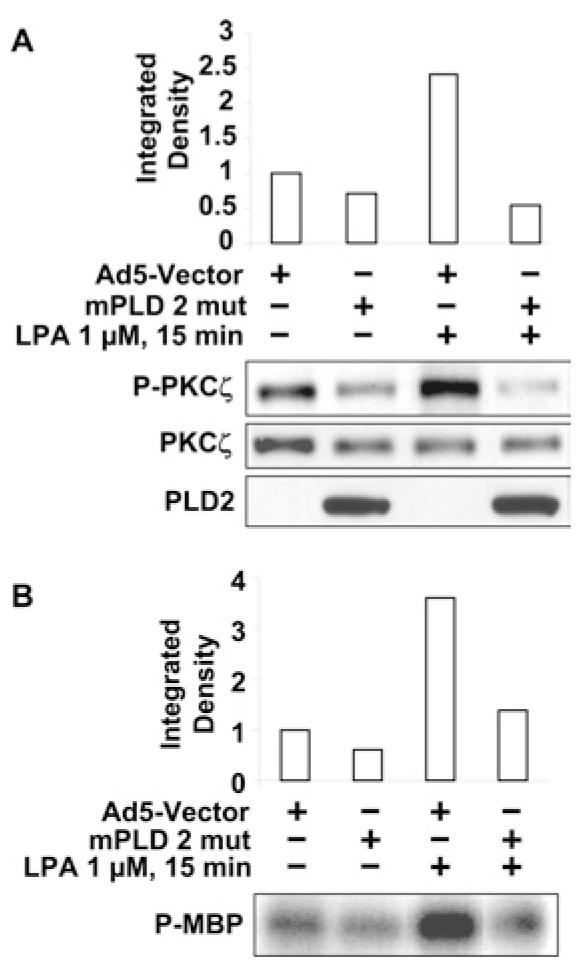
(A) HBEpCs (50–60% confluence) were infected with an adenoviral vector containing mPLD2 mutant (mPLD 2 mut) (10 MOI) or control empty vector (Ad5-Vector) (10 MOI) for 24 h, then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with phospho-PKCζ (P-PKCζ ) or total PKCζ antibodies. Quantitative analysis from representative blots of three independent experiments is shown above the representative blots. PLD2 mutant expression is also shown. (B) HBEpCs (50–60% confluence) were infected with adenoviral vector containing mPLD2 mutant (10 MOI) or control empty vector (10 MOI) for 24 h, then challenged with LPA (1 μM) for 15 min before cell lysates were immunoprecipitated with anti-PKCζ antibody. In vitro phosphorylation activity was tested with MBP as a substrate and 32P-labelled MBP was analysed after separation in SDS/PAGE (20% gel). Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot.
Activation of C/EBPβ is downstream of LPA-induced transactivation of EGFR
A previous study has demonstrated that cross-talk between LPARs and EGFR partly regulates LPA-induced IL-8 release [24]; however, signalling pathways downstream of transactivation of EGFR have not been established. Here we investigated whether transactivation of EGFR regulates activation of the transcription factors NF-κB, c-Jun and C/EBPβ in HBEpCs. LPA treatment (1 μM for 15 min) induced phosphorylation of IκB, whereas pretreatment of HBEpCs with a specific EGFR tyrosine kinase inhibitor, AG1478 (0.01, 0.1 and 1.0 μM for 1 h), had no effect on LPA-induced phosphorylation of IκB (Figure 8A). Furthermore, blocking the EGFR tyrosine kinase activity with AG1478 did not block LPA-induced (1 μM for 30 min) translocation of the NF-κB p65 (RelA) subunit from the cytoplasm to the nucleus, as determined by immunocytochemistry (Figure 8B). The effects of AG1478 on EGF- and LPA-induced tyrosine phosphorylation of EGFR [24] and COX-2 expression (Figure 4C) have been demonstrated. These results suggest that the NF-κB pathway is not downstream of transactivation of EGFRs.
Figure 8. NF-κB and JNK pathways are not dependent on EGFR trans-activation.
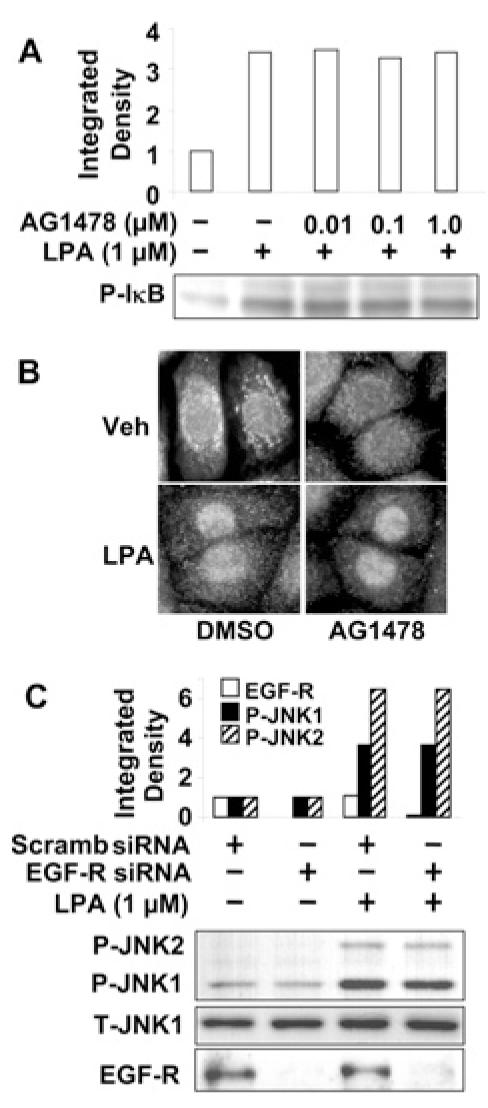
(A) HBEpCs (80–90% confluence) were pretreated with AG1478 (0.01, 0.1 and 1.0 μM) for 1 h, then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with antibody to phospho-IκB (P-IκB). Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot. (B) HBEpCs (80–90% confluence) grown on coverslips were pretreated with AG1478 (1 μM) for 1 h then challenged with vehicle (Veh) (medium alone) or LPA (1 μM) for 15 min. Cells were fixed and stained with the NF-κB p65 subunit antibody (RelA). Shown are representative images of three independent experiments. (C) HBEpCs (50–60% confluence) were transfected with scrambled (Scramb) siRNA (50 nM) or EGFR siRNA (50 nM) for 72 h, then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with phospho-JNK1 (P-JNK1), phospho-JNK2 (P-JNK2), total JNK1 (T-JNK1) or EGFR antibodies. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots.
Next, we examined the role of LPA-induced transactivation of EGFR in JNK/c-Jun signalling. HBEpCs, transfected with either scrambled or EGFR siRNA (50 nM for 72 h), were challenged with LPA (1 μM) for 15 min. As shown in Figure 8(C), down-regulation of EGFR with EGFR siRNA had no effect on LPA-induced phosphorylation of JNK1/2. However, EGFR siRNA did reduce EGFR expression (Figure 8C).
To ascertain further a role for EGFR transactivation by LPA in C/EBPβ activation, HBEpCs were transfected with EGFR siRNA (50 nM for 72 h), to down-regulate EGFR expression, or pretreated with AG1478 (0.1–1.0 μM for 1 h) prior to challenge with LPA (1 μM) or EGF (20 ng/ml) for 15 min, and phosphorylation of C/EBPβ and total C/EBPβ were examined by Western blotting with specific antibodies to phospho-threonine-C/EBPβ and C/EBPβ respectively. As shown in Figure 9(A), EGFR siRNA reduced EGFR expression and attenuated LPA-induced phosphorylation of C/EBPβ (from ∼3.7-fold to ∼1.8-fold after LPA treatment). Furthermore, HBEpCs pretreated with AG1478 (0.1, 0.5 and 1.0 μM) for 1 h blocked, in a dose dependent manner, LPA- and EGF-induced phosphorylation of C/EBPβ (Figure 9B). These results suggest that among the three transcriptional factors, only C/EBPβ is downstream of EGFR transactivation by LPA in HBEpCs.
Figure 9. C/EBPβ is regulated by EGFR transactivation.
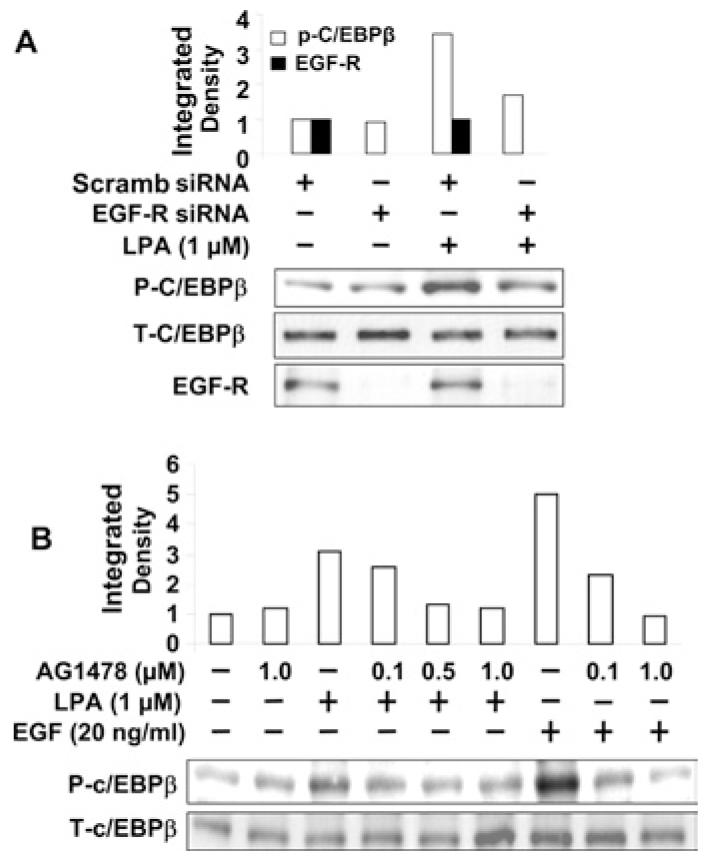
(A) HBEpCs (50–60% confluence) were transfected with scrambled (Scramb) siRNA (50 nM) or EGFR siRNA (50 nM) for 72 h, then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with phospho-C/EBPβ (P-C/EBPβ), total C/EBPβ (T-C/EBPβ) or EGFR antibodies. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots. (B) HBEpCs (80–90% confluence) were pretreated with AG1478 (0.1, 0.5 and 1.0 μM) for 1 h, then challenged with LPA (1 μM) or EGF (20 ng/ml) for 15 min. Cell lysates were analysed with phospho-C/EBPβ or total C/EBPβ antibodies. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blot.
Role of PLD2 and PKCζ in LPA-induced C/EBPβ activation
Having established a role for PLD2 and PKCζ in regulating LPA-induced transactivation of EGFR (Figures 5C and 6B) and COX-2 expression (Figures 5B and 6A), and that C/EBPβ is downstream of transactivation of EGFR in HBEpCs (Figures 9A and 9B), we next determined whether LPA-induced C/EBPβ activation was dependent on PLD2 and PKCζ. HBEpCs, infected with the adenoviral construct of mutant mPLD2 (10 MOI for 24 h) or pre-treated with the myristoylated PKCζ -inhibitor peptide (1, 5 and 10 μM for 1 h), were challenged with LPA (1 μM) for 15 min and cell lysates were analysed for increased phosphorylation of C/EBPβ by Western blotting with phospho-specific antibody. As shown in Figures 10(A) and 10(B), overexpression of mutant mPLD2 or pretreatment with myristoylated PKCζ -inhibitor peptide partially attenuated the LPA-induced threonine phosphorylation of C/EBPβ. These results suggest that PLD2 and PKCζ regulate C/EBPβ activation, which is downstream of EGFR transactivation by LPA in HBEpCs.
Figure 10. PLD2 and PKCζ regulate LPA-induced phosphorylation of C/EBPβ.
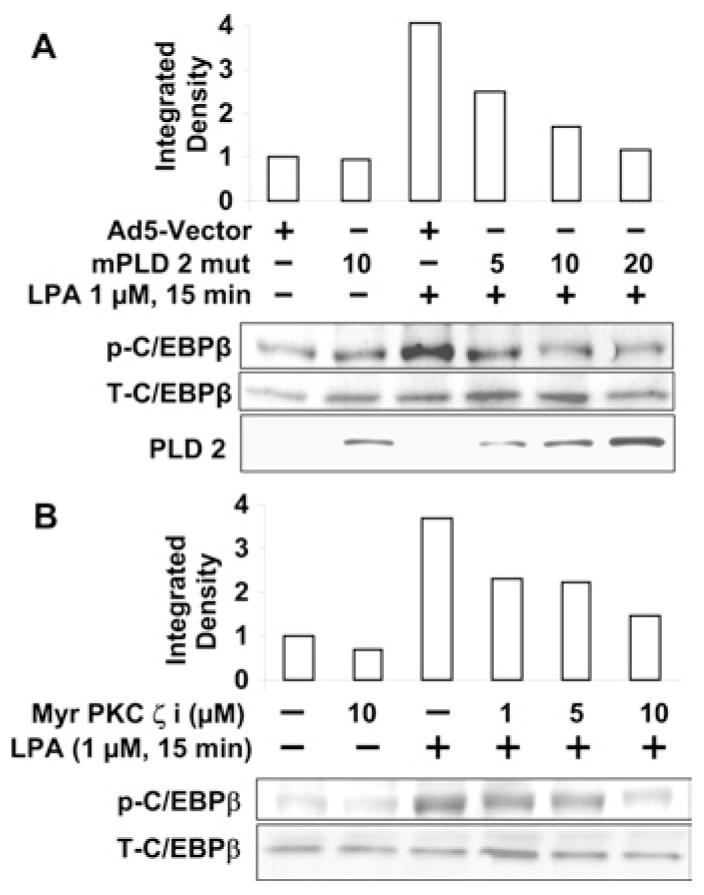
(A) HBEpCs (50–60% confluence) were infected with adenoviral vector containing mPLD2 mutant (mPLD 2 mut) (1, 10 or 20 MOI) or control empty vector (Ad5-Vector) (10 MOI) for 24 h, then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with phospho-C/EBPβ (p-C/EBPβ), total C/EBPβ (T-C/EBPβ) or PLD2 antibodies. Quantitative analysis from a representative blot of three independent experiments is shown above the representative blots. (B) HBEpCs (80–90% confluence) were pretreated with myr PKCζ i (1, 5 and 10 μM) for 1 h, then challenged with LPA (1 μM) for 15 min. Cell lysates were analysed with phospho-C/EBPβ or total C/EBPβ antibodies. Quantitative analysis from representative blots of three independent experiments is shown above a representative blot.
DISCUSSION
The results presented here demonstrate for the first time two independent mechanisms of regulation of COX-2 expression and PGE2 secretion by LPA in airway epithelium. The first pathway is dependent on the activation of C/EBPβ, which is downstream of transactivation of EGFR by LPA; whereas the second pathway involves regulation by NF-κB and c-Jun transcriptional factors, which are independent of EGFR transactivation by LPA. Furthermore, we have characterized the role of PLD2 and PKCζ in LPA-induced activation of C/EBPβ via EGFR transactivation. The current data also demonstrate that COX-2 and PGE2 play a critical role in LPA-induced IL-13Rα2 expression in airway epithelial cells.
COX comprises two isoforms, COX-1 and COX-2, which regulate biosynthesis of various PGs and thromboxanes. In many cell types, COX-2 is an inducible gene, while COX-1 is constitutively expressed [36-38]. In HBEpCs, our data show that both COX-1 and COX-2 were constitutively expressed (COX-1 > COX-2), and stimulation with LPA enhanced the mRNA and protein expression of COX-2 but not COX-1. Our results on basal COX-2 expression and PGE2 release are in agreement with earlier reports demonstrating a role for COX-2 in basal PG generation in airway epithelium [37,38]. Previous studies suggest that COX-2 is anti-inflammatory in the airway, as inhibition of COX-2 activity by selective inhibitors or down-regulation of COX-2 expression by gene knockout procedures [29-31] enhanced airway inflammation. The present findings on LPA-induced COX-2 expression and PGE2 secretion in this study extend our previous studies demonstrating that LPA plays a key regulatory role in airway inflammation and remodelling by stimulating expression and secretion of IL-8 [15-18] and IL-13Rα2 [19]. Furthermore, our observation that LPA-induced COX-2 gene and protein expression is dependent on Gi-coupled LPARs is in agreement with earlier studies on IL-8 secretion and IL-13Rα2 expression stimulated by LPA in HBEpCs [16,19].
LPA induces various cellular responses via transcriptional regulation of gene expression [15-19]. Previously, LPA-induced COX-2 expression has been demonstrated in renal mesangial [39], human colon cancer LoVo [40] and ovarian cancer cells [41]; however, mechanism(s) of regulation of COX-2 by LPA is unclear. The human COX-2 promoter region has been sequenced and contains a canonical TATA box and putative transcriptional regulatory elements such as AP-1/AP-2, Sp1 (stimulating protein-1), GATA, CRE (cAMP-response element), C/EBP and NF-κB [35]. The participation of NF-κB, AP-1 and C/EBPβ in changes of COX-2 expression with various stimuli in different cell types has been described [42-46]. Activation of C/EBPβ is essential for COX-2 expression in macrophages but not in fibroblasts [44]. In human lung epithelium, Streptococcus pneumonia-induced COX-2 expression was dependent on p38 MAPK-mediated NF-κB, but not JNK, activation [45]. However, the influenza virus-induced COX-2 expression was dependent on JNK, but not p38 MAPK-signalling pathway [46]. The present study, using specific inhibitors or siRNA for NF-κB, JNK/c-Jun or C/EBPβ,shows that activation of NF-κB, AP-1 and C/EBPβ pathways by LPA regulates COX-2 expression in HBEpCs. Although the involvement of NF-κB and AP-1 in LPA-induced COX-2 expression has been extensively demonstrated, the present study, for the first time, defines a novel pathway of C/EBPβ activation of COX-2 expression by LPA via PLD2 and PKCζ -mediated transactivation of EGFR in HBEpCs. Knockdown of EGFR with EGFR siRNA or inhibition of tyrosine phosphorylation of EGFR with AG1478 blocked LPA-induced phosphorylation of C/EBPβ, but not IκB or c-Jun phosphorylation, indicating that C/EBPβ, but not NF-κB and JNK/c-Jun, is the downstream signal of LPA-induced EGFR transactivation. In recent years, C/EBPβ has emerged as a key regulator of COX-2 expression by pro-inflammatory mediators [35,43,44,47]. The C/EBP family consists of six proteins that belong to the basic zipper transcriptional factors [48], and based on sequence homology are divided into two subgroups, namely C/EBPα, β and δ,andC/EBPγ , ε and ζ. While several of the proinflammatory mediators stimulate COX-2 expression via C/EBPβ and δ isoforms [47], the isoform(s) involved in COX-2 expression by LPA in HBEpCs remains to be determined.
Cross-talk between GPCRs and RTKs regulates the downstream signals of both receptors [14,18,20,23-25]. Though transactivation of EGFRs by GPCRs has been well documented in several cell types [18,23,24], the mechanism(s) regulating GPCR-mediated EGFR transactivation and the physiological relevance of this transactivation phenomenon has not been well established. We have shown previously that ligation of LPA to LPARs induces tyrosine phosphorylation of EGFRs [18] and PDGFRβ [14] as well as serine phosphorylation of c-Met [20] in HBEpCs. We also previously observed that secretion of the EGFR ligand HB-EGF, induced by LPA via Lyn kinase-dependent activation of MMP2/9, resulted in autocrine signalling through EGFR activation, and increased IL-8 expression and secretion [18]. Furthermore, the involvement of PLD2, but not PLD1, in transactivation of PDGFRβ by LPA in HBEpCs [14], and crosstalk between angiotensin II receptor and EGFR [49], has been demonstrated. In addition to the involvement of PKCδ in LPA-induced activation of Lyn, MMP2/9, HB-EGF release and tyrosine phosphorylation of EGFR [18], here we provide evidence for PLD2-dependent activation of PKCζ in EGFR transactivation and COX-2 expression by LPA in HBEpCs. Previously, transactivation of both c-Met and EGFR, and induction of COX-2 expression by LPA, have been demonstrated in human colon cancer LoVo cells; however, the link between LPARs and c-Met/EGFR in LPA-induced COX-2 expression was not established [40]. We have shown that LPA induces transinactivation of c-Met [20], whereas knockdown of c-Met with c-Met siRNA has no effect on LPA-induced COX-2 expression in HBEpCs (Y. Zhao, unpublished work). In contrast to c-Met transinactivation, transactivation of EGFR by LPA-induced COX-2 expression and PGE2 release was blocked by EGFR siRNA or the EGFR tyrosine kinase inhibitor AG1478. These results provide evidence supporting the role of LPA in regulating inflammation partly through cross-talk between LPARs and EGFR in airway epithelium. In addition, the PDGFRβ kinase inhibitor, AG1296 (1 μM) attenuated LPA-induced COX-2 expression (Y. Zhao, unpublished work), suggesting the cross-talk between LPARs and PDGFRβ contributed to LPA-induced COX-2 expression in HBEpCs. Future investigation will determine the mechanisms of the role of cross-talk between LPAR and PDGFRβ on COX-2 expression.
In summary, our results demonstrate that LPA induces COX-2 expression and PGE2 production through an EGFR transactivation-dependent C/EBPβ pathway and through EGFR transactivation-independent NF-κB and JNK/c-Jun pathways in HBEpCs. Furthermore, stimulation of PKCζ by PLD2 regulated LPA-induced EGFR transactivation, C/EBPβ activation and COX-2 expression (Figure 11). As COX-2 and PGE2 exhibit anti-inflammatory properties in the airway [29-31], our results indicate that enhanced COX-2 expression by LPA may have a protective role in airway inflammation and remodelling.
Figure 11. LPA induces transcriptional regulation of COX-2 expression through EGFR transactivation-dependent activation of C/EBPβ and transactivation-independent activation of NF-κB and JNK/c-Jun in HBEpCs.
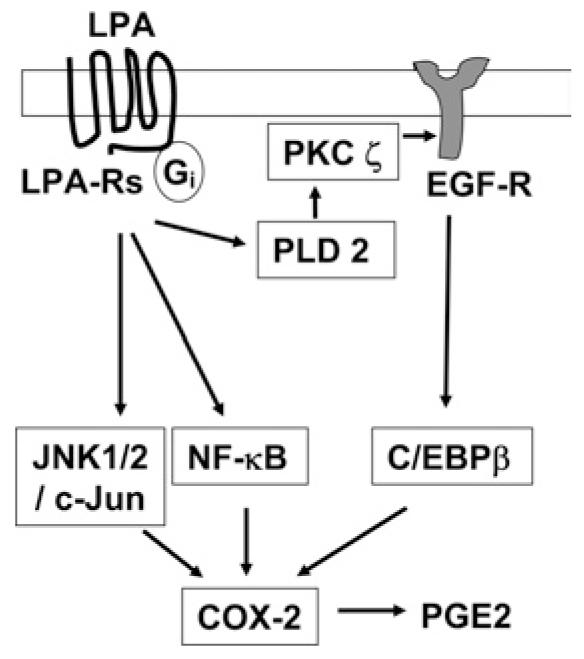
In the first pathway, LPA induces activation of C/EBPβ through EGFR transactivation involving PLD2 and PKCζ; whereas the second pathway involves LPA-induced regulation of NF-κB and c-Jun transcriptional factors, which are independent of EGFR transactivation. Both pathways go on to activate COX-2 expression and thus PGE2 is released.
Acknowledgments
This work was supported by an American Cancer Association Institutional Grant IRG-58-044-47 (to Y.Z.) and National Institutes of Health grants HL71152 and HL79396 (to V.N.). We thank Dr Motoi Ohba (Showa University, Japan) for providing the adenoviral dn-PKC isoforms.
Abbreviations used
- AP
activating protein
- BAL
bronchoalveolar lavage
- BEBM
bronchial epithelial basal medium
- BEGM
basal essential growth medium
- C/EBPβ
CCAAT/enhancer-binding protein β
- COX
cyclo-oxygenase
- dn
dominant negative
- EGF
epidermal growth factor
- EGFR
EGF receptor
- GPCR
G-protein-coupled receptor
- HB-EGF
heparin-binding EGF
- HBEpC
human bronchial epithelial primary cell
- IκB
inhibitory κB
- IL
interleukin
- IL-13Rα2
IL-13 receptor α2
- JNK
Jun N-terminal kinase
- LPA
lysophosphatidic acid
- LPAR
LPA receptor
- MAPK
mitogen-activated protein kinase
- MBP
myelin basic protein
- MMP2/9
matrix metalloproteinase 2 and 9
- MOI
multiplicity of infection
- NF-κB
nuclear factor-κB
- P0/1/2
passage zero/one/two
- PDGFR
platelet-derived growth factor receptor
- PG
prostaglandin
- PKC
protein kinase C
- PLD
phospholipase D
- hPLD1b
human PLD1b
- mPLD2
mouse PLD2
- PTx
pertussis toxin
- RTK
receptor tyrosine kinase
- RT—PCR
reverse transcription—PCR
- siRNA
small interference RNA.
REFERENCES
- 1.Patchell BJ, Dorscheid DR. Repair of the injury to respiratory epithelial cells characteristic of asthma is stimulated by Allomyrina dichotoma agglutinin specific serum glycoproteins. Clin. Exp. Allergy. 2006;36:585–593. doi: 10.1111/j.1365-2222.2006.02394.x. [DOI] [PubMed] [Google Scholar]
- 2.Sacco O, Silvestri M, Sabatini F, Sale R, Defilippi AC, Rossi GA. Epithelial cells and fibroblasts: structural repair and remodelling in the airways. Paediatr. Respir. Rev. 2004;5:S35–S40. doi: 10.1016/s1526-0542(04)90008-5. [DOI] [PubMed] [Google Scholar]
- 3.Hulsmann AR, Raatgeep HR, den Hollander JC, Stijnen T, Saxena PR, Kerrebijn KF, de Jongste JC. Oxidative epithelial damage produces hyperresponsiveness of human peripheral airways. Am. J. Respir. Crit. Care. Med. 1994;149:519–525. doi: 10.1164/ajrccm.149.2.8306055. [DOI] [PubMed] [Google Scholar]
- 4.Martin LD, Rochelle LG, Fischer BM, Krunkosky TM, Adler KB. Airway epithelium as an effector of inflammation: molecular regulation of secondary mediators. Eur. Respir. J. 1997;10:2139–2146. doi: 10.1183/09031936.97.10092139. [DOI] [PubMed] [Google Scholar]
- 5.Raeburn D, Webber SE. Proinflammatory potential of the airway epithelium in bronchial asthma. Eur. Respir. J. 1994;7:2226–2233. doi: 10.1183/09031936.94.07122226. [DOI] [PubMed] [Google Scholar]
- 6.Message SD, Johnston SL. Host defense function of the airway epithelium in health and disease: clinical background. J. Leukoc. Biol. 2004;75:5–17. doi: 10.1189/jlb.0703315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu SS, Chiu T, Rozengurt E. ANG II and LPA induce Pyk2 tyrosine phosphorylation in intestinal epithelial cells: role of Ca2+, PKC, and Rho kinase. Am. J. Physiol. Cell. Physiol. 2002;282:C1432–C1444. doi: 10.1152/ajpcell.00323.2001. [DOI] [PubMed] [Google Scholar]
- 8.Chou CH, Wei LH, Kuo ML, Huang YJ, Lai KP, Chen CA, Hsieh CY. Up-regulation of interleukin-6 in human ovarian cancer cell via a Gi/PI3K-Akt/NF-κB pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis. 2005;26:45–52. doi: 10.1093/carcin/bgh301. [DOI] [PubMed] [Google Scholar]
- 9.Xu YJ, Saini HK, Cheema SK, Dhalla NS. Mechanisms of lysophosphatidic acid-induced increase in intracellular calcium in vascular smooth muscle cells. Cell Calcium. 2005;38:569–579. doi: 10.1016/j.ceca.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 10.van Leeuwen FN, Giepmans BN, van Meeteren LA, Moolenaar WH. Lysophosphatidic acid: mitogen and motility factor. Biochem. Soc. Trans. 2003;31:1209–1212. doi: 10.1042/bst0311209. [DOI] [PubMed] [Google Scholar]
- 11.Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, Bittman R, Tigyi G, Aepfelbacher M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6931–6936. doi: 10.1073/pnas.96.12.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tokumura A. Physiological and pathophysiological roles of lysophosphatidic acids produced by secretory lysophospholipase D in body fluids. Biochim. Biophys. Acta. 2002;1582:18–25. doi: 10.1016/s1388-1981(02)00133-6. [DOI] [PubMed] [Google Scholar]
- 13.Georas S, Berdyshev E, Hubbard W, Morris A, Gorshkova IA, Usatyuk PV, Saatian B, Myers AC, Williams MA, Xiao HQ, et al. Lysophosphatidic acid is detectable in human bronchoalveolar lavage fluids at baseline and increased after segmental allergen challenge. Clin. Exp. Allergy. 2006;37:311–322. doi: 10.1111/j.1365-2222.2006.02626.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Cummings R, Zhao Y, Kazlauskas A, Sham JK, Morris A, Georas S, Brindley DN, Natarajan V. Involvement of phospholipase D2 in lysophosphatidate-induced transactivation of platelet-derived growth factor receptor-β in human bronchial epithelial cells. J. Biol. Chem. 2003;278:39931–39940. doi: 10.1074/jbc.M302896200. [DOI] [PubMed] [Google Scholar]
- 15.Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JGN, Watkins T, He D, Saatian B, Natarajan V. Protein kinase Cδ mediates lysophosphatidic acid-induced NF-κB activation and interleukin-8 secretion in human bronchial epithelial cells. J. Biol. Chem. 2004;279:41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Usatyuk PV, Cummings R, Saatian B, He D, Watkins T, Morris A, Spannhake EW, Brindley DN, Natarajan V. Lipid phosphate phosphatase-1 regulates lysophosphatidic acid-induced calcium release, NF-κB activation and interleukin-8 secretion in human bronchial epithelial cells. Biochem. J. 2005;385:493–502. doi: 10.1042/BJ20041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saatian B, Zhao Y, He D, Georas SN, Watkins T, Spannhake EW, Natarajan V. Transcriptional regulation of lysophosphatidic acid-induced interleukin-8 expression and secretion by p38 MAPK and JNK in human bronchial epithelial cells. Biochem. J. 2005;393:657–668. doi: 10.1042/BJ20050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase Cδ, Lyn kinase, and matrix metalloproteinases. J. Biol. Chem. 2006;28:19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y, He D, Zhao J, Wang L, Leff AR, Spannhake Wm., Georas S, Natarajan V. Lysophosphatidic acid induces interleukin-13 (IL-13) receptor α2 expression and inhibits IL-13 signaling in primary human bronchial epithelial cells. J. Biol. Chem. 2007;282:10172–10179. doi: 10.1074/jbc.M611210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, He D, Stern R, Usatyuk PV, Spannhake Wm., Salgia R, Natarajan V. Lysophosphatidic acid modulates c-Met redistribution and hepatocyte growth factor/c-Met signaling in human bronchial epithelial cells through PKC δ and E-cadherin. Cell. Signal. 2007;19:2329–2338. doi: 10.1016/j.cellsig.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ediger TL, Toews ML. Dual effects of lysophosphatidic acid on human airway smooth muscle cell proliferation and survival. Biochim. Biophys. Acta. 2001;1531:59–67. doi: 10.1016/s1388-1981(01)00084-1. [DOI] [PubMed] [Google Scholar]
- 22.Toews ML, Ediger TL, Romberger DJ, Rennard SI. Lysophosphatidic acid in airway function and disease. Biochim. Biophys. Acta. 2002;1582:240–250. doi: 10.1016/s1388-1981(02)00177-4. [DOI] [PubMed] [Google Scholar]
- 23.Shah BH, Baukal AJ, Shah FB, Catt KJ. Mechanisms of extracellularly regulated kinases 1/2 activation in adrenal glomerulosa cells by lysophosphatidic acid and epidermal growth factor. Mol. Endocrinol. 2005;19:2535–2548. doi: 10.1210/me.2005-0082. [DOI] [PubMed] [Google Scholar]
- 24.Mori K, Kitayama J, Shida D, Yamashita H, Watanabe T, Nagawa H. Lysophosphatidic acid-induced effects in human colon carcinoma DLD1 cells are partially dependent on transactivation of epidermal growth factor receptor. J. Surg. Res. 2006;132:56–61. doi: 10.1016/j.jss.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 25.Moughal NA, Waters CM, Valentine WJ, Connell M, Richardson JC, Tigyi G, Pyne S, Pyne NJ. Protean agonism of the lysophosphatidic acid receptor-1 with Ki16425 reduces nerve growth factor-induced neurite outgrowth in pheochromocytoma 12 cells. J. Neurochem. 2006;98:1920–1929. doi: 10.1111/j.1471-4159.2006.04009.x. [DOI] [PubMed] [Google Scholar]
- 26.Lin HY, Ballou LM, Lin RZ. Stimulation of the α1A adrenergic receptor inhibits PDGF-induced PDGF β receptor Tyr751 phosphorylation and PI 3-kinase activation. FEBS Lett. 2003;540:106–110. doi: 10.1016/s0014-5793(03)00233-3. [DOI] [PubMed] [Google Scholar]
- 27.Dempke W, Rie C, Grothey A, Schmoll HJ. Cyclooxygenase-2: a novel target for cancer chemotherapy? J. Cancer Res. Clin. Oncol. 2001;127:411–417. doi: 10.1007/s004320000225. [DOI] [PubMed] [Google Scholar]
- 28.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin. Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 29.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004;25:40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Oguma T, Asano K, Shiomi T, Fukunaga K, Suzuki Y, Nakamura M, Matsubara H, Sheldon HK, Haley KJ, Lilly CM, et al. Cyclooxygenase-2 expression during allergic inflammation in guinea-pig lungs. Am. J. Respir. Crit. Care. Med. 2002;165:382–386. doi: 10.1164/ajrccm.165.3.2103093. [DOI] [PubMed] [Google Scholar]
- 31.Nakata J, Kondo M, Tamaoki J, Takemiya T, Nohara M, Yamagata K, Nagai A. Augmentation of allergic inflammation in the airways of cyclooxygenase-2-deficient mice. Respirology. 2005;10:149–156. doi: 10.1111/j.1440-1843.2005.00687.x. [DOI] [PubMed] [Google Scholar]
- 32.Chun J, Rosen H. Lysophospholipid receptors as potential drug targets in tissue transplantation and autoimmune diseases. Curr. Pharm. Des. 2006;12:161–171. doi: 10.2174/138161206775193109. [DOI] [PubMed] [Google Scholar]
- 33.Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 34.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 35.Wu KK, Liou JY, Cieslik K. Transcriptional control of COX-2 via C/EBPβ. Arterioscler. Thromb. Vasc. Biol. 2005;25:679–685. doi: 10.1161/01.ATV.0000157899.35660.61. [DOI] [PubMed] [Google Scholar]
- 36.Chandrasekharan NV, Simmons DL. The cyclooxygenases. Genome Biol. 2004;5:241. doi: 10.1186/gb-2004-5-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asano K, Lilly CM, Drazen JM. Prostaglandin G/N synthase-2 is the constitutive and dominant isoform in cultured human lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 1996;217:L126–L131. doi: 10.1152/ajplung.1996.271.1.L126. [DOI] [PubMed] [Google Scholar]
- 38.Watkins DN, Peroni DJ, Lenzo JC, Knight DA, Garlepp MJ, Thompson PJ. Expression and localization of COX-2 in human airways and cultured airway epithelial cells. Eur. Respir. J. 1999;13:999–1007. doi: 10.1034/j.1399-3003.1999.13e12.x. [DOI] [PubMed] [Google Scholar]
- 39.Hahn A, Barth H, Kress M, Mertens PR, Goppelt-Struebe M. Role of Rac and Cdc42 in lysophosphatidic acid-mediated cyclo-oxygenase-2 gene expression. Biochem. J. 2002;362:33–40. doi: 10.1042/0264-6021:3620033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, Nagawa H. Lysophosphatidic acid transactivates both c-Met and epidermal growth factor receptor and induces cyclooxygenase-2 expression in human colon cancer LoVo cells. World J. Gastroenterol. 2005;11:5638–5643. doi: 10.3748/wjg.v11.i36.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Symowicz J, Adley BP, Woo MM, Auersperg N, Hudson LG, Stack MS. Cyclooxygenase-2 functions as a downstream mediator of lysophosphatidic acid to promote aggressive behavior in ovarian carcinoma cells. Cancer Res. 2005;65:2234–2242. doi: 10.1158/0008.5472.CAN-04-2781. [DOI] [PubMed] [Google Scholar]
- 42.Lee JC, Kundu JK, Hwang DM, Na HK, Surh YJ. Humulone inhibits phorbol ester-induced COX-2 expression in mouse skin by blocking activation of NF-κB and AP-1: IκB kinase and c-Jun-N-terminal kinase as respective potential upstream targets. Carcinogenesis. 2007;28:1491–1498. doi: 10.1093/carcin/bgm054. [DOI] [PubMed] [Google Scholar]
- 43.Faour WH, He Q, Mancini A, Jovanovic D, Antoniou J, Di Battista JA. Prostaglandin E2 stimulates p53 transactivational activity through specific serine 15 phosphorylation in human synovial fibroblasts. Role in suppression of c/EBP/NF-κB-mediated MEKK1-induced MMP-1 expression. J. Biol. Chem. 2006;281:19849–19860. doi: 10.1074/jbc.M601293200. [DOI] [PubMed] [Google Scholar]
- 44.Gorgoni B, Caivano M, Arizmendi C, Poli V. The transcription factor C/EBPβ is essential for inducible expression of the cox-2 gene in macrophages but not in fibroblasts. J. Biol. Chem. 2001;276:40769–40777. doi: 10.1074/jbc.M106865200. [DOI] [PubMed] [Google Scholar]
- 45.N’Guessan PD, Hippenstiel S, Etouem MO, Zahlten J, Beermann W, Lindner D, Opitz B, Witzenrath M, Rosseau S, Suttorp N, Schmeck B. Streptococcus pneumoniae induced p38 MAPK- and NF-κB-dependent COX-2 expression in human lung epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L1131–L1138. doi: 10.1152/ajplung.00383.2005. [DOI] [PubMed] [Google Scholar]
- 46.Mizumura K, Hashimoto S, Maruoka S, Gon Y, Kitamura N, Matsumoto K, Hayashi S, Shimizu K, Horie T. Role of mitogen-activated protein kinases in influenza virus induction of prostaglandin E2 from arachidonic acid in bronchial epithelial cells. Clin. Exp. Allergy. 2003;33:1244–1251. doi: 10.1046/j.1365-2222.2003.01750.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhu Y, Saunders MA, Yeh H, Deng WG, Wu KK. Dynamic regulation of cyclooxygenase-2 promoter activity by isoforms of CCAAT/enhancer-binding proteins. J. Biol. Chem. 2002;277:6923–6928. doi: 10.1074/jbc.M108075200. [DOI] [PubMed] [Google Scholar]
- 48.Wedel A, Ziegler-Heitbrock HW. The C/EBP family of transcription factors. Immunobiology. 1995;193:171–185. doi: 10.1016/s0171-2985(11)80541-3. [DOI] [PubMed] [Google Scholar]
- 49.Li F, Malik FU. Angiotensin II-induced Akt activation through the epidermal growth factor receptor in vascular smooth muscle cells is mediated by phospholipid metabolites derived by activation of phospholipase D. J. Pharmacol. Exp. Ther. 2005;312:1043–1054. doi: 10.1124/jpet.104.076588. [DOI] [PubMed] [Google Scholar]