

Abstract
Sphingosine 1-phosphate (S1P) regulates diverse cellular functions through extracellular ligation to S1P receptors, and it also functions as an intracellular second messenger. Human pulmonary artery endothelial cells (HPAECs) effectively utilized exogenous S1P to generate intracellular S1P. We, therefore, examined the role of lipid phosphate phosphatase (LPP)-1 and sphingosine kinase1 (SphK1) in converting exogenous S1P to intracellular S1P. Exposure of 32P-labeled HPAECs to S1P or sphingosine (Sph) increased the intracellular accumulation of [32P]S1P in a dose- and time-dependent manner. The S1P formed in the cells was not released into the medium. The exogenously added S1P did not stimulate the sphingomyelinase pathway; however, added [3H]S1P was hydrolyzed to [3H]Sph in HPAECs, and this was blocked by XY-14, an inhibitor of LPPs. HPAECs expressed LPP1–3, and overexpression of LPP-1 enhanced the hydrolysis of exogenous [3H]S1P to [3H]Sph and increased intracellular S1P production by 2–3-fold compared with vector control cells. Down-regulation of LPP-1 by siRNA decreased intracellular S1P production from extracellular S1P but had no effect on the phosphorylation of Sph to S1P. Knockdown of SphK1, but not SphK2, by siRNA attenuated the intracellular generation of S1P. Overexpression of wild type SphK1, but not SphK2 wild type, increased the accumulation of intracellular S1P after exposure to extracellular S1P. These studies provide the first direct evidence for a novel pathway of intracellular S1P generation. This involves the conversion of extracellular S1P to Sph by LPP-1, which facilitates Sph uptake, followed by the intracellular conversion of Sph to S1P by SphK1.
Sphingosine 1-phosphate (S1P)2 is a bioactive lipid mediator that plays an important role in regulating intracellular mobilization of Ca2+, cytoskeletal reorganization, cell growth, differentiation, motility, angiogenesis, and survival (1-5). In biological fluids such as plasma, S1P is present at 0.2–0.5 μM, whereas higher concentrations (1–5 μM) in serum are attributed to enhanced release from activated platelets (1, 5). S1P is generated by phosphorylation of free sphingosine (Sph) by two sphingosine kinases (SphKs) 1 and 2, which are highly conserved enzymes present in most of the mammalian cells and tissues (6-9). Cellular levels of S1P are regulated through its formation via SphKs and by its degradation by S1P lyase (SPL) (10-12), S1P phosphatases (SPPs) (13-15), and intracellular lipid phosphate phosphatases (LPPs) (16-18). Platelets lack S1P lyase (19), but in most cells the balance between S1P formation and degradation translates to low basal levels of intracellular S1P. S1P exerts dual actions in cells; it acts as an intracellular second messenger and functions extracellularly as a ligand for a family of five G-protein-coupled receptors formerly known as endothelial differentiation gene (Edg) receptors. To date, five G-protein-coupled receptors, S1P-1 (Edg-1), S1P-2 (Edg-5), S1P-3 (Edg-3), S1P-4 (Edg-6), and S1P-5 (Edg-8), have been identified. All these receptors bind to and are activated by extracellular S1P and dihydro-S1P (1, 5, 20-22). In the vessel wall extracellular S1P is a potent stimulator of angiogenesis (23, 24) and is a major chemotactic factor for endothelial cells (ECs). Recently, circulating S1P and the immunosuppressive drug FTY720, which is also phosphorylated by SphKs, have been implicated in lymphocyte homing and immunoregulation (25, 26). In addition to its extracellular action, S1P functions as an intracellular second messenger in the regulation of Ca2+ mobilization and suppression of apoptosis (27, 28).
Unlike platelets (29, 30), ECs do not secrete large amounts of S1P upon stimulation by agonists such as TNF-α or thrombin (1, 31). Although TNF-α stimulates endothelial SphK by ∼2-fold, it is unclear if intracellular S1P levels are increased in ECs (31). During studies on intracellular S1P formation, we observed that exogenously added S1P was rapidly converted to intracellular S1P in human lung ECs. This suggested the existence of a novel but yet to be defined pathway whereby S1P could be taken by ECs from the circulation and used for intracellular signaling. Recently, several LPPs have been described in mammalian cells, and they are partly expressed as ectoenzymes on the cell surface (32-35). The LPPs could hydrolyze S1P (16-18), which could facilitate the rapid uptake of Sph by ECs. Intracellular SphK1 and SphK2 could then synthesize intracellular S1P and influence angiogenesis, EC motility, or survival (23, 24, 36, 37). In this study we demonstrate that in lung ECs exogenous S1P is a preferred source for the intracellular production of S1P compared with several agonists that stimulate sphingomyelinase activity. Our results also show that the exogenous S1P is hydrolyzed by ecto-LPP-1 present on human lung ECs to Sph, which is subsequently converted by SphK1 to intracellular S1P.
EXPERIMENTAL PROCEDURES
Materials
HPAECs, EBM-2 basal media, and Bullet kit were obtained from Clonetics (San Diego, CA). Phosphate-buffered saline was from Biofluids (Rockville, MD). Ampicillin, fetal bovine serum (FBS), trypsin, MgCl2, EGTA, Tris-HCl, Triton X-100, sodium orthovanadate, aprotinin, Tween 20, Me2SO, antibodies to LPP-2, LPP-3, and c-Myc tag (9E10), and Bacillus cereus sphingomyelinase were from Sigma. D-erythro-C18 Sph, D-erythro-S1P, D-erythro-C17-S1P, and D-erythro-dihydro-S1P were from Avanti Polar Lipids (Alabaster, AL). N,N-Dimethylsphingosine (DMS) was from Biomol Research Laboratory (Plymouth Meeting, PA). XY-14 was obtained from Echelon (Salt Lake City, UT). SMART pool siRNA against human SphK1, SphK2, SPP1, SPL, and LPP-1 mRNA and scrambled siRNA were purchased from Dharmacon (Lafayette, CA). Antibodies to SphK1 and SphK2 and to FLAG tag were obtained from Oncogene Research Products and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. The antibody to LPP-1 was kindly provided by Dr. Andrew Morris (University of Kentucky). Horseradish peroxidase-conjugated goat anti-rabbit, anti-mouse were purchased from Invitrogen/Molecular Probes (Eugene, OR). The enhanced chemiluminescence (ECL) kit was from Amersham Biosciences. Silica gel 60TLC plastic sheets were from EM Chemicals Science (Gibbstown, NJ). [γ-32P]ATP in 10 mM Tricine buffer (specific activity 6000 Ci/mmol) was purchased from PerkinElmer Life Sciences. D-Ribophytosphingosine 1-phosphonate was synthesized as described previously (38).
Endothelial Cell Culture
For HPAECs, passages between 5 and 8 were grown to contact-inhibited monolayers with typical cobblestone morphology in EGM-2 complete media with 10% FBS, 100 units/ml penicillin and streptomycin in a 37 °C incubator under 5% CO2, 95% air atmosphere (39, 40). Cells from T-75 flasks were detached with 0.05% trypsin and resuspended in fresh complete medium and cultured in 35- or 60-mm dishes or on glass coverslips for immunofluorescence studies. All cells were starved overnight in EGM-2 medium containing 1% FBS before exposure to vehicle or agonists.
Generation of Adenoviral Vectors
The SphK1 complete cDNA (GI: 21361087) with FLAG-tag DNA at the C terminus, the SphK1 dominant negative G82D with FLAG-tag DNA at C terminus, the SphK2 complete cDNA (GI: 21361698) with c-Myc-tag DNA at C terminus, and mouse LPP-1 (GI: 45592927) with c-Myc-tag DNA at the N terminus were inserted into an adenoviral expression vector with cytomegalo-virus promoter. The recombinant plasmids were linearized and propagated in HEK 293 cells, and the high titer-purified preparations (∼1010 plaque-forming units/ml) were generated by the University of Iowa Gene Transfer Vector Core.
Infection of HPAECs with Adenoviral Vectors
Infection of HPAECs (∼60% confluence) with purified adenoviral empty vectors, adenoviral vectors containing cDNA for SphK1 wild type (wt) or SphK2 wt or SphK1 mutant, and wild type LPP-1 were carried out in 6-well plates as described previously (40, 41). After infection with different m.o.i. in 1 ml of EGM for 24 h, the virus containing medium was replaced with EBM, and the experiments were carried out.
Transfection of HPAECs with siRNA
HPAEC grown to ∼50% confluence in 6-well plates were transfected with Gene Silencer® (Gene Therapy System, San Diego, CA) transfecting agent with target specific siRNA (50 nM) and scrambled siRNA (50 nM) in serum-free EBM-2 medium according to the manufacturer’s recommendation. After 3 h post-transfection, 1 ml of fresh complete EGM-2 medium containing 10% FBS was added, and the cells were cultured for an additional 72 h for analysis of SphK1, SphK2, SPP1, SPL, LPP-1, LPP-2, and LPP-3 mRNA by real-time RT-PCR.
RNA Isolation
Total RNA was isolated from cultured HPAECs using TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions. RNA was quantified spectrophotometrically, and samples with an absorbance of ≥1.8 measured at 260/280 nm were analyzed by real-time RT-PCR.
Quantitative RT-PCR and Real-time RT-PCR
RNA (1 μg) was reverse-transcribed using a cDNA synthesis kit (Bio-Rad), and real-time PCR and quantitative PCR were performed to assess expression of the SphK1, SphK2, SPP1, SPL, LPP-1, LPP-2, and LPP-3 using primers designed for the human mRNA sequences. Amplicon expression in each sample was normalized to its 18 S RNA content. The relative abundance of target mRNA in each sample was calculated as 2 raised to the negative of its threshold cycle value times 106 after being normalized to the abundance of its corresponding 18 S (e.g. 2-(sphingosine kinase 1 threshold cycle)/2-(18 S threshold cycle) × 106).
Measurement of Intracellular [32P]S1P Generation
Control or HPAECs (35-mm dishes) infected with adenoviral vectors containing cDNA for wild type SphK1, SphK2 wild type, or LPP-1 (10 m.o.i. for 48 h) were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free Dulbecco’s modified Eagle’s medium (DMEM) media for 3 h. The media was then aspirated, and cells were challenged with 1 ml of minimum Eagle’s medium alone or media containing S1P or Sph (1 μM)in the presence of 0.1% BSA for 15–60 min. Lipid labeling was terminated by the addition of 100 μl of 12 M HCl followed by 2 ml of methanol. Cells were harvested with a cell scraper, the total extract was transferred to 15-ml glass tubes, and lipids were partitioned after vortexing into the chloroform phase by the addition of 2 ml of chloroform and 700 μl of 1 M HCl (to give a final ratio of 1:1:0.9 of chloroform:methanol:acidic aqueous phase). After vortexing, the lower (chloroform) phase was dried under nitrogen, and the lipid extracts were subjected thin layer chromatography (TLC). Lipid extracts were applied at 10 cm from the bottom of 20-cm plastic baked silica gel 60 plates. The plate was developed in chloroform/methanol/NH4OH (65:35:7.5, v/v/v), air-dried for 20 min, and then cut 2.0 cm above the origin. This removed neutral lipids and most of the zwitterionic phospholipids, whereas several acidic phospholipids such as phosphatidic acid, S1P, lysophosphatidate and ceramide 1-phosphate remained near the origin. The top part of the cut plate was discarded, and the bottom of the plate was then developed in the reverse direction with chloroform/methanol/glacial acetic acid/acetone/water (10:2:3:4:1, v/v/v/v). Dried plates were subjected to autoradiography, the area corresponding to labeled S1P was excised, and radioactivity was determined by liquid scintillation counting. The data were normalized to total radioactivity in the lipid extract or total cells on the monolayer (42).
Lipid Extraction and Sample Preparation for LC-MS/MS Analysis of S1P, Dihydro-S1P, and Sph
Cellular lipids were extracted by a modified Bligh and Dyer procedure under acidic conditions using 0.1 M HCl and C17-S1P (40 pmol) and C17-Sph (30 pmol), which were added as internal standards during the lipid extraction step. The lipid extracts were dissolved in ethanol (200 μl), and aliquots were analyzed for total lipid phosphate (40, 42) and then subjected to LC-MS/MS for quantification of S1P, dihydro-S1P, and Sph as described previously (40).
S1P Hydrolysis to Sph by Ecto LPP Activity in HPAECs
HPAECs were grown on 35-mm dishes to ∼90% confluence. S1P (1 μM) (unlabeled plus [3H]S1P, 100,000 dpm per dish; specific activity 2.2 × 103 dpm/pmol) complexed to 0.1% BSA in 1 ml of EGM medium was added to HPAECs, and the cells were incubated at 37 °C for various times (0–60 min). After the media (1 ml) were transferred to glass tubes, 2 ml of methanol, 1 M HCl (100:1 v/v) was added, and lipids were extracted by the addition of 2 ml of chloroform and 0.8 ml of H2O and centrifuged to separate the chloroform and methanol/aqueous phases. The lower (chloroform) phase was transferred to vials and evaporated under N2, and the hydrolysis of [3H]S1P to [3H]Sph was determined after separation by TLC in the presence of unlabeled sphingosine, added as carriers to the total lipid extracts (42). The plates were developed in chloroform/methanol/NH4OH (65:35:7.5, v/v/v) and exposed to iodine vapors to identify Sph band, and radioactivity associated with Sph was determined by liquid scintillation counting. The hydrolysis of [3H]S1P to [3H]Sph was expressed as dpm/dish or pmol/dish.
Surface Labeling of ECs with Biotin
HPAECs (passage 6) grown on T-75-cm2 flasks were infected for 24 h with adenoviral empty vector or adenoviral mLPP-1 on the C terminus with Myc (10 m.o.i.). Media were removed, and cells were washed twice with ice-cold phosphate-buffered saline. Surface labeling of cells with biotin was performed with the Cell Surface Protein Isolation kit (Pierce) as per the manufacturer’s recommendation. Briefly, HPAECs were incubated with 10 ml of ice-cold phosphate-buffered saline containing sulfo-NHS-SS-biotin (0.25 mg/ml) for 30 min at 4 °C with constant rocking, and cells were collected, and lysed by sonication on ice for 5 s using lysis buffer (Pierce). The cell lysates were incubated with Immobilized NeutrAvidin™ gel for 60 min at room temperature with end-over mixing, and surface proteins were boiled in 400 μl of SDS-PAGE sample buffer containing 50 mM dithiothreitol and analyzed by Western blotting with anti-Myc(10E9) or anti-LPP-1 antibodies.
Measurement of [3H]Ceramide Formation
HPAECs grown to ∼90% confluence in 35-mm dishes were labeled with L-[3H]serine (100 μCi/ml) in serine-free medium for 24 h to label sphingomyelin. L-[3H]Serine was removed by washing, and cells were challenged with medium alone or medium containing TNF-α (20 ng/ml), S1P (1 μM), H2O2 (100 μM), or bacterial sphingomyelinase (1 units/ml) for 30 min. The medium was aspirated, cells were scraped into 2 ml of methanol, 1 M HCl (100:1 v/v), and lipids were extracted by the addition of 2 ml of chloroform and 1.8 ml of 1 M HCl. Tubes were then centrifuged to separate the chloroform and methanol/aqueous phases. The lower (chloroform) phase was removed and dried under nitrogen, and the formation of [3H]ceramide was determined after separation by TLC with chloroform/methanol/glacial acetic acid/acetone/water (10:2:3:4:1 v/v/v/v). [3H]Ceramide was identified with an authentic standard visualized under I2 vapors, and radioactivity was determined by liquid scintillation counting.
Western Blotting
Cells were rinsed twice with ice-cold phosphate-buffered saline and lysed in 200 μl of buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EGTA, 5 mM β-glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Cell lysates were incubated at 4 °C for 10 min, sonicated on ice for 10 s, and centrifuged at 5000 × g for 5 min at 4 °C in a micro-centrifuge. Protein concentrations were determined with a BCA protein assay kit (Pierce) using BSA as standard. Equal amounts of protein (20 μg) or concentrated media (20 μl) were analyzed on 10% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes, blocked with 5% (w/v) BSA in TBST (25 mM Tris-HCl, pH 7.4, 137 mM NaCl, and 0.1% Tween 20) for 1 h, and incubated with primary antibodies in 5% (w/v) BSA in TBST for 1–2 h at room temperature. The membranes were washed at least 3 times with TBST at 15-min intervals and then incubated with mouse, rabbit, or goat horseradish peroxidase-conjugated secondary antibody (1:3000) for 1 h at room temp. The membranes were developed with the enhanced chemiluminescence detection system according to the manufacturer’s instructions.
Statistical Analyses
The results were analyzed by a Student-Newman-Keuls test. Data are expressed as the means ± S.D. of triplicate samples from two or more experimental groups, and statistical significance was taken to be p < 0.05.
RESULTS
Agonist-induced Generation of S1P in ECs
Although activated platelets generate and secrete micromolar levels of S1P, several other circulating and non-circulating cells have the ability to produce intracellular S1P. S1P is a key angiogenic factor in the endothelium; however, the ability of ECs to generate intracellular S1P and its signaling effects has not been well defined. Therefore, several agonists that activate EC signal transduction and their effects on intracellular S1P formation were tested. As shown in Fig. 1A, among numerous agonists such as thrombin, vascular endothelial growth factor, phorbol ester, the calcium ionophore A23187, and TNF-α, only TNF-α increased [32P]S1P production (∼1.4-fold increase over control) in HPAECs. Interestingly, incubation of 32P-labeled HPAECs with human PPP or lipids derived from PPP showed a statistically significant increase in intracellular S1P production (Fig. 1B). By contrast, charcoal-treated PPP, as compared with lipids from charcoal treated PPP, failed to show any significant change in intracellular S1P (Fig. 1B). Analysis of the lipid extracts derived from the PPP fraction by LC-MS/MS revealed the presence of substantial amounts of S1P (1973 ± 97 pmol/ml of PPP), whereas charcoal treatment of PPP reduced the S1P levels by ∼75% (597 ± 38 pmol/ml) (Fig. 1C). Sph levels in the PPP fraction were very low (23 ± 6 pmol/ml of PPP), and charcoal treatment of PPP reduced the sphingosine levels by ∼50% (10 ± 2 pmol/ml of PPP). These results indicate that circulating Sph and/or S1P could serve as a source of intracellular S1P in ECs.
FIGURE 1. Agonist-induced intracellular generation of S1P in HPAECs.
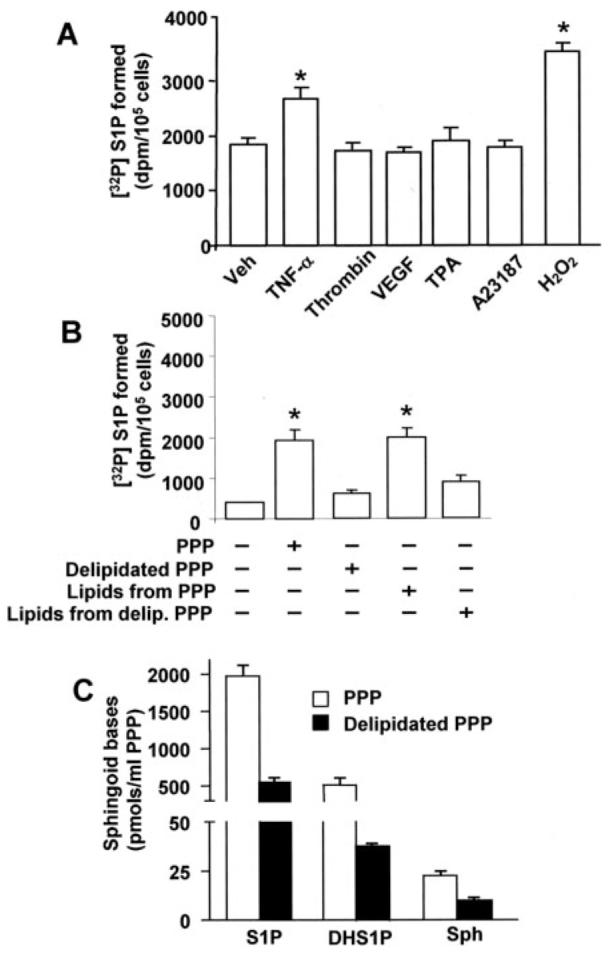
Panel A, HPAECs grown to ∼95% confluence in 35-mm dishes were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h. The radioactive medium was aspirated, and cells were rinsed in DMEM without serum and challenged with medium alone or medium containing TNF-α (20 ng/ml), thrombin (10 ng/ml), vascular endothelial growth factor (VEGF;20 ng/ml), phorbol ester (TPA,25nM), ionophore A23187 (1 μM), or H2O2 (250 μM). In panel B cells were treated with human PPP or lipids extracted from human PPP for 30 min. The reaction was terminated by the addition of 100 μl of 12 M HCl; lipids were extracted under acidic condition and separated by TLC for [32P]S1P generation. In panel C total lipids were extracted from PPP or prelipidated PPP, and sphingoid bases were quantified by LC-MS/MS as described under “Experimental Procedures.” Values are the means ± S.D. of six independent experiments. *, significantly different from cells exposed to medium alone (p < 0.05). Veh, vehicle.
Conversion of Exogenous Sph and S1P to Intracellular S1P
HPAECs were labeled with [32P]orthophosphate for 3 h and then exposed to either Sph or S1P, and the medium as well as the total cell lysates were analyzed to detect the relative [32P]S1P production. As shown in Fig. 2, A and B, exposure of cells to either Sph or S1P resulted in the accumulation of [32P]S1P in a dose- and time-dependent manner. At all concentrations of the exogenously added substrate, formation of [32P]S1P from Sph was higher than that of S1P (Figs. 2, A and B). Furthermore, analysis of the medium and cells exposed to either exogenous S1P or Sph showed that >95% of the [32P]S1P generated was recovered in the total cell lysates with statistically insignificant levels present in the medium (Fig. 2C). In independent experiments, cells were incubated with Sph, and total cell lysates and medium were analyzed for S1P using LC-MS/MS (40). As shown in Fig. 2D, no S1P was detected in the medium, and >95% of the S1P generated was recovered in total cell lysates. Next, we investigated the ability of various mammalian cells to convert exogenously added S1P to intracellular S1P. Among the various cell types we investigated, which included epithelial cells, monocytes, and macrophages, only the ECs from various vascular beds demonstrated high rates of intracellular S1P production; however, all the cell types investigated utilized exogenous Sph to generate intracellular S1P (Table 1). These results show that ECs from macro- and microvessels exhibit increased generation of S1P from extracellular S1P. Little of this S1P was released to external milieu.
FIGURE 2. Dose- and time-dependent formation of intracellular S1P from exogenous sphingosine or S1P.

HPAECs grown to ∼95% confluence in 35-mm dishes were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h, the radioactive medium was aspirated, and cells were rinsed in DMEM without serum. In panel A, cells were challenged with medium alone or medium containing different concentrations of Sph or S1P in the presence of 0.1% BSA for 30 min; in panel B cells were challenged with medium alone or medium Sph (1 μM) or S1P (1 μM) in the presence of 0.1% BSA for 5, 10, 15, 30, and 60 min. The medium was removed, cell lipids were extracted, and intracellular [32P]S1P was quantified. In panel C, HPAECs labeled with [32P]orthophosphate (20 μCi/ml) for 3 h as described in panel A were exposed to either Sph (1 μM) or S1P (1 μM) for 30 min, and the lipids were extracted and quantified from the media and cells. The distribution of [32P]S1P in medium and cells was measured. Panel D shows LC-MS/MS quantification of S1P in media and HPAECs after exposure to Sph (100 nM) for 30 min. The values are the means ± S.D. of six independent experiments. *, significantly different compared with cells exposed to medium alone (p < 0.05); **, significantly different compared with cells exposed to medium alone (p < 0.01). NS, not significant; Veh, vehicle.
TABLE 1. Intracellular [32P]S1P formation in different cell types.
Confluent cells were labeled with [32P]orthophosphate (20 μCi/ml) in DMEM phosphate-free medium for 3 h, radioactive medium was aspirated, and cells were rinsed with medium and challenged with medium alone or medium containing Sph (1 μM) or S1P (1 μM) complexed to 0.1% BSA for 30 min. Lipids were extracted, and radioactivity associated with S1P was determined. Values are the means ± S.E. of three independent experiments in triplicate. HLMVECs, human lung microvascular endothelial cells; HBEpCs, human bronchial epithelial primary cells.
| Cell type | [32P]S1P formed |
||
|---|---|---|---|
| Vehicle | Sphingosine | S1P | |
| dpm/105 cells | |||
| HPAECs | 754 ± 66 | 2960 ± 193 | 1843 ± 26 |
| HLMVECs | 577 ± 108 | 1572 ± 216 | 1988 ± 69 |
| HUVECs | 518 ± 35 | 2096 ± 138 | 1751 ± 163 |
| HBEpCs | 650 ± 146 | 1853 ± 260 | 984 ± 70 |
| A549 | 404 ± 58 | 1140 ± 90 | 868 ± 66 |
| H441 | 988 ± 94 | 2992 ± 170 | 1340 ± 260 |
Specificity of Sphingoid Bases on Intracellular Formation of Sphingoid Phosphates in HPAECs
Next we investigated the ability of HPAECs to utilize different sphingoid bases provided exogenously in the intracellular generation of the corresponding sphingoid phosphates. As shown in Table 2, exogenous S1P was a better substrate than dihydro-S1P and phyto-S1P, whereas the non-hydrolysable analog, D-Ribophytosphingosine phosphonate (PHS-C-P) could not be converted to phytosphingosine. Conversion of Sph to S1P was higher than that of dihydro-Sph to dihydro-S1P. These results indicate that among the various sphingoid phosphates investigated, S1P is a preferred substrate, and hydrolysis of the sphingoid phosphate to a free sphingoid base is a necessary step in the intracellular production of S1P by HPAECs.
TABLE 2. Intracellular formation of sphingoid phosphates in HPAECs.
HPAECs (∼90% confluence) were labeled with [32P]orthophosphate (20 μCi/ml) in DMEM phosphate-free medium for 3 h, medium was aspirated, and the cells were rinsed in medium before challenge with medium alone or medium containing Sph (1 μM), dihydro-Sph (1 μM), D-ribophytosphingosine 1-phosphonate (1 μM), S1P (1 μM), dihydro-S1P (1 μM), or phyto-S1P (1 μM) complexed to 0.1% BSA for 30 min. Lipids were extracted, and accumulation of sphingoid phosphates was quantified. Values are the means ± S.E. of three independent experiments in triplicate.
| Substrate |
32P-Labeled sphingoid phosphate formed |
% Control |
|---|---|---|
| dpm/105 cells | ||
| Vehicle | 768 ± 74 | 100 |
| Sph | 5519 ± 215a | 719 |
| Dihydro-Sph | 3752 ± 117a | 489 |
| 4-D-Ribophytosphingosine 1-phosphonate |
858 ± 199 | 112 |
| S1P | 3023 ± 164a | 394 |
| Dihydro-S1P | 2152 ± 76a | 280 |
| Phyto-S1P | 1367 ± 165a | 178 |
Significantly different from cells treated with medium alone (p < 0.05).
Extracellular S1P Does Not Stimulate Ceramide Formation in HPAECs
The intracellular generation of S1P from extracellular S1P could arise via activation of sphingomyelinase (generating ceramide and subsequently Sph via ceramidase and S1P via SphK) or via LPPs (forming Sph and then S1P via SphK) in ECs. To determine whether extracellular S1P stimulated sphingomyelinase to generate ceramide, HPAECs were labeled with 10 μML-[3H]serine (100 μCi/ml) in EGM-2 medium containing growth factors and 10% FBS for 24 h. Cells were rinsed in complete medium before being exposed to either vehicle, S1P (1 μM), TNF-α (100 ng/ml), H2O2 (250 μM), or neutral sphingomyelinase (B. cereus, 1 unit/ml) for 30 min. After the lipids were extracted, [3H]ceramide formed was separated by TLC, and the radioactivity was quantified. Although TNF-α, H2O2, and sphingomyelinase treatment resulted in increased [3H]ceramide formation from cells labeled in sphingomyelin with L-[3H]serine, cells challenged with S1P showed no change in [3H]ceramide accumulation (Fig. 3). These results indicate that exogenous S1P does not stimulate hydrolysis of sphingomyelin to ceramide via sphingomyelinase in HPAECs.
FIGURE 3. Agonist-induced generation of [3H]ceramide in HPAECs.
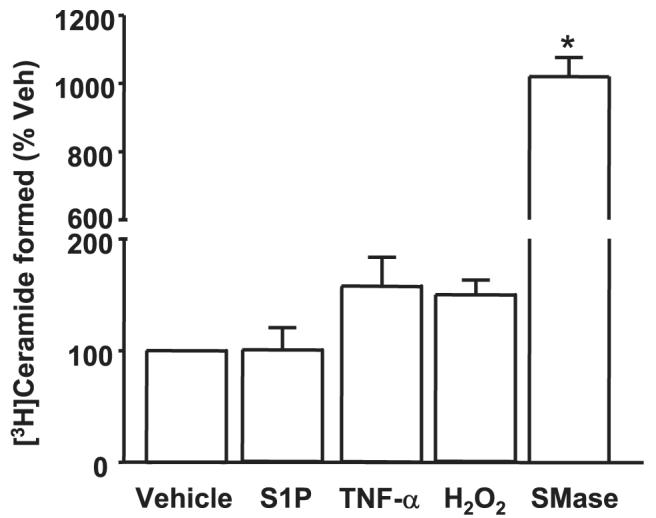
HPAECs grown to ∼95% confluence in 35-mm dishes were labeled with L-[3H]serine (100 μCi/ml) in serine-free DMEM for 24 h. The radioactive medium was aspirated, and cells were rinsed and challenged with medium alone or medium containing S1P (1μM), TNF-α (20 ng/ml), H2O2 (250μM), or sphingomyelinase (SMase; 1 units/ml) for 30 min. Lipids were extracted, and accumulation of [3H]ceramide was calculated from the averages of two independent experiments in triplicate. Veh, vehicle.
Hydrolysis of Exogenous [3H]S1P to [3H]Sph Is Mediated by LPP Activity
We investigated if exogenous S1P was hydrolyzed to Sph by HPAECs. Cells (∼95% confluent) were exposed to [3H]S1P (105 dpm, specific activity 100 dpm/pmol) for varying times periods, cells plus medium were extracted with 1-butanol under acidic conditions, and the radioactivity associated with [3H]Sph and non-hydrolyzed [3H]S1P was determined after separation by TLC. The addition of [3H]S1P to HPAECs resulted in the generation of [3H]Sph; ∼12% of the added [3H]S1P was hydrolyzed to [3H]Sph in 1 h (Fig. 4A). The generation of Sph was correlated with the loss of [3H]S1P that was added to the cells. A small percent of [3H]Sph formed (<0.1%) was incorporated into sphingomyelin via the de novo pathway (data not shown). These results show that exogenous S1P is hydrolyzed to free Sph, most likely by LPPs present on the cell surface of HPAECs. This was confirmed by using the compound XY-14 as an inhibitor of LPPs (43). HPAECs were treated with 10 μM XY-14 for 5 min before the addition of [3H]S1P (1 μM, specific activity (SA) = 100 dpm/pmol) and then incubated for 5, 30, and 60 min. Fig. 4B shows that XY-14 inhibited the hydrolysis of [3H]S1P to [3H]Sph.
FIGURE 4. Time course and effect of XY-14 on hydrolysis of [3H]S1P in HPAECs.

In panel A, HPAECs (∼90% confluence) in 35-mm dishes were incubated with [3H]S1P (1 μM; specific radioactivity, 100 dpm/pmol) complexed with 0.1% BSA in BEGM medium (Clonetics) for up to 60 min. At each time point the medium was removed, and lipids were extracted and analyzed for [3H]Sph and [3H]S1P by TLC. Values are from two independent experiments in triplicate and expressed as dpm/dish. In panel B, HPAECs were pretreated with XY-14 (10 μM) before exposure to [3H]S1P (1 μM; specific activity 100 dpm/pmol) for 5, 30, and 60 min. At each time point, medium was removed, and the formation of [3H]Sph from [3H]S1P was quantified. Values are from three independent experiments in triplicate and are expressed as a percentage of total radioactivities (dpm) in the lipid extract.
To further investigate the role of LPPs in intracellular generation of S1P from exogenous S1P, we designed primers for LPP-1, -2, and -3 based on previous work on human LPPs (35). Using these primers, we found that RT-PCR of total RNA from HPAECs showed expression of LPP-1, -2, and -3 and SPP-1 transcripts, with β-actin as an internal standard (data not shown). The RT-PCR data were quantified by real-time RT-PCR (Fig. 5A). Western blotting of cell lysates with specific LPP antibodies showed protein expression of all the three LPPs in HPAECs (Fig. 5B). These results show that HPAECs express all three LPP isoforms.
FIGURE 5. Detection of LPPs by real-time RT-PCR and Western blotting in HPAECs.
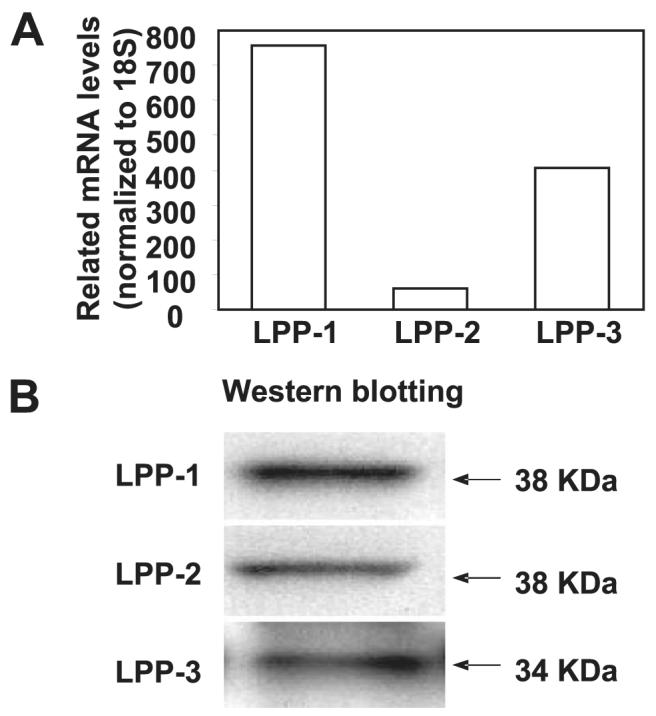
In panel A total RNA was extracted from HPAECs, and expression of LPPs was normalized to 18 S and quantified by real-time RT-PCR. Values are the average of three independent experiments. In panel B cell lysates (30 μg of protein) were subjected to SDS-PAGE and analyzed by Western blotting with anti-LPP-1, -LPP-2, and -LPP-3 antibodies. The figure shows a representative Western blot of three independent experiments.
Next, the role of LPPs in intracellular production of S1P was investigated by overexpression of wild type LPP-1 followed by exposure of cells to exogenous [3H]S1P. HPAECs were infected with cDNA for Myc-tagged human LPP-1 (10 m.o.i.) using adenoviral constructs for 24 h before the addition of [3H]S1P (1 μM, SA = 100 dpm/pmol) for 30 min. As shown in Fig. 6A, Myc-tagged mLPP-1 was efficiently overexpressed in HPAECs after 24 h of adenoviral infection, as evidenced by Western blotting. In unstimulated cells, the overexpressed mLPP-1 wild type was localized at the cell surface and also in intracellular organelles, including the perinuclear membrane, as evidenced by confocal immunocytochemistry with anti-Myc antibody (Fig. 6B). To further evaluate localization of LPP-1 to the cell surface, we examined susceptibility of the protein to labeling with a cell-impermeant biotin derivative. As shown in Fig. 6C, in resting Myc-tagged LPP-1 overexpressing or control cells, we detected Myc or LPP-1 in Western blots of avidin-captured proteins. These results indicate that the overexpressed and native LPP-1 are present at the surface of resting HPAECs. Having established the surface localization of LPP-1, we investigated the effect of overexpression of Myc-tagged LPP-1 on hydrolysis of [3H]S1P. Overexpression of LPP-1 enhanced the hydrolysis of exogenously added S1P to Sph (by ∼2-fold) compared with vector-infected cells (Fig. 6D). These results confirm a role for LPPs in the hydrolysis of exogenous S1P to Sph in HPAECs.
FIGURE 6. Effect of overexpression of adenoviral construct of LPP-1 wt on hydrolysis of [3H]S1P in HPAECs.

HPAECs (∼60% confluence in 35-mm dishes) were infected with adenoviral construct (10 m.o.i.) for the empty vector or vector containing cDNA for Myc-tagged mLPP-1 for 24 h. Panel A, cell lysates were subjected to SDS-PAGE and Western blotting with anti-LPP-1 antibody (Ab). Panel B, HPAECs grown on glass coverslips to ∼60% confluence were infected with cDNA for Myc-tagged mLPP-1 for 24 h, and cells were subjected to immunostaining with anti-Myc antibody (9E10) and examined by confocal fluorescent microscopy. Panel C, HPAECs (passage 6) grown on T-75 cm2 flasks were infected with adenoviral construct (10 m.o.i.) for the empty vector or vector containing cDNA for Myc-tagged mLPP-1 for 24 h. Surface labeling of cells with biotin was performed with the Cell Surface Protein Isolation kit (Pierce) as described under “Experimental Procedures.” Surface proteins were analyzed by Western blotting with anti-Myc (10E6) or anti-LPP-1 antibodies. Panel D, [3H]S1P (1 μM; specific activity 100 dpm/pmol) complexed with 0.1% BSA in BEGM was added to each dish, and hydrolysis was examined at the end of a 60-min incubation. Lipids were extracted and separated by TLC. Values for sphingosine production are the means ± S.D. of three independent experiments. *, significantly different compared with empty vector infected cells (p < 0.05). IB, immunoblot.
Overexpression of LPP-1 Potentiates Intracellular [32P]S1P Formation
Because the above results established a role for LPPs in hydrolyzing exogenous S1P, we examined the effect of overexpressing LPP-1 wt on the intracellular production of S1P. LPP-1-overexpressing HPAECs were labeled with [32P]orthophosphate (20 μCi/ml) for 3 h and exposed to either S1P (1 μM) or phyto-S1P (1 μM) for 15 min. As shown in Fig. 7, overexpression of LPP-1 potentiated the formation of intracellular [32P]S1P or 32P-labeled phyto-S1P (vector: vehicle, 1470 ± 95; S1P, 3447 ± 143; phyto-S1P, 2662 ± 92; LPP-1 wt: vehicle, 2047 ± 343; S1P, 6140 ± 100; phyto-S1P, 5586 ± 71). The effect of overexpressed LPP-1 was specific to exogenously added S1P because it did not alter either TNF-α- or Sph-mediated production of intracellular S1P. These results show that overexpression of LPP-1 specifically enhanced the intracellular production of S1P or phyto-S1P from exogenous S1P or phyto-S1P, respectively, in HPAECs.
FIGURE 7. LPP-1 wt overexpression enhances intracellular accumulation of [lsqb]32P]S1P and [32P]phyto-S1P.
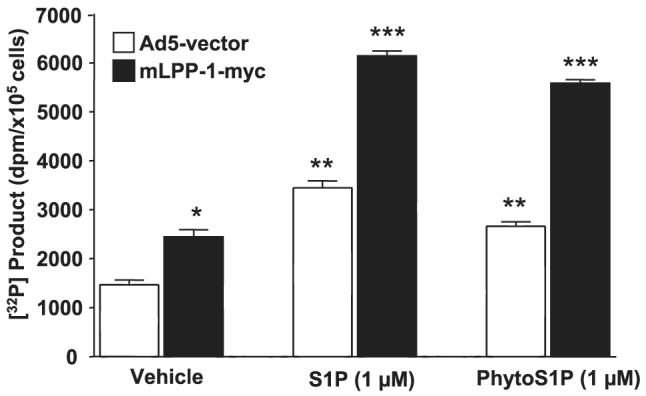
HPAECs (∼60% confluence) were infected with adenoviral constructs (10 m.o.i.) that contained cDNA for the empty vector or Myc-tagged LPP-1 for 24 h. Cells were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h before exposure to either S1P (1 μM) or phyto-S1P (1 μM) for 30 min. The formation of 32P-labeled S1P or phyto-S1P was determined after separation by TLC. Values are expressed as the means ± S.D. of three independent experiments in triplicate. *, significantly different compared with cells infected with empty vector adenoviral construct (p < 0.05); **, significantly different compared with empty vector infected cells (p < 0.05); ***, significantly different compared with Myc-tagged LPP-1 wt-infected cells without S1P or phyto-S1P treatment (p < 0.01).
Gene Silencing of LPP-1 Attenuates Intracellular S1P Formation
Because LPP-1 enhanced intracellular S1P production, we examined whether gene silencing of LPP-1 affects intracellular S1P formation from extracellular S1P and Sph. Transfection of HPAECs with double-stranded RNAs targeted at the human LPP-1 mRNA sequence decreased LPP-1 mRNA and protein expression to greater than 90% without reducing LPP-2 or LPP-3 mRNA or protein levels (Fig. 8, A and B). Furthermore, transfection of cells with LPP-1 siRNA was accompanied by a decrease of ∼60% of total LPP activity as measured by activity assays employing LPP-1 immunoprecipitates and [3H]S1P as substrate (data not shown). Down-regulation of LPP-1 mRNA partially attenuated intracellular S1P generation from extracellular S1P, whereas the conversion of exogenous Sph to S1P was not altered (Fig. 8C). These results demonstrate that LPP-1 is essential for the hydrolysis and subsequent conversion of exogenous S1P to intracellular S1P in HPAECs.
FIGURE 8. Knock down of LPP-1 by siRNA decreases intracellular generation of [32P]S1P from exogenous S1P but not from Sph.
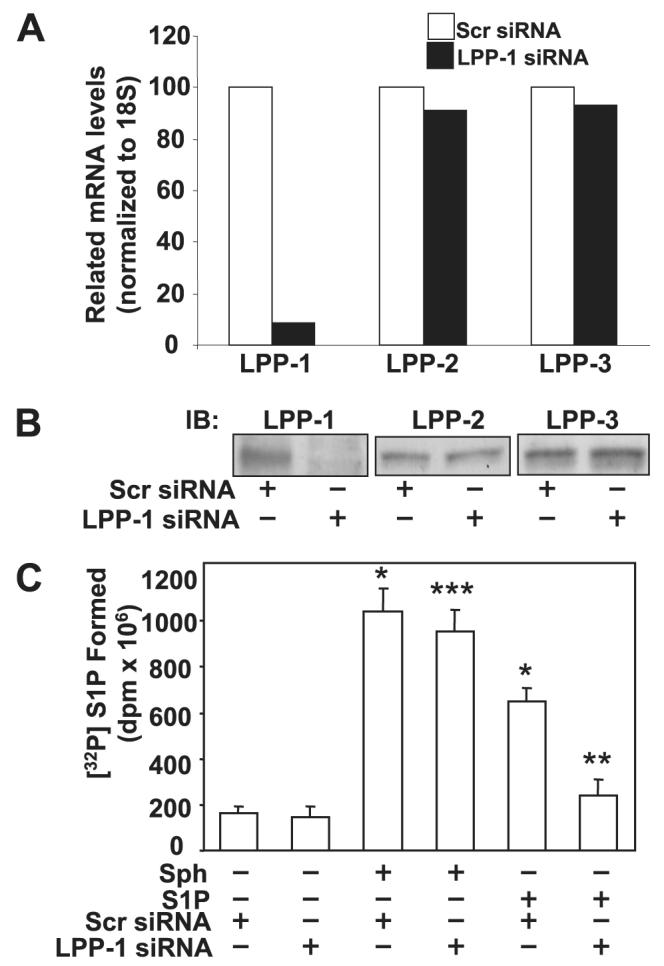
Panel A, total RNA was extracted from HPAECs transfected with scrambled siRNA or LPP-1 siRNA for 72 h, and transcription of the mRNA was determined by real-time RT-PCR. Panel B, cell lysates from scrambled and LPP-1 siRNA-transfected cells were analyzed by Western blotting (IB) for LPP-1, LPP-2, and LPP-3 protein expression using LPP-specific antibodies. Panel C, HPAECs were transfected with scrambled siRNA or LPP-1 siRNA for 72 h as described under “Experimental Procedures.” The transfected media was aspirated, and cells were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM before exposure to exogenous S1P (1 μM) or Sph (1 μM) complexed with 0.1% BSA for 30 min. The accumulation of [32P]S1P was determined after separation by TLC. Values are the means ± S.D. of six independent experiments. *, significantly different compared with scrambled siRNA transfected cells (p < 0.05); **, significantly different compared with cells exposed to scrambled siRNA + S1P (p, 0.05); ***, not significantly different compared with cells transfected with scrambled siRNA + sphingosine (p > 0.05).
Role of SphK1 wt, SphK2 wt, or SphK1 mutant on the Production of Intracellular S1P and Dihydro-S1P Production in HPAECs
mRNA expressions for SphK1 and SphK2 in HPAECs were detected by RT-PCR (Fig. 9A). Additionally, real-time PCR analysis suggested that the relative expression of SphK1 message was higher compared with that of SphK2 (Fig. 9B), and Western blotting with specific antibodies revealed expression of both SphK1 and SphK2 in HPAECs (Fig. 9C). Having established the presence of SphK1 and SphK2 in HPAECs, we next investigated the role of SphK1 and SphK2 in intracellular production of S1P. First, DMS, an inhibitor of SphK, partially blocked S1P- and Sph-dependent formation of [32P]S1P in HPAECs labeled with [32P]orthophosphate, indicating the involvement of SphK in S1P generation (Fig. 10). To further establish a role for SphK1 in S1P formation, HPAECs were infected with adenoviral FLAG-tagged SphK1 or Myc-tagged SphK2 (25 m.o.i.) for 24 and 48 h. Analysis of the cells by immunocytochemistry or cell lysates by Western blotting showed increased expression of the proteins (Fig. 11, A—D). The effect of overexpression of SphK1 and SphK2 wt and SphK1 mutant on SphK activity was examined in the 100,000 × g cytosol fraction. In vitro phosphorylation of Sph (5 μM)by [γ-32P]ATP for 30 min was ∼4 and ∼2-fold higher in SphK1- and SphK2 overexpressing cells, respectively, compared with vector controls (Fig. 12A). However, the cytosol fraction from cells infected with the mutant SphK1 exhibited a marked reduction (∼50%) in phosphorylation activity (Fig. 12A). In intact cells, overexpression of SphK1 increased [32P]S1P formation when exogenous S1P was added, whereas the catalytically inactive mutant of SphK1 did not increase S1P formation significantly (Fig. 12B).
FIGURE 9. Detection of SphK1 and SphK2 by RT-PCR, real-time RT-PCR, and Western blotting in HPAECs.
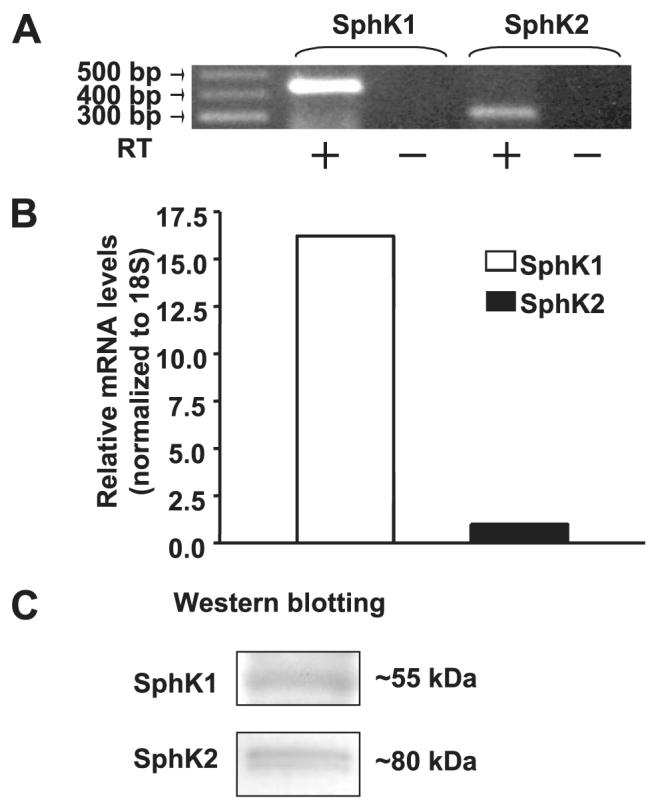
Panel A, total RNA was extracted from ∼95% confluent HPAECs and transcription of the genes encoding SphK1 and SphK2 was assessed by RT-PCR (- indicates the absence of reverse transcriptase, and + indicates the presence of reverse transcriptase during RT reaction) with primers indicated to SphKs. Panel B, one-step real-time RT-PCR was performed with total RNA from HPAECs. The relative abundance of target mRNA was calculated as 2 raised to the negative of its threshold cycle value multiplied by 106 normalized to the abundance of 18 S. Panel C, cell lysates (30 μg of protein) from HPAECs were subjected to SDS-PAGE and analyzed by Western blotting with anti-SphK1 and anti-SphK2 antibodies. Each Western blot is representative of three independent experiments.
FIGURE 10. Effect of DMS on intracellular generation of [32P]S1P.
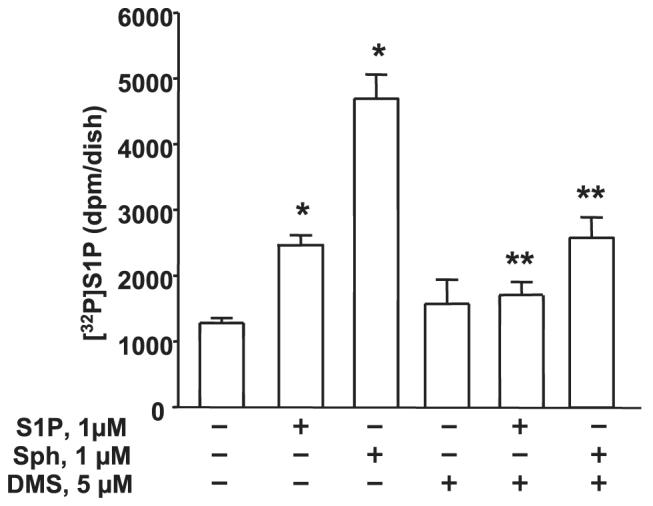
HPAECs (∼95% confluence) were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h before pretreatment with DMS (5 μM) for 1 h. The medium was aspirated, and cells were exposed to medium alone or medium containing either S1P (1 μM) or Sph (1 μM) complexed with 0.1% BSA for 30 min. Cellular lipids were extracted, and accumulation of [32P]S1P was measured after separation by TLC. Values are given as the means ± S.D. of three independent experiments in triplicate. *, significantly different compared with vehicle treatment (p < 0.05); **, significantly different compared either S1P or Sph exposed cells without DMS (p < 0.01).
FIGURE 11. Overexpression of adenoviral constructs of FLAG-tagged SphK and Myc-tagged SphK2 in HPAECs.
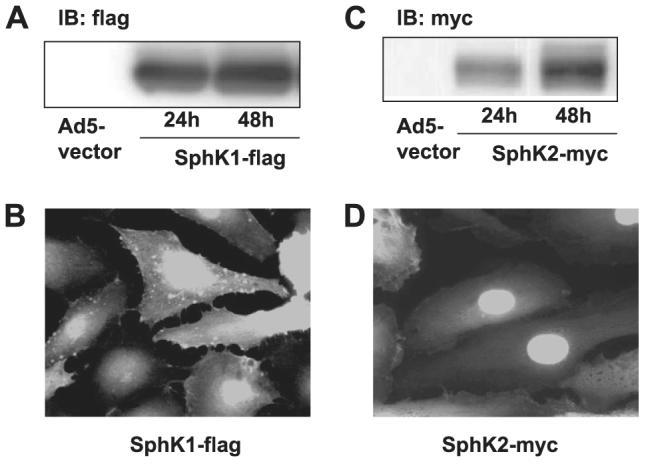
Panels A and C, HPAECs (∼60% confluence in 35-mm dishes) were infected with empty vector or with cDNA for FLAG-tagged SphK1 or Myc-tagged SphK2 adenoviral constructs (10 m.o.i.) in complete BEGM for 24 and 48 h. At the indicated time points cell lysates were prepared and subjected to SDS-PAGE and Western blotting (IB) with anti-FLAG or anti-Myc (9E10) antibodies; Panels B and D, HPAECs grown on glass coverslips to ∼70% confluence were infected with cDNA for FLAG-tagged SphK1 or Myc-tagged SphK2 adenoviral constructs (10 m.o.i.) for 24 h. Cells were subjected to immunostaining with anti-FLAG or anti-Myc (9E10) antibody and examined by fluorescent microscopy. The Western blots and immunofluorescence images are representative of three independent experiments.
FIGURE 12. Effect of overexpression of SphK1 or SphK2 or SphK1 mutant on S1P formation in vitro and in vivo.

HPAECs (∼70% confluence in 100-mm dishes) were infected with cDNA for FLAG-tagged SphK1, Myc-tagged SphK2 or FLAG-tagged SphK1 mutant (10 m.o.i.) for 24 h. Panel A, aliquots of 100 μg of protein were incubated with Sph (1 μM) complexed to 0.1% BSA and [γ-32P]ATP (10 μM, specific activity 1 × 104 dpm/pmol) for 30 min at 37 °C. The formation of [32P]S1P was determined by TLC. Results are expressed as pmol of S1P formed/μg of protein/min. In panel B, cells were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h before challenge with S1P (1 μM) for 30 min. The intracellular accumulation of [32P]S1P was measured after separation by TLC. Values are the means ± S.D. of three independent experiments. *, significantly different from empty vector without S1P addition (p < 0.05); **, significantly different compared with empty vector (p < 0.01); ***, significantly different compared with cells infected with empty vector plus S1P (p < 0.05). In panels C and D cells were infected with SphK1 wt, SphK2 wt, or sphK1 mutant as described in panel A. Cells were challenged with DMEM or DMEM plus C18-sphingosine (1 μM) for 30 min, and intracellular accumulation of C18-S1P or C18-dihydro (DH)-S1P was measured by LC-MS/MS as described under “Experimental Procedures.” Values are the means ± S.D. of three independent experiments. *, significantly different from vector control in the absence or presence of sphingosine (p < 0.05); **, significantly different from vector control in the absence or presence of sphingosine (p < 0.01). Veh, vehicle.
Because overexpression of SphK1, but not SphK2, resulted in predominant up-regulation of de novo biosynthesis of dihydro-S1P (6-9, 40), we evaluated the effects of overexpression of SphK1 wt, SphK2 wt, or SphK1 mutant on intracellular accumulation of S1P and dihydro-S1P by LC-MS/MS in the absence or presence of exogenous sphingosine. Overexpression of SphK1, but not SphK2, revealed a significant accumulation of C18-S1P (∼10-fold increase over vector control) and C18 dihydro-S1P (∼900-fold increase over vector control) without exogenous addition of sphingosine (Fig. 12, C and D). In the presence Sph, overexpression of SphK1wt and SphK2 wt further enhanced intracellular S1P formation as compared with without sphingosine addition (Fig. 12C); however, sphingosine addition had no effect on accumulation of dihydro-S1P in SphK1- or SphK2 wt-expressing cells (Fig. 12D). Overexpression of SphK1 mutant blocked intracellular conversion of sphingosine to S1P in HPAECs but had no effect on dihydro-S1P formation (Fig. 12D).
Effect of SphK, S1P Phosphatase, and S1P Lyase siRNA on Intracellular S1P and Dihydro-S1P Formation in HPAECs
Accumulation of S1P in cells is a balance between its formation via SphK and catabolism catalyzed by SPP and SPL (10-12). To examine the relative role of these enzymes in intracellular S1P formation from exogenous sphingosine or S1P, HPAECs were transfected with siRNA specific for SphK1, SphK2, SPP1, or SPL. As shown in Fig. 13A, the mRNA and protein expressions of SphK1 and SphK2 were down-regulated by SphK1 or SphK2 siRNA, as determined by real-time PCR and Western blotting. Similarly, siRNA for SPP and SPL also reduced the mRNA levels of SPP and SPL (Fig. 13B). Down-regulation of SphK1, but not SphK2, expression by siRNA significantly attenuated intracellular [32P]S1P formation with Sph or S1P as an extracellular substrate (Fig. 13C). In contrast to SphK1 siRNA, down-regulation of SPP1 and SPL with siRNA increased accumulation of C18-S1P and C18-dihydro-S1P from exogenous sphingosine as compared with scrambled siRNA transfected cells (Fig. 13, D and E). These results suggest a significant role for SphK1, but not SphK2, in the generation of intracellular S1P by HPAECs exposed to exogenous S1P or Sph. Furthermore, experiments with SPP1 and SPL siRNA indicate that the intracellularly generated S1P is degraded by SPP and SPL in HPAECs.
FIGURE 13. Effect of down-regulation of SphK1, SphK2, SPP1, and SPL on intracellular generation of S1P in HPAECs.

HPAECs (∼70% confluence in 35-mm dishes) were transfected with scrambled, SphK1, SphK2, SPP1, or SLP siRNA (100 nM) for 72 h. In panels A and B, total RNA was isolated, and mRNA expression of SphK1, SphK2, SPP1, and SPL, under different transfection, was evaluated by real-time RT-PCR and normalized to 18 S. Values are the averages of six independent experiments. The efficacy of SphK1 and SphK2 siRNA was also evaluated by Western blotting (IB, panel A). In panel C transfected cells were labeled with [32P]orthophosphate (20 μCi/ml) in phosphate-free DMEM for 3 h before exposure to either Sph (1 μM) or S1P (1 μM) for 30 min. The accumulation of intracellular [32P]S1P was quantified by TLC. Values given are the means ± S.D. of six independent experiments. *, significantly different compared with scrambled siRNA transfection alone (p < 0.05); **, significantly different compared with scrambled siRNA (p < 0.01); ***, significantly different compared with scrambled siRNA plus S1P or scrambled siRNA + Sph (p < 0.05). In panels D and E, cells were transfected with scrambled, SphK1, SphK2, SPP1, or SPL siRNA for 72 before exposure to medium or medium plus C18-sphingosine (1 μM) for 30 min. The accumulation of intracellular C18-S1P or C18- dihydro-S1P was quantified by LC-MS/MS as described under “Experimental Procedures.” *, significantly different compared with scrambled siRNA transfection (p < 0.05); **, significantly different compared with scrambled siRNA plus Sph (p < 0.01). Veh, vehicle.
DISCUSSION
The present study provides the first evidence that both human lung ECs and ECs from other vascular beds utilize and convert extracellular S1P to intracellular S1P. Conversion of exogenous S1P to intracellular S1P required the hydrolysis of the added S1P to Sph, a process that was mediated by LPPs and subsequent phosphorylation of Sph by intracellular SphK1, but not SphK2, in HPAECs. Our conclusions about the roles of LPPs and SphK1 in intracellular S1P production are supported by experiments in which we increased or decreased LPP-1 and SphK1 activities using LPP-1 wt/LPP-1 and SphK1 adenoviral constructs or siRNA.
Generation of Sph is the rate-limiting step in S1P production catalyzed by SphK1 or SphK2 in mammalian cells (1, 2, 7). Several earlier studies have documented agonist-dependent generation of S1P in PC12, HEK 293, and NG108 cells (44-46). We showed that in HPAECs only TNF-α, but not vascular endothelial growth factor, 12-O-tetradecanoylphorbol-13-acetate, thrombin, or A23187, increased [32P]S1P accumulation (∼1.5-fold) compared with vehicle treatment (Fig. 1A). Although TNF-α (31, 39, 47), angiotensin II (48), or growth factors (49-51) increase intracellular S1P levels via the sphingomyelinase/ceramide pathway, S1P did not activate sphingomyelinase in HPAECs (Fig. 3), indicating participation of a sphingomyelinase-independent pathway in the production of Sph. Interestingly, incubation of HPAECs with human PPP or lipids isolated from PPP stimulated intracellular production of [32P]S1P (Fig. 1C). Analysis of human PPP by LC-MS/MS revealed the presence of S1P and Sph at levels of 1973 and 23 pmol/ml, respectively, suggesting that circulating S1P in plasma could act as a source of intracellular S1P for ECs lining the vessel walls. The source of circulating plasma S1P is unclear; however, platelets can convert Sph to S1P and release it into the blood (29, 30). Platelets and cells such as HEK 293, mast cells, or NG108 secrete part of S1P (44-46), whereas the S1P that is generated intracellularly in HPAECs from either Sph or extracellular S1P is not released into the medium (Fig. 2, C and D). Although platelets or other circulating cells could serve as a major source of S1P in the blood, the possibility that small amounts of free Sph is phosphorylated to S1P by extracellular SphKs cannot be ruled out. In this context, release of overexpressed SphK1 wild type, but not SphK2 wild type, into the cell culture medium was observed in human umbilical vein ECs (51) and in HPAECs.3 In recent studies of the five SphK isoforms expressed in ECs, only SphK1a isoform was selectively secreted in HEK 293, and HUVECs and human plasma were shown to contain SphK1 activity (45, 51, 52). Thus, ECs generate intracellular S1P and also secrete into circulation SphK1a (51, 52), which could generate small but significant levels of S1P from Sph (51, 52).
S1P in the plasma seems to be metabolically stable because of its interaction with albumin and lipoprotein fractions (29, 53); however, the level of this bioactive lipid is regulated by ecto-LPPs that degrade S1P to Sph. The addition of [3H]S1P to HUVECs or whole blood resulted in marked degradation of the added substrate with concomitant formation of [3H]Sph, which was blocked by the phosphatase inhibitor, vanadate; however, a definitive role for LPPs in the metabolism of [3H]S1P to [3H]Sph was not demonstrated, although HUVECS were shown to express mRNAs for LPP1–3 (54). The present study provides the first compelling evidence that LPPs present on the surface of HPAECs regulate the degradation of exogenously added S1P to Sph, which subsequently transported into the cell and phosphorylated by SphK1 to intracellular S1P. In support of this conclusion, exogenously added [3H]S1P was hydrolyzed in a time-dependent fashion primarily to [3H]Sph, and the reaction was blocked by XY-14, an inhibitor of the LPPs (43, 55). The involvement of LPPs in the generation of intracellular S1P from exogenous S1P was also confirmed by decreasing LPP-1 activity using siRNA, which attenuated intracellular accumulation of S1P in HPAECs (Fig. 9C). Overexpression of LPP-1 enhanced the hydrolysis of [3H]S1P to [3H]Sph by 2–3-fold compared with cells infected with control adenoviral vector (Fig. 6C). In addition to LPP-1, HPAECs also expressed LPP-2 and LPP-3 as determined by real-time PCR and Western blotting (Fig. 5). However, unlike LPP-1 that is expressed on the cell surface and internal organelles (Fig. 6B), it is unclear if LPP-2 and LPP-3 are also localized on the extracellular side of the plasma membrane or if they are expressed only within intracellular cytoplasmic organelles of HPAECs. Our results with LPP-1 siRNA (Fig. 9C) indicate that LPP-2 and LPP-3 may be localized on the cell surface to degrade S1P or other lipid phosphate substrates. Therefore, down-regulation of LPP-1 alone may not be sufficient to completely block the hydrolysis of exogenous S1P and its conversion to intracellular S1P in HPAECs. Furthermore, the importance of LPPs in mediating the hydrolysis of exogenous S1P and generation of intracellular S1P is evident from experiments in which HPAECs were provided with a phosphonate analog of phyto-S1P (PHS-C-P), which failed to generate intracellular phyto-S1P. Interestingly, PHS-C-P was a weak inhibitor of LPP activity and partly attenuated intracellular S1P or phyto-S1P production from extracellular S1P without affecting the utilization of Sph. Exposure of HPAECs to PHS-C-P (1 μM) for 30 min partly attenuated S1P-induced ERK activation, suggesting a role for intracellular S1P in signal transduction.3 Further studies are necessary to address the mechanism(s) of action of PHS-C-P in attenuating LPP activity and signaling in ECs.
LPPs influence physiological responses mediated by lipid phosphates such as S1P or lysophosphatidate through regulating the availability of the extracellular ligand and also by controlling the accumulation of bioactive lipid phosphates downstream of G-protein receptor activation (35). Recent studies show that changing the expression of different LPPs modulates the S1P- or lysophosphatidate-mediated activation of extracellular signal-regulated kinase 1/2, phospholipase D, DNA synthesis, cell migration, changes in [Ca2+]i, IκB phosphorylation, and translocation of NF-κB to the nucleus from the cytoplasm and interleukin-8 secretion (32, 35, 41, 56). However, part of S1P- or lysophosphatidate-mediated signal transduction does not depend upon the ecto-LPP activity (41, 18, 58, 59). As a further function for the LPPs, we show that the hydrolysis of extracellular S1P by ecto-LPP activity is one of the critical steps involved in the intracellular generation of S1P and, consequently, its signaling effects. This work compliments that of Morris and co-workers (60) who showed that increasing LPP activity enhances the uptake of diacylglycerol by cells treated with external phosphatidate. Thus, the LPPs convert lipid phosphates that have very limited ability to enter cells into products that more readily traverse the plasma membrane and which can then signal directly or after phosphorylation.
In the present study we also demonstrated that SphK1, but not SphK2, increases intracellular S1P production from exogenous S1P and Sph in HPAECs. Analysis of total RNA by real-time RT-PCR revealed that both the isoforms were expressed in HPAECs; however, the relative distribution of SphK1 mRNA was relatively higher compared with SphK2 (Fig. 9, A and B). In HPAECs, in vitro phosphorylation of Sph to S1P was higher after overexpression of SphK1 compared with SphK2, whereas overexpression of a SphK1 mutant inhibited phosphorylation of Sph in vitro (Fig. 12A) and intracellular generation of S1P from exogenous S1P in HPAECs (Fig. 12B). A role for SphK1, but not SphK2, in utilizing exogenous S1P as a source for intracellular S1P generation was confirmed by when SphK1 or SphK2 expression was knocked down with siRNA (Fig. 13C). Agonist-mediated activation of SphK exhibited a biphasic response with an early initial phase (∼2-fold increase) that was independent of new protein synthesis and a late second phase that was dependent on new protein synthesis (50). Our current study shows that S1P did not stimulate sphingomyelinase; however, it is not known if SphK1 is activated by exogenous S1P and, if so, which mechanism is involved in the activation. Numerous agonists stimulate SphK1 and increase in intracellular S1P; however, no stimulus has been described that specifically affects SphK2 activity (2). Although SphK1 efficiently phosphorylates both Sph and dihydro-Sph to S1P and dihydro-S1P (62), phyto-Sph and the Sph analog FTY720 are poor substrates for SphK1 (25, 26). Although most cells express both SphK1 and SphK2, they seem to have opposing functions. It is now well established that SphK1 promotes cell growth and pro-survival, whereas SphK2 inhibits cell growth and enhances apoptosis (63, 64). Furthermore, overexpression of SphK1 decreased incorporation of [3H]palmitate into C16-ceramide, whereas SphK2 increased it (65). The mechanism(s) responsible for opposing functions of SphK1 and sphK2 in ceramide production in NIH 3T3 fibroblasts and HEK 293 cells is unclear; however, distinct localization of the two proteins or their translocation to different compartments within the cell may explain for the opposing effects (65). Accumulation of intracellular S1P is a balance between its synthesis through SphK and degradation by SPPs and SPL. Analyses of total RNA revealed expression of SPP-1 and SPL in HPAECs (Fig. 13B). Knocking down the expression of SPP-1 or SPL in HPAECs with siRNA increased the accumulation of C18-S1P in the presence of Sph (Fig. 13D), suggesting a role for these two enzymes in regulating S1P levels. Our present results show that overexpression of LPP-1 increased the accumulation of intracellular S1P in response to exogenous S1P because of the ecto-activity of LPP-1.
Recent studies by Giussani et al. (66) show that overexpression of SPP-1 decreased the levels of S1P and dihydro-S1P in HEK 293 cells because of the internal activity of SPP-1, but there were no concomitant increases in sphingosine and dihydrosphingosine. The present study suggests that the effect of LPP-1 is coupled to SphK1 in intracellular S1P formation in HPAECs, although in HEK 293 cells, overexpression of SPP-1 appears to be uncoupled to SphK1 (66). Indeed, LPPs co-localize with SphK1 in Chinese hamster ovary cells (17), further suggesting linkage between these two enzymes. Thus, overexpression of LPP-1 appears to have the opposite effects on the conversion of exogenous S1P to intracellular S1P in HPAECs compared with the effects reported with SPP-1 in HEK 293 cells (66). Furthermore, overexpression of SPP-1 increased ceramide levels (61, 66), whereas down-regulating this phosphatase increased S1P and reduced Sph levels (57). Therefore, it appears that SPP-1 regulates endoplasmic reticulum-to-Golgi trafficking of ceramide and proteins in HEK 293 cells (66). However, the effect of overexpression of LPP-1 on ceramide levels and regulation of membrane transport of ceramide and proteins from endoplasmic reticulum to Golgi apparatus has not been studied. In agreement with our findings, overexpression of SPP-1 increased ceramide levels (66), whereas down-regulating this phosphatase increased S1P and reduced Sph levels (61). Further studies are necessary to understand the physiological role of SPPs and SLP in regulating intracellular S1P signaling and EC activation.
In summary, this study has examined mechanisms for intracellular production of S1P. We have demonstrated that LPP-1 (and probably other LPPs) plays a critical role in facilitating the uptake by ECs of Sph formed from circulating S1P. The concentration of circulating S1P is about 10 times higher than that of Sph, and thus, S1P can provide significant quantities of intracellular Sph. This Sph is then converted to S1P by SphK1, but not SphK2, in ECs. Thus, LPPs can modify the balance of signaling by S1P by three different mechanisms. First, they can decrease extracellular S1P concentrations, thereby lowering the activation of cell surface receptors. Second, they have been shown to attenuate signaling downstream of the activation of surface S1P receptors. Third, by promoting the formation of intracellular S1P, they increase intracellular signaling by this agonist. These combined observations add to our understanding of the complex interplay between the roles of S1P as an extracellular versus intracellular signaling molecule.
Acknowledgment
We thank Dr. Nigel Pyne for helpful discussions.
Footnotes
This work was supported by NHLBI, National Institutes of Health Grants RO1 HL 79396 (to V. N.) and RO1 HL 803187 (to R. B.) and Canadian Institute of Health Research Grants MOP 49491 and 81137 (to D. N. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- S1P
- sphingosine 1-phosphate
- DMS
- N,N-dimethylsphingosine
- EC
- endothelial cell
- HPAEC
- human pulmonary artery endothelial cell
- HUVEC
- human umbilical vein endothelial cell
- LPP
- lipid phosphate phosphatase
- mLPP
- mouse LPP
- PPP
- platelet poor plasma
- Sph
- D-erythro-C18-sphingosine
- SphK
- sphingosine kinase
- SPP
- sphingosine 1-phosphate phosphatase
- SPL
- sphingosine 1-phosphate lyase
- PHSC-P
- D-ribophytosphingosine 1-phosphonate
- XY-14
- [(3S)-1,1-difluoro-3,4-bis(oleoyloxy)butyl]phosphonate
- DMEM
- Dulbecco’s modified Eagle’s medium
- Edg
- endothelial differentiation gene
- TNF
- tumor necrosis factor
- FBS
- fetal bovine serum
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- wt
- wild type
- siRNA
- small interfering RNA
- RT
- reverse transcription
- m.o.i.
- multiplicity of infection
- LC-MS/MS
- liquid chromatograph-tandem mass spectroscopy
- BSA
- bovine serum albumin
- HEK cells
- human embryonic kidney cells
V. Natarajan, unpublished data.
REFERENCES
- 1.Pyne S, Pyne NJ. Biochem. J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spiegel S, Milstien S. Nat. Rev. Mol. Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 3.Hla T. Pharmacol. Res. 2003;47:401–407. doi: 10.1016/s1043-6618(03)00046-x. [DOI] [PubMed] [Google Scholar]
- 4.Ozaki H, Hla T, Lee MJ. J. Atheroscler. Thromb. 2003;10:125–131. doi: 10.5551/jat.10.125. [DOI] [PubMed] [Google Scholar]
- 5.Kluk MJ, Hla T. Biochim. Biophys. Acta. 2002;1582:72–80. doi: 10.1016/s1388-1981(02)00139-7. [DOI] [PubMed] [Google Scholar]
- 6.Liu H, Chakravarty D, Maceyka M, Milstien S, Spiegel S. Prog. Nucleic Acid Res. Mol. Biol. 2002;71:493–511. doi: 10.1016/s0079-6603(02)71049-0. [DOI] [PubMed] [Google Scholar]
- 7.Olivera A, Spiegel S. Prostaglandins Other Lipid Mediat. 2001;64:123–134. doi: 10.1016/s0090-6980(01)00108-3. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. J. Biol. Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 9.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J. Biol. Chem. 1998;273:23722–23728. doi: 10.1074/jbc.273.37.23722. [DOI] [PubMed] [Google Scholar]
- 10.Kihara A, Ikeda M, Kariya Y, Lee EY, Lee YM, Igarashi Y. J. Biol. Chem. 2003;278:14578–14585. doi: 10.1074/jbc.M211416200. [DOI] [PubMed] [Google Scholar]
- 11.Le Stunff H, Milstien S, Spiegel S. J. Cell. Biochem. 2004;92:882–899. doi: 10.1002/jcb.20097. [DOI] [PubMed] [Google Scholar]
- 12.Kariya Y, Kihara A, Ikeda M, Kikuchi F, Nakamura S, Hashimoto S, Choi CH, Lee YM, Igarashi Y. Genes Cells. 2005;10:605–615. doi: 10.1111/j.1365-2443.2005.00862.x. [DOI] [PubMed] [Google Scholar]
- 13.Le Stunff H, Peterson C, Thornton R, Milstien S, Mandala SM, Spiegel S. J. Biol. Chem. 2002;277:8920–8927. doi: 10.1074/jbc.M109968200. [DOI] [PubMed] [Google Scholar]
- 14.Le Stunff H, Mikami A, Giussani P, Hobson JP, Jolly PS, Milstien S, Spiegel S. J. Biol. Chem. 2004;279:34290–34297. doi: 10.1074/jbc.M404907200. [DOI] [PubMed] [Google Scholar]
- 15.Mechtcherialova D, Wlachos S, Sobanov J, Kopp T, Reuschel R, Bornancin F, Cai R, Zemann B, Urtz N, Stingl G, Zlabinger G, Woisetschlager M, Baumruker T, Billich A. Cell. Signal. 2007;19:748–760. doi: 10.1016/j.cellsig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Brindley DN, English D, Pilquil C, Buri K, Ling ZC. Biochim. Biophys. Acta. 2002;1582:33–44. doi: 10.1016/s1388-1981(02)00135-x. [DOI] [PubMed] [Google Scholar]
- 17.Sciorra VA, Morris AJ. Biochim. Biophys. Acta. 2002;1582:45–51. doi: 10.1016/s1388-1981(02)00136-1. [DOI] [PubMed] [Google Scholar]
- 18.Long J, Darroch P, Wan KF, Kong KC, Ktistakis N, Pyne NJ, Pyne S. Biochem. J. 2005;391:25–32. doi: 10.1042/BJ20050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yatomi Y, Yamamura S, Ruan F, Igarashi Y. J. Biol. Chem. 1997;272:5291–5297. doi: 10.1074/jbc.272.8.5291. [DOI] [PubMed] [Google Scholar]
- 20.Anliker B, Chun J. Semin. Cell Dev. Biol. 2004;15:457–465. doi: 10.1016/j.semcdb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Rosen H, Goetzl EJ. Nat. Rev. Immunol. 2005;5:560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 22.Pyne S, Pyne NJ. Biochim. Biophys. Acta. 2002;1582:121–131. doi: 10.1016/s1388-1981(02)00146-4. [DOI] [PubMed] [Google Scholar]
- 23.Singleton PA, Dudek SM, Chiang ET, Garcia JG. FASEB J. 2005;19:1646–1656. doi: 10.1096/fj.05-3928com. [DOI] [PubMed] [Google Scholar]
- 24.Argraves KM, Obeid LM, Hannun YA. Adv. Exp. Med. Biol. 2002;507:439–444. doi: 10.1007/978-1-4615-0193-0_68. [DOI] [PubMed] [Google Scholar]
- 25.Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. FEBS Lett. 2003;554:189–193. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- 26.Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. J. Biol. Chem. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- 27.zu Herigndorf D. Meyer, Liliom K, Schaefer M, Danneberg K, Jaggar JH, Tigyi G, Jakobs KH. FEBS Lett. 2003;554:443–449. doi: 10.1016/s0014-5793(03)01219-5. [DOI] [PubMed] [Google Scholar]
- 28.Zhi L, Leung BP, Melendez AJ. J. Cell. Physiol. 2006;208:109–115. doi: 10.1002/jcp.20646. [DOI] [PubMed] [Google Scholar]
- 29.Yatomi Y, Ruan F, Hakomori S, Igarashi Y. Blood. 1995;86:193–202. [PubMed] [Google Scholar]
- 30.English D, Welch Z, Kovala AT, Harvey K, Volpert OV, Brindley DN, Garcia JG. FASEB J. 2000;14:2255–2265. doi: 10.1096/fj.00-0134com. [DOI] [PubMed] [Google Scholar]
- 31.Xia P, Wang L, Gamble JR, Vadas MA. J. Biol. Chem. 1999;274:34499–34505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 32.Jasinska R, Zhang QX, Pilquil C, Singh I, Xu J, Dewald J, Dillon DA, Berthiaume LG, Carman GM, Waggoner DW, Brindley DN. Biochem. J. 1999;340:677–686. [PMC free article] [PubMed] [Google Scholar]
- 33.Kai M, Wada I, Imai S, Sakane F, Kanoh H. J. Biol. Chem. 1997;272:24572–24578. doi: 10.1074/jbc.272.39.24572. [DOI] [PubMed] [Google Scholar]
- 34.Nanjundan M, Possmayer F. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:1484–1493. doi: 10.1152/ajplung.2001.281.6.L1484. [DOI] [PubMed] [Google Scholar]
- 35.Brindley DN. J. Cell. Biochem. 2004;92:900–912. doi: 10.1002/jcb.20126. [DOI] [PubMed] [Google Scholar]
- 36.Bernatchez PN, Tremblay F, Rollin S, Neagoe PE, Sirois MG. J. Cell. Biochem. 2003;90:719–731. doi: 10.1002/jcb.10686. [DOI] [PubMed] [Google Scholar]
- 37.Limaye V, Li X, Hahn C, Xia P, Berndt MC, Vadas MA, Gamble JR. Blood. 2005;105:3169–3177. doi: 10.1182/blood-2004-02-0452. [DOI] [PubMed] [Google Scholar]
- 38.Sun C, Bittman R. J. Org. Chem. 2004;69:7694–7699. doi: 10.1021/jo0487404. [DOI] [PubMed] [Google Scholar]
- 39.Usatyuk PV, Natarajan V. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289:999–1010. doi: 10.1152/ajplung.00211.2005. [DOI] [PubMed] [Google Scholar]
- 40.Berdyshev EV, Gorshkova IA, Usatyuk P, Zhao Y, Saatian B, Hubbard W, Natarajan V. Cell. Signal. 2006;18:1779–1792. doi: 10.1016/j.cellsig.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Usatyuk PV, Cummings R, Saatian B, He D, Watkins T, Morris A, Spannhake EW, Brindley DN, Natarajan V. Biochem. J. 2005;385:493–502. doi: 10.1042/BJ20041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin A, Duffy PA, Liossis C, Gomez-Munoz A, O’Brien L, Stone JC, Brindley DN. Oncogene. 1997;14:1571–1580. doi: 10.1038/sj.onc.1200987. [DOI] [PubMed] [Google Scholar]
- 43.Smyth SS, Sciorra VA, Sigal YJ, Pamuklar Z, Wang Z, Xu Y, Prestwich GD, Morris AJ. J. Biol. Chem. 2003;278:43214–43223. doi: 10.1074/jbc.M306709200. [DOI] [PubMed] [Google Scholar]
- 44.Misasi R, Sorice M, Di Marzio L, Campana WM, Molinari S, Cifone MG, Pavan A, Pontieri GM, O’Brien JS. FASEB J. 2001;15:467–474. doi: 10.1096/fj.00-0217com. [DOI] [PubMed] [Google Scholar]
- 45.zu Heringdorf D. Meyer, Lass H, Alemany R, Laser KT, Neumann E, Zhang C, Schmidt M, Rauen U, Jakobs KH, van Koppen CJ. EMBO J. 1998;17:2830–2837. doi: 10.1093/emboj/17.10.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chin TY, Hwang HM, Chueh SH. Mol. Pharmacol. 2002;61:486–494. doi: 10.1124/mol.61.3.486. [DOI] [PubMed] [Google Scholar]
- 47.De Palma C, Meacci E, Perrotta C, Bruni P, Clementi E. Arterioscler. Thromb. Vasc. Biol. 2006;26:99–105. doi: 10.1161/01.ATV.0000194074.59584.42. [DOI] [PubMed] [Google Scholar]
- 48.Mulders AC, Hendriks-Balk MC, Mathy MJ, Michel MC, Alewijnse AE, Peters SL. Arterioscler. Thromb. Vasc. Biol. 2006;26:2043–2048. doi: 10.1161/01.ATV.0000237569.95046.b9. [DOI] [PubMed] [Google Scholar]
- 49.Culmsee C, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JH, Mattson MP, Krieglstein J. Neuroscience. 2002;115:1089–1108. doi: 10.1016/s0306-4522(02)00539-0. [DOI] [PubMed] [Google Scholar]
- 50.Doll F, Pfeilschifter J, Huwiler A. Biochim. Biophys. Acta. 2005;1738:72–81. doi: 10.1016/j.bbalip.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Grananta R, Trovato L, Garbarino G, Taliano M, Ponti R, Sala G, Ghidoni R, Ghigo E. FASEB J. 2004;18:1456–1458. doi: 10.1096/fj.04-1618fje. [DOI] [PubMed] [Google Scholar]
- 52.Venkataraman K, Thangada S, Michaud J, Oo ML, Ai Y, Lee YM, Wu M, Parikh NS, Khan F, Proia RL, Hla T. Biochem. J. 2006;397:461–471. doi: 10.1042/BJ20060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, Ui M, Okajima F. J. Biol. Chem. 2001;276:31780–31785. doi: 10.1074/jbc.M104353200. [DOI] [PubMed] [Google Scholar]
- 54.Aoki S, Yatomi Y, Ohta M, Osada M, Kazama F, Satoh K, Nakahara K, Ozaki Y. J. Biochem. (Tokyo) 2005;138:47–55. doi: 10.1093/jb/mvi100. [DOI] [PubMed] [Google Scholar]
- 55.Prestwich GD, Xu Y, Qian L, Gajewiak J, Jiang G. Biochem. Soc. Trans. 2005;33:1357–1361. doi: 10.1042/BST0331357. [DOI] [PubMed] [Google Scholar]
- 56.Pilquil C, Singh I, Zhang Q, Ling Z, Buri K, Stromberg LM, Dewald J, Brindley DN. Prostaglandins Other Lipid Mediat. 2001;64:83–92. doi: 10.1016/s0090-6980(01)00101-0. [DOI] [PubMed] [Google Scholar]
- 57.Johnson KR, Johnson KY, Becker KP, Bielawski J, Mao C, Obeid LM. J. Biol. Chem. 2003;278:34541–34547. doi: 10.1074/jbc.M301741200. [DOI] [PubMed] [Google Scholar]
- 58.Pyne S, Long JS, Ktistakis NT, Pyne NJ. Biochem. Soc. Trans. 2005;33:1370–1374. doi: 10.1042/BST0331370. [DOI] [PubMed] [Google Scholar]
- 59.Pilquil C, Dewald J, Cherney A, Gorshkova I, Tigyi G, English D, Natarajan V, Brindley DN. J. Biol. Chem. 2006;281:38418–38429. doi: 10.1074/jbc.M601670200. [DOI] [PubMed] [Google Scholar]
- 60.Sigal YJ, Mcdermott MI, Morris AJ. Biochem. J. 2005;387:281–293. doi: 10.1042/BJ20041771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Le Stunff H, Galve-Roperh I, Peterson C, Milstien S, Spiegel S. J. Cell Biol. 2002;158:1039–1049. doi: 10.1083/jcb.200203123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nava VE, Lacana E, Poulton S, Liu H, Sugiura M, Kono K, Milstien S, Kohama T, Spiegel S. FEBS Lett. 2000;473:81–84. doi: 10.1016/s0014-5793(00)01510-6. [DOI] [PubMed] [Google Scholar]
- 63.Taha TA, Hannun YA, Obeid LM. J. Biochem. Mol. Biol. 2006;39:113–131. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- 64.Spiegel S, Milstien S. J. Biol. Chem. 2007;282:2125–2129. doi: 10.1074/jbc.R600028200. [DOI] [PubMed] [Google Scholar]
- 65.Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr., Milstien S, Spiegel S. J. Biol. Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 66.Giussani P, Maceyka M, Le Stunff H, Mikami A, Lepine S, Wang E, Kelly S, Merrill AH, Jr., Milstein S, Spiegel S. Mol. Cell. Biol. 2006;26:5055–5069. doi: 10.1128/MCB.02107-05. [DOI] [PMC free article] [PubMed] [Google Scholar]