

Abstract
The gastrin releasing peptide receptor (GRP-R) is over-expressed on a number of tumors and cancer cell lines including pancreas, prostate, breast, gastrointestinal and small cell lung cancer (SCLC). Radiolabeled bombesin (BBN) analogues have exhibited high binding affinity and specificity to the GRP-R. A bombesin analogue with an antagonist targeting-vector at the C-terminus, DOTA-aminohexanoyl-[D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN[6-14] (1, “Bomproamide”), has been synthesized and displays high binding affinity (IC50 = 1.36 ± 0.09 nM) against 125I-Tyr4-BBN in in vitro competitive assays using PC-3 cells. Maximum internalization of 111In-1 reached 14% in PC-3 cells after 45 minutes of incubation. Rapid (0.25-hour PI) and high (12.21 ± 3.2 %ID/g) pancreatic uptake of 111In-1 was observed in healthy CF-1 mice and 90% of the activity was blocked by co-injection of 100 μg of BBN. Rapid (0.25-hour PI) and high uptake (6.90 ± 1.06 %ID/g) was observed in PC-3 prostate cancer xenografts in SCID mice, as well as visualized clearly in a SPECT/CT study. These results support the use of a bombesin construct with an antagonist C-terminal vector as a candidate of choice for specific in vivo imaging of tumors over-expressing GRP-receptors.
Keywords: Antagonist, bombesin, prostate cancer, imaging
Introduction
Receptor-avid radiopharmaceuticals have attracted great attention as efficient biomarkers. Molecular selectivity is achieved with the appropriate choice of a biomolecule, whether it is a monoclonal antibody or fragment, small molecule, or a peptide. For many imaging or radiotherapeutic applications, introduction of the radioactive nuclide is addressed through the bifunctional chelate approach. Regulatory peptides such as the bombesin (BBN), gastrin-releasing peptide (GRP) and neuromedin-B are a group of promising small bio-vectors. BBN contains 14 amino acid residues and is the amphibian analogue of the mammalian counterpart GRP which contains 27 amino acid residues (1, 2). BBN, originally isolated and purified from the skin of European frog “bombina bombina,” shows marked binding affinity and specificity to the GRP receptor subtype BB2 (3). GRP receptors are normally expressed in the non-neurocrine tissues of the pancreas and breast as well as in neuroendocrine tissue of the prostate, lung, colon, anterior pituitary cells as well as central and peripheral nervous system (4-6). Over-expression of several regulatory peptide receptors, such as BB2, has been detected in various human neoplasia (7-12) and may act as a prognostic marker for prostate, breast and lung carcinogenesis (13-15).
Agonist-based peptide constructs have been studied as receptor targeting agents (16-21) due to their ability to induce receptor-mediated internalization, most likely by endocytosis. Accumulation of the radioconjugate within the targeted cells is a prerequisite to triggering efficient internalization. For therapeutic purposes, efficient internalization of the radioconjugate appears to be important in delivering the highest radioactivity dose to the targeted cells.
Structural modifications to these radiopharmaceuticals have included variation in the peptide sequence, the nature of the chelate and radiometal, and the length of the spacer attaching the two components (Figure 1). For example, an agonist BBN analogue, DO3A-CH2CO-G-4-aminobenzoyl-Q-W-A-V-G-H-L-M-NH2 (AMBA) was synthesized and labeled with 177Lu for prostate cancer targeting (22). 177Lu-AMBA shows a high affinity for the GRP-R (IC50 = 2.50 ± 0.50 nM) and 76.8% internalization. The in vitro binding properties of 177Lu-AMBA were subsequently investigated in human neoplastic and non-neoplastic tissues known for their expression of GRP-receptors (16). Interestingly, in contrast to the labeling observed in GRP-positive mouse pancreas, the authors reported that the human pancreas shows no labeling with 177Lu-AMBA unless chronic pancreatitis was present.
Figure 1.
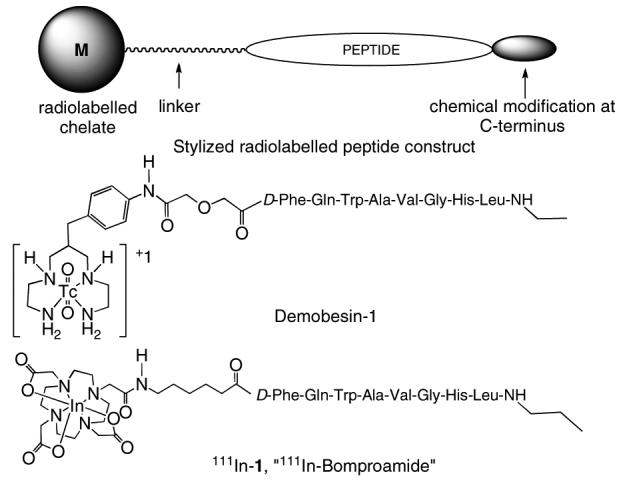
General structure for a radiolabelled peptide construct, structure of Demobesin-1 and 111In-Bomproamide (111In-1).
A novel series of DTPA coupled BBN analogues with a shortened peptide sequence also containing non-natural amino acid residues was recently reported (19). Among this series, DTPA-ACMpip-Tha6-BAla11-Tha13-Nle14-BBN[6-14] showed two to three times higher uptake for CA20948 pancreas tumor and 1.3 times for PC-3 prostate tumors when compared to DTPA-BBN[1-14]. The authors concluded that shortening the amino acid sequence and replacing specific amino acids in the BBN sequence can improve the receptor binding affinity (19).
A method of radiolabeling with technetium or rhenium via the metal precursor [M(H2O)3(CO)3]1 was tested with an agonist BBN analogue containing an appended 2,3-diaminopropanoyl moiety. The radiolabelled construct, (DPr)-Ser-Ser-Ser-Gln-Trp-AlaVal-Gly-His-Leu-Met-(NH2), exhibits excellent binding affinity to the GRP-receptors in the human PC-3 cell line (IC50 ∼ 0.86 nM). In addition, high uptake (16 ± 1.3 ID/g) in normal pancreas and extensive internalization 80% after 90 min incubation with PC-3 cells was reported (23). Studies on compounds reported by Nock et al. in 2005 utilized the open chain tetraamine-functionalized BBN analogue based on the agonist sequence [Pro1, Tyr4, Nle14] BBN, Demobesin-4 (15). Demobesin-4 has binding affinity for the GRP-receptors in the PC-3 cells with (IC50 ∼ 0.15 nM) (15). Demobesin-4 was recently evaluated using the intercellular calcium mobilization-based signaling assay in PC-3 and HEK-GRPR cells and an immunofluorescence/ELISA-based internalization assay in HEK-GRPR cells (24). Its behavior as an agonist was confirmed.
Two chelation systems, CB-TE2A (1,4,8,11-tetraazabicyclo [6.6.2] hexadecane-4,11-diacetic acid) and NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid), have been compared to DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) for the stable incorporation of 64Cu for PET imaging of prostate cancer using the construct BBN[7-14]NH2.(18, 25). The authors reported that the lack of retention of NOTA complex radioactivity in collateral tissues such as liver at 4 and 24 hr time points suggests a high degree of in vivo stability of NOTA complex as compared with peptide conjugates comprised of similar polyaminocarboxylate chelators such as DOTA (25). The images of the 64Cu-CB-TE2A radioconjugate demonstrate considerably reduced activity in the kidneys and gastrointestinal tract compared to 64Cu-DOTA radioconjugate. The authors reported that the high stability of 64Cu-CB-TE2A radioconjugate in vivo contributes to less retention of the radioactivity as opposed to the 64Cu-DOTA radioconjugate (18). Kidney uptake and retention of 64Cu-NOTA conjugate was lower than the 64Cu-DOTA-conjugate and very similar to 64Cu-CB-TE2A-Y3-TATE at 1 h post injection (25).
The effect of the length of the hydrocarbon spacer [0-carbon, β-Ala, 5-Ava, 8-Aoc and 11-Aun] between the agonist BBN [7-14]NH2 moiety and the radiochelate on the in vitro binding affinity toward GRP-Receptors was studied (26). The results illustrated that analogues containing either no spacer or the 11 carbon spacer (11-Aun) exhibited GRP-receptor specificity, but the measured affinity was less than one percent of the 8-carbon spacer analogue. The authors concluded that the length of the hydrocarbon spacer could be varied from three to eight carbon atoms without adversely affecting the resultant binding affinity.
Structural modifications at the C-terminus of bombesin yield peptides that retain their superb binding affinity for GRP-receptors, but also confer antagonist properties (27, 28). The rationale of developing antagonist agents is their capability of binding the receptors regardless of their limited capability to trigger internalization. The superb binding characteristics of antagonist radioconjugates may be due to their ability to bind a higher number of receptor binding sites than agonists (24, 29). These antagonist-based peptide constructs are now gaining attention as efficient targeting agents for diagnostic applications (30, 31). The in vitro biological results (32) as well as the solid-phase synthesis (33) of some of these parent peptides have been reported. One BBN analogue, [D-Phe6, Leu-NHEt13, des Met14] BBN[6-14], was derivatized with a benzylaminodiglycolic acid linker and the open-chain tetraamine chelate to incorporate technetium to yield Demobesin-1 (Figure 1) (33). Results show that Demobesin-1 has a high binding affinity (IC50 = 0.7 nM) in human prostate PC-3 cancer cells. Pancreatic uptake of 58.7 % ID/g at 30 minute post-injection was also reported. Demobesin-1 internalized within the cells to approximately 25 %. During the course of our work, Demobesin-1 was tested for its internalization properties using the intercellular calcium mobilization-based signaling assay in PC-3 and HEK-GRPR cells and an ELISA-based internalization assay in HEK-GRPR cells. Based upon these results, the authors confirm that their construct was acting as an antagonist. In addition, Demobesin-1 exhibited a much higher in vivo uptake than the aforementioned Demobesin-4, an agonist; thus, the authors concluded that GRP-receptor antagonists may be preferable agents to GRP-receptor agonists for tumor targeting (30).
In this study, we report the solid-phase synthesis of an antagonist bombesin analogue, DOTA-aminohexanoyl-[D -Phe6 , Leu-NHCH2CH2CH313, des Met14] BBN [6-14] (1, “Bomproamide”, Figure 1), as a targeting vector for radioimaging human prostate cancer (PC-3) over-expressing GRP-receptors. This peptide was chosen because the reported IC50 of [D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN [6-14] to guinea pig acini was higher (1.6 nM) than that observed for [D-Phe6, Leu-NHCH2CH313, des Met14] BBN [6-14] (7.0 nM) (27). The peptide is first synthesized and then equipped with a 6-carbon linker to a DOTA chelate for the efficient incorporation of 111In. The initial in vitro and in vivo evaluation of this radioconjugate in CF1 mice and those containing PC-3 prostate cancer xenografts in SCID mice are described.
Materials and Methods
All reagents used were of analytical reagent or HPLC grade. All syntheses were conducted in a manual solid phase peptide synthesis (SPPS) reaction vessel. HPLC analysis and separations were performed were performed with a C-18 Kromosil column (5 μm, 100 Å, 150 mm × 4.5 mm, Keystone Scientific Inc). HPLC solvents consisted of 0.1% aqueous trifluoroacetic acid (TFA), (solvent A) and acetonitrile (ACN) containing 0.1% TFA (solvent B) at a 1.0 mL/min flow rate and UV detection at 214 nm and 280 nm.
A TSQ700 mass spectrometer (Thermo Finnigan) was coupled to an HPLC equipped with a NovaPak C18 column (3.9 × 300mm, Waters) for LC-MS analyses.
HPLC analysis and isolation of the 111In-radiolabeled constructs were performed with a C-18 Jupiter (RP-18) HPLC analytical column, (5 μm, pore size 300 Å, 4.6 × 250 mm, (Phenomenex). HPLC solvents consisted of water (H2O) containing 0.1% TFA, (solvent A) and of ACN containing 0.1% TFA (solvent B). Elution system of 25% to 40% ACN in 60 minutes was used for purification. The flow rate maintained at 1.5 mL/min for all the analytical and separation runs at 280 nm.
111InCl3 in 0.05 M HCl (pH ∼1.48) was obtained from Mallinckrodt Inc., St. Louis, MO. Radioactivity was counted using a well counter equipped with a sodium Iodide (Thallium), NaI(Tl), scintillation detector. Gamma ray radioactivity of the in vitro binding affinity (IC50) studies was counted using a gamma counter.
All biodistribution studies performed during the course of this research were conducted using protocols approved by the Institutional Animal Care and in accordance with US Public Health Service Guidelines.
The micro single photon emission tomography (microSPECT) imaging was performed using a microCAT II® SPECT/CT (Siemens Medical Solutions) small animal system composed of dual SPECT system. Each microSPECT detector contains 10,000 NaI(Tl) crystals (1.25 × 1.25 × 5 mm elements) with a detector pitch of 1.45 mm and a field of view of 145 mm × 145 mm. The detectors are coupled to a 3 × 3 array of position sensitive photomultiplier tubes (Hamamatsu H8500), each of which has a 49 × 49 mm nominal active area and housing dimensions of 52 × 52 mm. The crystal arrays are separated by approximately 0.20 mm with the region between the crystals filled with an opaque white reflective compound material. The imaging was performed using dual pinhole collimators with interchangeable pinhole apertures of 2 mm in diameter. The pinhole focal length or detector–to-aperture distance was 90 mm. The pinhole apertures were fabricated as knife-edge apertures to minimize photon penetration and are fabricated of Tungsten.
The micro computed tomography (CT) images were performed using Micro CT unit (microCAT II® MicroCT scanner, CTI). The system consists of a fully shielded cabinet x-ray system with a maximum voltage of 80 kV, maximum current of 500 μA and a Tungsten anode. The X-ray detector has a 4096 × 4096 pixel charge coupled device (CCD) array that is couple to a phosphor screen via a fiber-optic device. The scanner is a third generation “step and shoot” system in which the low-power X-ray source and CCD array detector rotate about the specimen. Computer algorithms were use for reconstructing images of < 50 μm isotropic voxel dimension using the Cobra 4.1 reconstruction engine.
The human immortalized prostate PC-3 cancer cell lines (American Type Culture Collection) used in the course of this study were grown in custom PRMI medium 1640 (Invitrogen Corp.) with 10% fetal bovine serum (US Bio-Technologies Inc.) and gentamicin (American Pharmaceutical Partners, Inc.). The PC-3 cell lines were grown and maintained by the University of Missouri Cell and Immunology Core Facility.
DOTA-Aminohexanoyl-[D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN[6-14] (1), “Bomproamide”
The D-Phe6, Leu-NHCH2CH2CH313, des Met14 BBN[6-14]-HMBA resin precursor, 2, was synthesized as previously described (34). Three equivalents, relative to the resin loading, of Fmoc-6-aminohexanoic acid (0.520 mmol, 183 mg) was dissolved in 9 mL NMP. HBTU (666 μmol, 177 mg) and HOBt (518 μmol, 79.0 mg) were dissolved in 3 mL DMF. These two solutions were mixed and three equivalents (518 μmol, 67.0 mg) of DIPEA were added and vortexed thoroughly for 10-15 min and added directly to 2 in a manual SPPS reaction vessel and agitated gently for 30 minutes under N2 at rt. The Fmoc group was removed by adding 3-4 mL of 25% (v/v) piperidine/DMF to yield 6-aminohexanoyl-[D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN [6-14]-HMBA resin, 3. Three equivalents, relative to the resin loading, of DOTA tris (t-Bu ester) (0.520 mmol, 296 mg) were dissolved in 9 mL of NMP. The carboxylic acid moiety was activated as above and was mixed with 3 and agitated gently for 30 minutes under N2 at rt to yield 4. The acid-sensitive sidechain protecting groups of histidine, tryptophan and glutamine, as well as the three BOC protecting groups on DOTA were removed by adding a deprotection-scavenger cocktail (10-20 mL cocktail per 1 g peptidyl resin) consisting of 95%TFA:2.5%H2O:2.5%TIS for 3 – 4 h at rt with gentle agitation using a stream of N2. DOTA-aminohexanoyl-[D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN [6-14]-HMBA resin was separated by filtration, washed twice with TFA, five times with NMP and then dried. Subsequently, a sample was swelled with THF (12 mL/g) for one hour and a cleavage solution of TEA/propylamine/THF (1:5:5 v/v), 50 mL of cleavage cocktail/g peptidyl resin, was added to the mixture was stirred overnight (∼18 hour) at 50 °C. The solution containing 1 was filtered and reduced in volume to ∼1 mL of yellowish oil. 1 was precipitated by the addition of cold diethyl ether to yield 180 mg (87.4% crude recovery) as white granules in 76% purity; k' = 8.11. Crude 1 was dissolved in 4 mL DMSO and 7 mL of ACN:H2O (50:50 v/v) and purified by semi-preparative HPLC using 10% ACN/H2O at starting flow rate of 4 mL/min with a gradient from 30 to 50% ACN in 60 minutes at a flow rate of 10 mL/minute. Fractions containing 1 with the same purity were collected together and lyophilized to yield 23 mg (39.5%) as a white powder: k' = 8.07, > 90% purity. ESI-MS showed the molecular ions (m/z): Theory: C72H108O17N18 = 1496.81; actual: (M+H)+ = 1497.7 and (M+2H)2+/2 = 749.7. The conjugate was stored at 4 °C.
In Vitro Competitive Cell Binding-Affinity Assay
The binding affinity of 1 to immortalized human prostate carcinoma PC-3 cells was examined by competitive displacement of 125I-Tyr4 BBN. In brief, 22,000 cpm of 125I-Tyr4-BBN and eight increasing concentrations (from 3.33 × 10−13 to 3.33 × 10−6 M) of 1 were incubated for 30 minutes at 37 °C in humidified air containing 5% CO2 with 3 × 104 PC-3 cells suspended at pH 7.4 in Roswell Park Memorial Institute (RPMI) medium 1640, containing 4.8 mg/mL N-(2-hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid), HEPES, 0.1 μg/mL Bacitracin and 2 mg/mL bovine serum albumin (BSA). At the end of the incubation period, the medium was aspirated and the cells were washed three times with fresh medium solution to remove any unbound radioactivity. The remaining cell-bound radioactivity was counted using a Packard Riastar gamma-counter. The 50% inhibitory concentration (IC50) value of was determined by plotting the percentage of the 125I-Tyr4-BBN radioactivity bound to the PC-3 cells' surface receptors, versus the concentration of 1.
Labeling with natIn
A sample of 1 (1.5 mg, 1.0 μmol) was dissolved in NH4OAc buffer (5 mg/mL, 65 mM, 200 μL) and mixed with a solution of natInCl3 (2.2 mg, 10 μmol) in NH4OAc buffer (200 μL). The pH of the NH4OAc buffer solution was adjusted to ∼5.5 - 6.0 using 0.01 M aqueous HCl. The solution was incubated for 60 minutes at 80 °C. After cooling to rt, natIn-1 was obtained as a white powder (1.3 mg, 81% crude recovery); k'=9.03, 87.5% purity. ESI-MS showed the molecular ions (m/z): Theory: C72H105InO17N18 = 1608.69; actual: (M+H)+ = 1610.1, and (M+2H)2+/2 = 805.5.
Radiolabeling with 111In
A sample of conjugate 1 (110 μg, 67.0 nmol) was dissolved in ammonium acetate (NH4OAc) buffer (200 μL, 0.4 M, pH 7.0) and mixed with ascorbic acid (∼20 mg). 111InCl3 (5.55 MBq, 150 μCi) as 111InCl3 in 0.05 M HCl (pH ∼1.48) was added to the solution of 1 (resultant pH ∼5.5) and incubated for 60 minutes at 80 °C. Ammonium acetate buffer solution, pH 7.0, was used to adjust the pH of the radiolabeling mixture. EDTA (50 μL, 0.002 M) was added to chelate any remaining 111In3+ and the reaction mixture was allowed cool to the rt. A few crystals of CuSO4·5H2O were added to complex excess 1. Aliquots (25 μL) of the radiolabeling mixture solution were subjected for HPLC analysis. The HPLC radiochromatogram indicates that 74% of the detected radioactivity was located at tR= 18.86 min corresponding to natIn-1. The HPLC UV chromatogram indicates the absence of 1, while The UV-peak corresponding to natCu-DOTA-peptide conjugate retention time appeared (tR = 20 min).
111In-1 was purified by RP-HPLC with a gradient consisting of 25% to 40% ACN in 60 minutes (1.0% ACN/4.0 min). The HPLC eluent containing pure 111In-1, was diluted with 3 mL of water and concentrated on an Empore C18-High Performance Extraction Disk (C18-HD) Cartridge 4215. The retained radioactivity was eluted using a 400 μL of ethanol/saline (6:4 v/v) mixture. A Capintec CRC®-15W Dose Calibrator/ well counter was used to measure the collected radioactivity of 111In-1 before and after concentration. Approximately 90%-94% of the original radioactivity was recovered and the radiochemical purity of the formulated 111In-DOTA-peptide conjugates was found to be ≥98% by HPLC.
For the in vitro internalization and efflux studies as well as in vivo pharmacokinetic studies, 111In-1 was diluted with an adequate volume of sterile normal saline to achieve 0.148-0.185 MBq (4-5 μCi)/100 μL solution. For the SPECT/CT imaging studies, the ethanol concentration was reduced to < 5% by evaporation under a stream of nitrogen. Quality control runs were performed using an HPLC separation gradient of 25% to 40% ACN in 60 minutes. Only samples with radiochemical purity >99% were used for the subsequent in vivo and imaging studies.
In Vitro Internalization and Efflux Studies
The degree of residualization in the PC-3 human prostate cancer cells was tested by incubating ∼20,000 cpm of 111In-1 with 3 × 104 PC-3 cells at 37 °C in an environment of 5% CO2 for various time periods. The PC-3 cells were suspended in an ice-cooled RPMI- medium comprised of 2.8 mg/mL HEPES and 2 mg/mL BSA (pH 7.4). After 15, 30, 45, 60, 90, and 120-minute incubation periods, the interaction between 111In-1 and the PC-3 cells was stopped by aspiration of the incubation medium and washing the cells three times with fresh ice-cooled cell culture medium. The PC-3 cell-surface-bound radioactivity was removed by incubating the PC-3 cells with an acidic buffer solution (pH 2.5) comprised of 0.2 M acetic acid and 0.5 M sodium chloride for five minutes. At the end of the incubation period, the PC-3 cells were precipitated from the buffer solution by centrifugation. The supernatant, containing the removed PC-3 cell-surface-bound radioactivity, was aspirated and the precipitated PC-3 cells were washed two times with fresh pH 2.5 buffer solution. Both the supernatant and the PC-3 cells were counted for their associated radioactivity and thereafter the percentage of both the internalized and the cell-surface-bound radioactivity was calculated.
The efflux studies were performed using the PC-3 cells at the time identified above with the highest percentage of 111In-1 internalization. The PC-3 cells after 45-minute incubation were allowed to efflux at 15, 30, 45, 60 and 90-minute periods. The PC-3 cells were treated and counted for their associated radioactivity as described above. The percentage of both the cell surface bound and the radioactivity effluxed was calculated.
Biodistribution Studies in Healthy CF-1 Mice
Twenty healthy CF-1 mice mice, with an average weight of 27 gm (five mice per time point), were injected with 100 μL of 0.148 MBq (4 μCi) aliquots of 111In-1 via the tail vein. The mice were sacrificed by neck dislocation at 0.25, 1, 4-, and 24 hours postinjection. Blood and urine were collected immediately after the mice were sacrificed. The organs and tissues of interest were excised and weighed, and their associated radioactivity was counted using an NaI(Tl) well counter. The small and large intestines as well as the stomach were weighed and counted without evacuating their contents. The % ID of the whole blood weight and % ID/g blood were calculated assuming that the whole blood volume constituted 6.5% of the whole mouse body weight. The distribution of radioactivity was calculated as a percentage of the injected dose per organ (% ID/organ) and as a percentage of the injected dose per gram (% ID/g), using an in-house program.
Blocking Studies
Three mice were injected with 111In-1 (100 μL, 0.185 MBq, 0.5 μCi) aliquots via the tail vein. Three mice were injected with 111In-1 (100 μL, 0.185 MBq, 5 μCi) along with 100 μg BBN via the tail vein. The mice were sacrificed by neck dislocation one hour postinjection. The organs and tissues of interest were excised, weighed and their associated radioactivity was counted. Distribution of radioactivity was calculated as % ID/organ, and % ID/g using an in-house program.
Pharmacokinetic Studies in PC-3 Tumor-Bearing SCID Mice
PC-3 cells were suspended in the culture media, centrifuged and the culture media was decanted. The resultant PC-3 cells' pellets were suspended in Dulbecco's phosphate-buffered saline solution. The concentrations of the PC-3 cells were adjusted to approximately 5 million cells per 100 μL of Dulbecco's phosphate-buffered saline solution. Four-to-five-week old ICR (Institute of Cancer Research) severely compromised immunodeficient (SCID) female mice were obtained from Taconic Farms. The mice were fed with the supplied irradiated rodent chow and water (Ralston Purina Corporation) ad libitum. Each group of five mice was housed in a sterile microisolator cage (Alternative Design) in temperature-and humidity-controlled rooms with alternating twelve-hour periods of light and darkness. The mice were anesthetized for PC-3 cells inoculation with a gas composed of ∼3.5% isoflurane (Baxter Healthcare Corp.) and ∼1 L/min oxygen. The gas anesthesia was administrated to SCID mice using a non-rebreathing multi-terminal anesthesia vaporizer apparatus (Summit Medical Equipment Co.). Approximately five million PC-3 cells suspended in 100 μL of normal sterile saline were inoculated subcutaneously in the bilateral flank of each SCID mice under gas anesthesia. The tumors were allowed to grow (ranging from 0.2 to 1.0 g) in the flanks of the SCID mice for 3-4 weeks after inoculation. The biodistribution was studied by injecting twenty mice, of average weight of 23.4 g, (five mice per time point) with 100 μL (0.185 MBq, 5 μCi) aliquots of 111In-1 via the tail vein. The mice were sacrificed by neck dislocation at 0.25, 0.5, 1, and 4 hours postinjection. Blood and urine were collected immediately after the mice were sacrificed. The organs and tissues of interest were processed as described above.
MicroSPECT/MicroCT Imaging Studies
The microSPECT imaging was performed using a microCAT II® SPECT/CT (Siemens Medical Solutions) small animal system composing of dual SPECT system. The microCT images performed using MicroCT unit (microCAT II® MicroCT scanner, CTI, Knoxville, TN). The tomographic SPECT imaging of a PC-3 tumor bearing SCID mouse was performed at 1-hr postinjection with 111In-1 (11.1 MBq, 300 μCi). The mouse was sacrificed at 1-hr after administration of 111In-1, and scanned for γ-ray acquisition. The mouse was aligned at the center of the scanner using a laser beam. The image was collected with the detectors fully calibrated and normalized for 111In. The mouse was scanned for 60 projections (rotation steps) over 360°. Reconstructed SPECT mouse data generated with a 3D-OSEM algorithm with geometrical corrections. The reconstructed images were smoothed post reconstruction with a 3D Gaussian kernel. A concurrent microCT scan was performed on the same mouse for future anatomical co-registration of the SPECT data.
Results and discussion
Synthesis of Construct 1
Bombesin and GRP most likely exert their effects through binding to the membrane surface G-protein bound receptors characterized by their similar seven trans-membrane domains which cluster to form the peptide binding pocket (2). The bombesin analogue, [D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN[6-14], was developed as a potential antagonist binding motif for targeting GRP-receptors (35, 36). Placing D-Phe in position 6 was based on the reported results indicating that placing D- or an unnatural amino acid in the peptide increases the resistance to proteolysis (35, 36). The readily oxidized Met14 residue was removed so that leucine became the C-terminal residue in position 13. This terminus was derivatized upon cleavage from the resin with propylamine, resulting in a structural change that confers antagonist behavior on the peptide. DOTA was selected as a bifunctional chelating agent to confer high thermodynamic stability and kinetic inertness to the formed 111In chelate. Since it is reported that the length of the hydrocarbon spacer could be varied from at least 3 to 8 carbon atoms without interrupting the binding affinity to the GRP-receptors (26), we linked DOTA to the peptide through an aminohexanoyl moiety. The construct, DOTA-aminohexanoyl-[D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN[6-14] (1), “Bomproamide,” was synthesized on HMBA resin as shown in Scheme 1 using conventional solid phase peptide synthesis strategies and our published protocols (34). Briefly, coupling of Fmoc-6-aminohexanoic acid and DOTA tris-(t-Bu ester) were performed sequentially by conventional in situ carboxy activation using HBTU and HOBT to afford 3 and 4, respectively. Side chain deprotection using TFA yielded DOTA-hexanoyl-D-Phe6-BBN [6-13]-HMBA resin. Treatment with TEA/propylamine/THF (1:5:5 v/v) overnight at 50 °C achieved cleavage of the peptide from the resin with concomitant formation of the C-terminal propylamide to afford 1. After purification of 1 by RP-HPLC, 23 mg (∼13%) in > 92 % purity was recovered and was characterized using ESI-MS.
Scheme 1.

In Vitro Competitive Cell Binding–Affinity Assay
The in vitro competitive cell binding affinity experiments were performed with 1 against 125I-Tyr4-BBN as the GRP-receptor radioligand. The experiments were performed with immortalized human androgen-insensitive prostate cancer PC-3 cells that are known to over-express the GRP-receptors predominantly. 1 retained high affinity (1.36 ± 0.09 nM) toward the GRP-receptor subtype.
Labeling with natIn and 111In and Characterization
The metallation of 1 was performed under standard conditions and was analyzed by LC-MS. Results indicate that the HPLC peak at tR= 23.5 min (k′ = 8.7) corresponds to the natIn-DOTA-peptide, natIn-1, in 89% purity. The radiolabeling of 1 with 111In was performed and the HPLC chromatograms show the coincidence of the major HPLC radio-peak with the UV-peak of natIn-1, offset only by the order of respective detectors of the HPLC. The isolation of pure 111In-1 was achieved by two modifications of a standard radiolabeling protocol prior to preparative HPLC. First, EDTA was added at the end of the radiolabeling period to chelate any remaining non-complexed (“free”) 111In3+. In addition, a few crystals of CuSO4·5H2O were added to complex excess 1. Aliquots (25 μL) of the radiolabeling mixture were analyzed by HPLC. The chromatogram depicts that most of the detected radioactivity was located at tR= 18.86 min corresponding to 111In-DOTA-peptide conjugate, 111In-1, while the UV-peak corresponding to the unlabeled DOTA-peptide conjugate, 1 was absent. The UV peak at longer retention time (tR = 20 min) is assigned as natCu-1. Pure 111In-1 was obtained through preparative HPLC in 75%-80% radiochemical yield in high radiochemical purity. The corresponding UV chromatogram confirms the absence of 1 or natCu-1. This result is important as the presence of excess 1 or natCu-1 could saturate the receptors in vivo. Only samples with radiochemical purity >99% were used for the subsequent in vivo studies. Concentration of the HPLC eluent containing 111In-1 was achieved through solid phase extraction. Approximately 90%-94% of the original radioactivity could be recovered after the formulation.
In Vitro Internalization and Efflux Studies
Internalization is the process of radioconjugate accumulation over time within the cells expressing the targeting receptors. The internalization behavior of 111In-1 in the human PC-3 prostate cancer cells was studied by incubation at 37 °C for 15-, 30-, 45-, 60-, 90-, and 120- minutes. The percentage of 111In-1 trapping within the cells was characterized by a steady and gradual increasing rate of accumulation within the cells up to 45 minutes of incubation (Figure 2). Internalization reaches its maximum value, ∼14%, after 45 minutes of incubation. The percentage of surface-bound radioactivity exceeded the percentage of the internalized activity over the entire investigated time points of incubation. The internalization does not increase by extending the incubation period beyond 45 minutes. The percentage of the internalized radioligand decreases to 12% at 60 minutes of incubation and then plateaus to 120 minutes of incubation. The level of internalization of 111In-1 is consistent with its antagonist-based structure.
Figure 2.
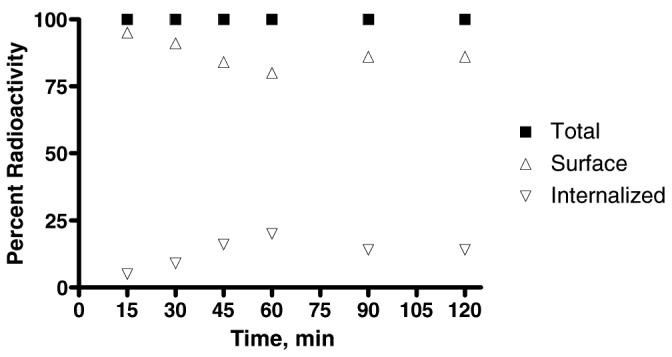
Internalization of 111In-1 in Prostate Cancer PC-3 Cells as a function of time. (n = 2; trials conducted in duplicate).
Efflux, on the other hand, is the process of radioconjugate externalization over time from the cells expressing the targeted receptors. The externalization behavior of 111In-1 from the PC-3 cells was observed after a 45 minute incubation, the time at which the highest internalization percentage (14%) had been noted. Efflux of radioactivity was studied over 15-, 30-, 45-, 60-, and 90-minutes. The internalized radioactivity remained almost constant over the investigated efflux time points (data not shown). This behavior is consistent with prior trapping a small amount of 111In-1 within the cells, and a significantly slow washout.
In Vivo Biodistribution Studies
The usefulness of bombesin-based constructs for in vivo GRP-Receptor targeting has been primarily assessed using agonists. Very little is known about the potential of using antagonist-based constructs as suitable biomarkers for in vivo GRP-Receptor imaging (33). Thus, the in vivo targeting behavior of 111In-1 for GRP-receptors was studied using healthy (CF-1) and tumor-bearing SCID mice.
Pharmacokinetic Studies in Healthy CF-1 Mice
The biodistribution behavior of 111In-1 in healthy CF-1 mice at 0.25-, 1-, 4-, and 24-hour post-injection are presented in Table 1. The pancreas is the primary non-malignant tissue that normally expresses a high density of GRP cell-surface receptors (37). It is well established that accumulation of the radioactivity in the pancreatic tissue in preference to other tissues is a direct reflection of the efficiency of the radioconjugate under evaluation for in vivo targeting of GRP-cell surface receptors (13, 26, 33). The results indicate that 111In-1 demonstrates rapid (0.25-hour PI) and high (12.21 ± 3.2 % ID/g) uptake in the pancreatic tissue. This pancreatic uptake was reduced to 0.95 ± 0.26 % ID/g at 1-hr PI and diminished to 0.01 ± 0.01 %ID/g at 4-hr PI. The observed decrease of pancreatic uptake was consistent with the suspected uptake behavior of an antagonist-based construct that exhibits limited internalization. It has been reported that the pancreatic uptake of Demobesin-1, decreases from 33.13 ± 3.63 % ID/g at 1-hr PI to 4.59 ± 0.63 % ID/g at 4-hr PI (33). Marginal uptake was observed in the liver (1.33 ± 0.24 % ID/g), and abdominal area at 0.25-hr PI. The liver uptake diminishes significantly (0.18 ± 0.02 % ID/g) at 1-hr PI. Similarly, the abdominal uptake decreased markedly [small (0.35 ± 0.03 % ID/g) and large (0.20 ± 0.05 % ID/g) intestines] at 1-hr PI. 111In-1 demonstrated rapid clearance from the blood (0.17 ± 0.05 % ID/g at 1-hr PI) mainly by renal extraction (1.74 ± 0.32 % ID/g at 1-hr PI) and urinary tract excretion (0.86 ± 0.46 % ID/g at 1-hr PI) into the urine (94.70 ± 0.62 % ID at 1-hr PI).
Table 1.
Biodistribution of 111In-1 in Healthy CF-1 Mice at 0.25-, 1-, 4- and 24-hour PI, (n = 5).a
| Organ/Tissue | 0.25-hr PI | 1-hr PI | 4-hr PI | 24-hr PI |
|---|---|---|---|---|
| Bladder | 3.34 ± 0.59 | 0.86 ± 0.46 | 0.05 ± 0.06 | 0.04 ± 0.6 |
| Heart | 1.52 ± 0.49 | 0.10 ± 0.06 | 0.03 ± 0.01 | 0.00 ± 0.01 |
| Lung | 2.00 ± 0.50 | 0.30 ± 0.09 | 0.29 ± 0.19 | 0.08 ± 0.06 |
| Liver | 1.33 ± 0.24 | 0.18 ± 0.02 | 0.07 ± 0.01 | 0.02 ± 0.01 |
| Kidneys | 7.56 ± 0.89 | 1.74 ± 0.32 | 0.90 ± 0.11 | 0.09 ± 0.03 |
| Spleen | 0.83 ± 0.27 | 0.10 ± 0.02 | 0.03 ± 0.01 | 0.01 ± 0.01 |
| Stomach | 0.75 ± 0.13 | 0.17 ± 0.03 | 0.06 ± 0.02 | 0.00 ± 0.00 |
| S. Intestine | 1.74 ± 0.30 | 0.35 ± 0.03 | 0.14 ± 0.05 | 0.01 ± 0.00 |
| L. Intestine | 1.37 ± 0.41 | 0.20 ± 0.05 | 0.65 ± 0.08 | 0.04 ± 0.01 |
| Muscle | 0.81 ± 0.19 | 0.07 ± 0.01 | 0.01 ± 0.01 | 0.00 ± 0.00 |
| Bone | 1.07 ± 0.35 | 0.11 ± 0.08 | 0.02 ± 0.01 | 0.02 ± 0.02 |
| Brain | 0.20 ± 0.04 | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 |
| Pancreas | 12.21 ± 3.2 | 0.95 ± 0.26 | 0.31 ± 0.04 | 0.01 ± 0.01 |
| Blood | 3.30 ± 2.98 | 0.17 ± 0.05 | 0.01 ± 0.00 | 0.00 ± 0.00 |
| Carcass | 1.61 ± 0.11 | 0.17 ± 0.04 | 0.03 ± 0.00 | 0.00 ± 0.00 |
| Excretion (% ID ± SD) | 52.65 ± 4.65 | 94.70 ± 0.62 | 97.96 ± 0.16 | 98.80 ± 0.05 |
All data, except excretion, are presented as an average % ID/g ± SD.
Blocking Studies
The effect of blocking the GRP-receptors on the pancreatic uptake of 111In-1 was investigated in CF-1 mice (Table 2). More than 90% of the GRP-Receptors mediated radioconjugate uptake in the pancreatic tissue was blocked by co-injection of 100 μg of BBN. The pancreatic uptake in the control mice was 2.04 ± 0.17 % ID/g at 1-hr post-injection and was significantly decreased by a factor of ten to 0.20 ± 0.01 % ID/g by blocking the binding sites with BBN. The remaining activity in the pancreas may be a reflection of GRP-Receptor subtype mediated trapping of 111In-1. Uptake studies in healthy CF-1 mice at 1-hr PI showed pancreatic uptake of 0.95 ± 0.26 % ID/g and lung uptake of 0.30 ± 0.09 % ID/g (Table 1). However, the blocking studies in healthy CF-1 control mice at 1-hr post injection showed pancreatic uptake of 2.04 ± 0.17 %ID/g and lung uptake of 1.37 ± 0.08 % ID/g (Table 2). The observed difference in the pancreas and lung uptakes between these two studies might be due to the differences in the density of normal GRP-receptor expression. This expectation is strongly confirmed by the substantial decrease, ∼75% less, in uptake (0.35 ± 0.05 % ID/g) of the blocked lung tissue.
TABLE 2.
Biodistribution of 111In-1 in Healthy CF-1 Mice at 1-hr Post-Injection in the presence and absence of 100 μg BBN (n = 3).a
| Organ/Tissues | Blocked | Control |
|---|---|---|
| Bladder | 1.96 ± 1.39 | 0.99 ± 0.69 |
| Heart | 0.13 ± 0.02 | 0.09 ± 0.04 |
| Lung | 0.35 ± 0.05 | 1.37 ± 0.08 |
| Liver | 0.24 ± 0.01 | 0.26 ± 0.07 |
| Kidneys | 1.44 ± 0.26 | 1.54 ± 0.16 |
| Spleen | 0.11 ± 0.04 | 0.17 ± 0.03 |
| Stomach | 0.11 ± 0.03 | 0.38 ± 0.07 |
| S. Intestine | 0.69 ± 0.11 | 0.90 ± 0.28 |
| L. Intestine | 0.13 ± 0.03 | 0.44 ± 0.31 |
| Muscle | 0.06 ± 0.01 | 0.04 ± 0.01 |
| Bone | 0.09 ± 0.03 | 0.07 ± 0.01 |
| Brain | 0.02 ± 0.00 | 0.01 ± 0.00 |
| Pancreas | 0.20 ± 0.01 | 2.04 ± 0.17 |
| Blood | 0.24 ± 0.02 | 0.16 ± 0.09 |
| Carcass | 0.20 ± 0.02 | 0.16 ± 0.01 |
| Excretion (% ID ± SD) | 91.25 ± 0.44 | 90.97 ± 1.08 |
All data, except excretion, are presented as an average %ID/g ± SD.
Pharmacokinetic Studies in PC-3 Tumor-Bearing SCID Mice
The pharmacokinetic studies in healthy CF-1 mice reflect a significant decrease of 111In-1 in the tissues with time. At 24 hours PI, the investigated tissues retained insignificant radioactivity. Accordingly, 0.5-hr PI was selected as a new time point for investigating the pharmacokinetic behavior of 111In-1 in PC-3 tumor-bearing SCID mice, and the 24-hr time point was excluded. The biodistribution behavior of 111In-1 in immortalized prostate cancer PC-3 tumor bearing mice at 0.25-, 0.5-, 1-, and 4-hour post-injection are presented in Table 3. Rapid (15 minutes post-injection) and significantly high uptake (6.90 ± 1.06 % ID/g) as observed in the tumors. The tumor uptake decreased over time to a value at 4-hr PI that was approximately 30% that found at 15 minutes (2.15 ± 0.36 % ID/g). In contrast to the prolonged uptake of 111In-1 in the tumor tissue, fast clearance from the other tissues was observed. This behavior is consistent with the reported data of antagonist-based Demobesin 1 (33). In these animals, 111In-1 demonstrated rapid (0.25-hour PI) and significantly high (14.88 ± 1.8 % ID/g) uptake in the pancreatic tissue. This pancreatic uptake showed a steep reduction to 3.99 ± 0.49 % ID/g at 0.5-hr PI and to 0.96 ± 0.18 % ID/g at 1-hr PI. The pancreatic uptake behavior in PC-3 tumor bearing mice mirrored its behavior in healthy CF-1 mice. The pancreatic uptake of 111In-1 at 4-hr PI decreased by ∼57% of its uptake at 1-hr PI. This behavior is comparable with the reported decrease (∼65%) of Demobesin-1 (33). These results are consistent with the marked internalization behavior of the agonist-based constructs and the limited internalization capabilities of the antagonist-based constructs. The observed high retention of 111In-1 in the tumor tissue over the investigated time points may be due to in part to that the human tumor xenografts are supported by murine vasculature architecture (26). It is also reported that antagonists form slowly dissociating antagonist-receptor complexes that result in long-lasting in vivo receptor binding (38). On the other hand, the low retention of 111In-1 in the pancreatic tissue may be due to excessive enzymatic activity of the pancreatic tissue as well as the limited internalization capabilities of the antagonist-based constructs (33). The high uptake and retention in the tumors and pancreas cells over the entire investigated time points is a direct reflection of the efficiency of antagonist-based constructs for in vivo targeting of the GRP cell surface receptors. Therefore, antagonist-based constructs seem to be the candidate of choice for in vivo imaging purposes, while for in vivo therapy purposes, the agonist-based constructs (trigger the internalization process that confer the basis for an efficient accumulation of the radioconjugate within the cells over the time) seem to be the candidate of choice.
Table 3.
Biodistribution of 111In-1 in PC-3 Tumor Bearing SCID Mice (n = 5).a
| Organ/Tissue | 0.25-hr PI | 0. 5-hr PI | 1-hr PI | 4-hr PI |
|---|---|---|---|---|
| Bladder | 14.09 ± 21.1 | 7.03 ± 8.68 | 1.67 ± 1.06 | 0.26 ± 0.14 |
| Heart | 1.85 ± 0.27 | 1.00 ± 0.30 | 0.16 ± 0.06 | 0.03 ± 0.02 |
| Lung | 4.45 ± 0.82 | 3.56 ± 1.72 | 1.78 ± 1.64 | 1.48 ± 2.54 |
| Liver | 2.14 ± 0.14 | 0.88 ± 0.20 | 0.27 ± 0.07 | 0.12±0.02 |
| Kidneys | 11.84 ± 1.97 | 5.88 ± 1.91 | 3.06 ± 0.92 | 2.24±1.02 |
| Spleen | 0.98 ± 0.37 | 0.59 ± 0.27 | 0.15 ± 0.08 | 0.06 ± 0.02 |
| Stomach | 1.91 ± 0.29 | 1.17 ± 0.37 | 0.34 ± 0.07 | 0.05 ± 0.03 |
| S. Intestine | 3.41 ± 0.54 | 1.46 ± 0.35 | 0.62 ± 0.19 | 0.18 ± 0.11 |
| L. Intestine | 1.64 ± 0.29 | 0.59 ± 0.07 | 0.54 ± 0.68 | 0.88 ± 0.39 |
| Muscle | 1.00 ± 0.10 | 0.40 ± 0.13 | 0.07 ± 0.04 | 0.02 ± 0.01 |
| Bone | 1.21 ± 0.42 | 0.78 ± 0.40 | 0.08 ± 0.02 | 0.05 ± 0.07 |
| Brain | 0.17 ± 0.06 | 0.12 ± 0.04 | 0.02 ± 0.01 | 0.00 ± 0.00 |
| Pancreas | 14.88 ± 1.86 | 3.99 ± 0.49 | 0.96 ± 0.18 | 0.41 ± 0.14 |
| Tumor#1 | 7.33 ± 1.18 | 5.41 ± 1.98 | 3.91 ± 0.46 | 2.02 ± 0.34 |
| Tumor#2 | 6.47 ± 0.82 | 6.11 ± 1.04 | 3.52 ± 0.70 | 2.29 ± 0.36 |
| Blood | 4.28 ± 0.59 | 1.85 ± 0.47 | 0.33 ± 0.15 | 0.02 ± 0.02 |
| Carcass | 2.26 ± 0.34 | 1.16 ± 0.41 | 0.39 ± 0.38 | 0.05 ± 0.03 |
| Excretion (% ID ± SD) | 40.86 ± 4.60 | 68.70 ± 7.55 | 89.15 ± 7.11 | 96.16 ± 1.06 |
| Tumors | 6.90 ± 1.06 | 5.76 ± 1.54 | 3.72 ± 0.60 | 2.15 ± 0.36 |
All data, except excretion, are presented as an average % ID/g ± SD.
Significant uptake of 4.45 ± 0.82 %ID/g was observed in the lung tissue at 0.25-hr PI and retained over the entire investigated time points. This lung uptake may be in part due to the normal expression of the GRP-Receptors in this tissue (39). Lung tissue is one of the Gastro-Entero-Pancreatic-Neuroendocrine tissues (GEP-NET) that is characterized by normal expression of GRP-Receptors in the interface between the endocrine hormonal cells and nervous cells (37). It is also reported that the three bombesin family receptors are expressed with various degrees in overlapping subsets of lung cancer cell lines (40, 41). The committed lung uptake at the various investigated time points may act as a prognostic biomarker for early detection of small cell lung cancer (SCLC) and non-small lung cells (NSLC) carcinogenesis. More research would have to be conducted to explore this hypothesis further.
Low uptake of 111In-1 was observed in the liver (2.14 ± 0.14 % ID/g), and abdominal area. The liver uptake decreased (0.88 ± 0.2 % ID/g) at 0.5-hr post-injection. Similarly, the abdominal uptake decreased markedly [small (1.46 ± 0.35 % ID/g) and large (0.59 ± 0.07 % ID/g) intestines] at 0.5-hr PI. 111In-1 demonstrated rapid clearance from the blood (1.85 ± 0.47 % ID/g at 0.5-hr PI) mainly by renal extraction (5.88 ± 1.91 % ID/g at 0.5-hr PI) and urinary tract excretion (7.03 ± 8.68 % ID/g at 0.5-hr PI) into the urine (68.70 ± 7.55 % ID at 0.5-hr PI).
Tumor to Nontumor Uptake Ratios of 111In-1
The main pharmacokinetic consideration in development of peptide-based imaging agents is to achieve diagnostically useful tumor to nontumor uptake ratios (∼ 5) with minimal time required to achieve them. Table 4 shows the tumor/nontumor uptake ratios of 111In-1 at 0.25-, 0.5-, 1-, 4-hours post-injection. At the same time, significantly high (> 10) and diagnostically useful ratios were achieved in most of the tissues after 1-hr post injection. The tumor/nontumor uptake ratios in certain tissues (muscle, spleen) were significantly high, > 20, at both 1- and 4-hrs PI. Comparable (difference less than five) target-to-nontarget uptake ratios were achieved at 1- and 4-hr PI in certain tissues (liver and small intestine). Higher tumor/nontumor uptake ratios were achieved at 1-hr PI, rather than 4-hr, in certain tissues (kidneys, lung and large intestine). For prostate cancer radioimaging, there is high demand for achieving minimal possible noise in the abdominal area (large intestine, small intestine, stomach, liver, kidneys and bladder) in a minimal possible time. It is clear from the tumor/nontumor uptake ratios that diagnostically useful ratios in mice could be achieved in most of the abdominal tissues (except bladder) at 1-hr PI. Accordingly, microSPECT imaging of PC-3 tumor xenografts in SCID mouse was performed at 1-hr PI.
Table 4.
Target-to-Nontumor uptake ratios of 111In-1 in PC-3 tumor-bearing SCID mice (n = 5).
| Tumor/Nontumor Tissue | 0.25-hr PI | 0.5-hr PI | 1-hr PI | 4-hr PI |
|---|---|---|---|---|
| Tumor/Blood | 1.62 | 2.87 | 10.76 | 108.5 |
| Tumor/Muscle | 6.92 | 13.28 | 50.71 | 108.5 |
| Tumor/Liver | 3.23 | 6.03 | 13.15 | 18.08 |
| Tumor/Kidneys | 0.58 | 0.90 | 1.16 | 0.97 |
| Tumor/Lung | 1.56 | 1.49 | 1.99 | 1.47 |
| Tumor/Bladder | 0.49 | 0.76 | 2.13 | 8.35 |
| Tumor/Spleen | 7.06 | 9.00 | 23.67 | 36.17 |
| Tumor/Stomach | 3.62 | 4.54 | 10.44 | 43.4 |
| Tumor/L. Intestine | 4.33 | 9.00 | 6.57 | 2.47 |
| Tumor/S. Intestine | 2.03 | 3.64 | 5.73 | 12.06 |
MicroSPECT/Micro CT Imaging Studies
MicroSPECT/CT images of immortalized prostate cancer-xenografts, Figure 3, was taken over 16 hours after 1-hr PI of 11.1 MBq (300 μCi) of 111In-1 in a PC-3 tumor-bearing SCID mouse. The camera head was positioned at multiple angles around the mouse body accumulating as many as 360° of views at specific angular intervals. The radioactivity associated with the images was acquired over time due to the rapid clearance properties of 111In-1. Reconstruction process was performed to retrieve the radiotracer spatial distribution from the projection data using a fully 3D OSEM algorithm. Regions where backprojection lines from different angles intersect represent areas that contain a higher concentration of radiopharmaceutical. The PC-3 tumor-xenografts in the SCID mouse were visualized clearly in the images. Delineating the kidneys in the image is an indication of the radioconjugate clearance via renal excretion. The images revealed the absence of the liver's anatomical boundaries and delineated the insignificance of radioactivity uptake in the gastrointestinal tract. The radioactivity distribution and accumulation in the microSPECT/CT images are consistent with the biodistribution data obtained from the pharmacokinetic studies. The power of the presented diagnostic imaging lies in its external detection of the in vivo radioactivity delineating anatomical boundaries and having a clue of status of the organs being imaged.
Figure 3.
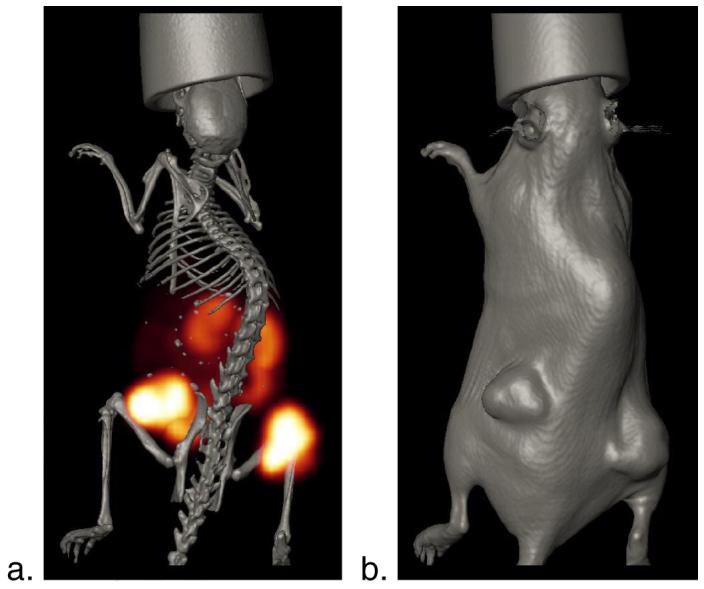
a. Axial MicroSPECT/CT Image of PC-3 Xenografts in a SCID mouse after 1 hour of 111In-1 administration; b. Skin CT image. Details of acquisition are found in the Materials and Methods.
Conclusion
The bombesin antagonist, [D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN [6-14], was synthesized and developed into 111In-DOTA-aminohexanoyl-[D-Phe6, Leu-NHCH2CH2CH313, des Met14] BBN [6-14] (111In-1), “Bomproamide,” a high affinity imaging agent targeting GRP-receptor. This study demonstrates the potential of using the bombesin analogues possessing antagonist C-terminal vectors as a diagnostic candidate of choice for specific in vivo targeting of the tumors over-expressing GRP-receptors.
Acknowledgements
The authors acknowledge the National Cancer Institute (P50 CA 103130: Center for Single Photon-Emitting Cancer Imaging Agents, W.A. Volkert, PI) for partial support of this research. This work was supported with the resources and the use of facilities at the Chemistry Department, University of Missouri-Columbia and Harry S. Truman Memorial VA Hospital.
Contributor Information
Abd-Elgaliel Wael, University of Missouri - Columbia, Chemistry.
Garrison Jered, University of Missouri - Columbia, Internal Medicine.
Rold Tammy, University of Missouri - Columbia, Internal Medicine.
Sieckman Gary, Harry S Truman Veterans' Hospital, Research.
Daibes Figueroa Said, University of Missouri - Columbia, Internal Medicine.
Hoffman Timothy, University of Missouri-Columbia, Radiopharmaceutical Sciences Institute.
Lever Susan, University of Missouri.
References
- 1.Anastasi A, Erspamer V, Bucci M. Isolation and structure of bombesin and alytesin, 2 analogous active peptides from the skin of the European amphibians Bombina and Alytes. Experientia. 1971;27:166–7. doi: 10.1007/BF02145873. [DOI] [PubMed] [Google Scholar]
- 2.McDonald TJ, Jornvall H, Nilsson G, Vag-Bloom SR, Mutt V. Characterization of a gastrin releasing peptide from porcine non-antral gastric tissue. Biochem. Biophys. Comm. 1979;90:227–233. doi: 10.1016/0006-291x(79)91614-0. [DOI] [PubMed] [Google Scholar]
- 3.Ohki-Hamazaki H, Iwabuchi M, Maekawa F. Development and function of bombesin-like peptides and their receptors. Int. J. Dev. Biol. 2005;49:293–300. doi: 10.1387/ijdb.041954ho. [DOI] [PubMed] [Google Scholar]
- 4.Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging. 2003;30:781–93. doi: 10.1007/s00259-003-1184-3. [DOI] [PubMed] [Google Scholar]
- 5.Reubi JC, Korner M, Waser B, Mazzucchelli L, Guillou L. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur. J. Nucl. Med. Mol. Imaging. 2004;31:803–10. doi: 10.1007/s00259-004-1476-2. [DOI] [PubMed] [Google Scholar]
- 6.Reubi JC, Gugger M, Waser B. Co-expressed peptide receptors in breast cancers as molecular basis for in vivo multireceptor tumor targeting. Eur. J. Nucl. Med. Mol. Imaging. 2002;29:855–862. doi: 10.1007/s00259-002-0794-5. [DOI] [PubMed] [Google Scholar]
- 7.Darker JG, Brough SJ, Heath J, Smart D. Discovery of potent and selective peptide agonists at the GRP-preferring bombesin receptor (BB2) J. Pept. Sci. 2001;7:598–605. doi: 10.1002/psc.359. [DOI] [PubMed] [Google Scholar]
- 8.Fleischmann A, Laderach U, Friess H, Buechler MW, Reubi JC. Bombesin receptors in distinct tissue compartments of human pancreatic diseases. Lab. Invest. 2000;80:1807–17. doi: 10.1038/labinvest.3780192. [DOI] [PubMed] [Google Scholar]
- 9.Reubi JC. In vitro identification of vasoactive intestinal peptide receptors in human tumors: implications for tumor imaging. J. Nucl. Med. 1995;36:1846–53. [PubMed] [Google Scholar]
- 10.Rogers BE, Parry JJ, Andrews R, Cordopatis P, Nock BA, Maina T. MicroPET imaging of gene transfer with a somatostatin receptor-based reporter gene and (94m)Tc-Demotate 1. J. Nucl. Med. 2005;46:1889–97. [PubMed] [Google Scholar]
- 11.Reubi JC. Neuropeptide receptors in health and disease: the molecular basis for in vivo imaging. J. Nucl. Med. 1995;36:1825–35. [PubMed] [Google Scholar]
- 12.Van de Wiele C, Dumont F, Vanden Broecke R, Oosterlinck W, Cocquyt V, Serreyn R, Peers S, Thornback J, Slegers G, Dierckx RA. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: a feasibility study. Eur. J. Nucl. Med. 2000;27:1694–9. doi: 10.1007/s002590000355. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Chen J, Waldherr C, Hinni K, Waser B, Reubi JC, Maecke HR. Synthesis and evaluation of bombesin derivatives on the basis of pan-bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for targeting bombesin receptor-expressing tumors. Cancer Res. 2004;64:6707–15. doi: 10.1158/0008-5472.CAN-03-3845. [DOI] [PubMed] [Google Scholar]
- 14.Lin KS, Luu A, Baidoo KE, Hashemzadeh-Gargari H, Chen MK, Brenneman K, Pili R, Pomper M, Carducci MA, Wagner HN., Jr. A new high affinity technetium-99m-bombesin analogue with low abdominal accumulation. Bioconjugate Chem. 2005;16:43–50. doi: 10.1021/bc049820h. [DOI] [PubMed] [Google Scholar]
- 15.Nock BA, Nikolopoulou A, Galanis A, Cordopatis P, Waser B, Reubi JC, Maina T. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: a preclinical study. J. Med. Chem. 2005;48:100–10. doi: 10.1021/jm049437y. [DOI] [PubMed] [Google Scholar]
- 16.Waser B, Eltschinger V, Linder K, Nunn A, Reubi JC. Selective in vitro targeting of GRP and NMB receptors in human tumours with the new bombesin tracer 177Lu-AMBA. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:95–100. doi: 10.1007/s00259-006-0229-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Schuhmacher J, Waser B, Wild D, Eisenhut M, Reubi JC, Maecke HR. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for the imaging and targeted radionuclide treatment of bombesin receptor-positive tumours. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:1198–208. doi: 10.1007/s00259-006-0347-4. [DOI] [PubMed] [Google Scholar]
- 18.Garrison JC, Rold TL, Sieckman GL, Figueroa SD, Volkert WA, Jurisson SS, Hoffman TJ. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using 64Cu bombesin analogs: side-by-side comparison of the CB-TE2A and DOTA chelation systems. J. Nucl. Med. 2007;48:1327–37. doi: 10.2967/jnumed.107.039487. [DOI] [PubMed] [Google Scholar]
- 19.de Visser M, Bernard HF, Erion JL, Schmidt MA, Srinivasan A, Waser B, Reubi JC, Krenning EP, de Jong M. Novel 111In-labelled bombesin analogues for molecular imaging of prostate tumours. Eur. J. Nucl. Med. Mol. Imaging. 2007;34:1228–38. doi: 10.1007/s00259-006-0356-3. [DOI] [PubMed] [Google Scholar]
- 20.Maina T, Nock B, Mather S. Targeting prostate cancer with radiolabelled bombesins. Cancer Imaging. 2006;6:153–7. doi: 10.1102/1470-7330.2006.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma L, Yu P, Veerendra B, Rold TL, Retzloff L, Prasanphanich A, Sieckman G, Hoffman TJ, Volkert WA, Smith CJ. In vitro and in vivo evaluation of Alexa Fluor 680-bombesin[7-14]NH2 peptide conjugate, a high-affinity fluorescent probe with high selectivity for the gastrin-releasing peptide receptor. Mol. Imaging. 2007;6:171–80. [PubMed] [Google Scholar]
- 22.Lantry LE, Cappelletti E, Maddalena ME, Fox JS, Feng W, Chen J, Thomas R, Eaton SM, Bogdan NJ, Arunachalam T, Reubi JC, Raju N, Metcalfe EC, Lattuada L, Linder KE, Swenson RE, Tweedle MF, Nunn AD. 177Lu-AMBA: Synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J. Nucl. Med. 2006;47:1144–52. [PubMed] [Google Scholar]
- 23.Smith CJ, Sieckman GL, Owen NK, Hayes DL, Mazuru DG, Kannan R, Volkert WA, Hoffman TJ. Radiochemical investigations of gastrin-releasing peptide receptor-specific [(99m)Tc(X)(CO)3-Dpr-Ser-Ser-Ser-Gln-Trp-Ala-Val-Gly-His-Leu-Met-(NH2)] in PC-3, tumor-bearing, rodent models: syntheses, radiolabeling, and in vitro/in vivo studies where Dpr = 2,3-diaminopropionic acid and X = H2O or P(CH2OH)3. Cancer Res. 2003;63:4082–8. [PubMed] [Google Scholar]
- 24.Perrin MH, Sutton SW, Cervini LA, Rivier JE, Vale WW. Comparison of an agonist, urocortin, and an antagonist, astressin, as radioligands for characterization of corticotropin-releasing factor receptors. J. Pharmacol. Exp. Ther. 1999;288:729–34. [PubMed] [Google Scholar]
- 25.Prasanphanich AF, Nanda PK, Rold TL, Ma L, Lewis MR, Garrison JC, Hoffman TJ, Sieckman GL, Figueroa SD, Smith CJ. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12462–7. doi: 10.1073/pnas.0705347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffman TJ, Gali H, Smith CJ, Sieckman GL, Hayes DL, Owen NK, Volkert WA. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J. Nucl. Med. 2003;44:823–31. [PubMed] [Google Scholar]
- 27.Wang LH, Coy DH, Taylor JE, Jiang NY, Moreau JP, Huang SC, Frucht H, Haffar BM, Jensen RT. des-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J. Biol. Chem. 1990;265:15695–703. [PubMed] [Google Scholar]
- 28.Coy DH, Taylor JE, Jiang NY, Kim SH, Wang LH, Huang SC, Moreau JP, Gardner JD, Jensen RT. Short-chain pseudopeptide bombesin receptor antagonists with enhanced binding affinities for pancreatic acinar and Swiss 3T3 cells display strong antimitotic activity. J. Biol. Chem. 1989;264:14691–7. [PubMed] [Google Scholar]
- 29.Sleight AJ, Stam NJ, Mutel V, Vanderheyden PM. Radiolabelling of the human 5-HT2A receptor with an agonist, a partial agonist and an antagonist: effects on apparent agonist affinities. Biochem. Pharmacol. 1996;51:71–6. doi: 10.1016/0006-2952(95)02122-1. [DOI] [PubMed] [Google Scholar]
- 30.Cescato R, Maina T, Nock B, Nikolopoulou A, Charalambidis D, Piccand V, Reubi JC. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008;49:318–26. doi: 10.2967/jnumed.107.045054. [DOI] [PubMed] [Google Scholar]
- 31.Ginj M, Zhang H, Waser B, Cescato R, Wild D, Wang X, Erchegyi J, Rivier J, Macke HR, Reubi JC. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16436–41. doi: 10.1073/pnas.0607761103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coy DH, Jensen RT, Jiang NY, Lin JT, Bogden AE, Moreau JP. Systematic development of bombesin/gastrin-releasing peptide antagonists. J. Natl. Cancer Inst. Monogr. 1992:133–9. [PubMed] [Google Scholar]
- 33.Nock B, Nikolopoulou A, Chiotellis E, Loudos G, Maintas D, Reubi JC, Maina T. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur J Nucl Med Mol Imaging. 2003;30:247–58. doi: 10.1007/s00259-002-1040-x. [DOI] [PubMed] [Google Scholar]
- 34.Abd-Elgaliel WR, Gallazzi F, Lever SZ. Total solid-phase synthesis of bombesin analogs with different functional groups at the C-terminus. J. Pept. Sci. 2007;13:487–92. doi: 10.1002/psc.878. [DOI] [PubMed] [Google Scholar]
- 35.Lister-James J, Moyer BR, Dean RT. Pharmacokinetic considerations in the development of peptide-based imaging agents. Q. J. Nucl. Med. 1997;41:111–8. [PubMed] [Google Scholar]
- 36.Arano Y. Strategies to reduce renal radioactivity levels of antibody fragments. Q. J. Nucl. Med. 1998;42:262–70. [PubMed] [Google Scholar]
- 37.Gonzalez N, Moody TW, Igarashi H, Ito T, Jensen RT. Bombesin-related peptides and their receptors: recent advances in their role in physiology and disease states. Curr. Opin. Endocrinol. Diabetes Obes. 2007;15:58–64. doi: 10.1097/MED.0b013e3282f3709b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vauquelin G, Van Liefde I. Slow antagonist dissociation and long-lasting in vivo receptor protection. Trends Pharmacol. Sci. 2006;27:356–9. doi: 10.1016/j.tips.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Pelosi G, Volante M, Papotti M, Sonzogni A, Masullo M, Viale G. Peptide receptors in neuroendocrine tumors of the lung as potential tools for radionuclide diagnosis and therapy. Q. J. Nucl. Med. Mol. Imaging. 2006;50:272–87. [PubMed] [Google Scholar]
- 40.Corjay MH, Dobrzanski DJ, Way JM, Viallet J, Shapira H, Worland P, Sausville EA, Battey JF. Two distinct bombesin receptor subtypes are expressed and functional in human lung carcinoma cells. Bio. Chem. 1991;266:18771–18779. [PubMed] [Google Scholar]
- 41.Hohla F, Schally AV, Kanashiro CA, Buchholz S, Baker B, Kannadka C, Moder A, Aigner E, Datz C, Halmos G. Growth inhibition of non-small-cell lung carcinoma by BN/GRP antagonist is linked with suppression of K-Ras, COX-2, and pAkt. Proc. Natl. Acad. Sci. U.S.A. 2007;104:18671–6. doi: 10.1073/pnas.0709455104. [DOI] [PMC free article] [PubMed] [Google Scholar]