

Abstract
Glial cells secrete proinflammatory mediators in the brain in response to exogenous stimuli such as infection and injury. Previously, we documented that systemic maternal lipopolysaccharide (LPS)-exposure at E18 causes oligodendrocyte (OL)-injury/hypomyelination in the developing brain which can be attenuated by N-acetyl cysteine (NAC; precursor of glutathione). The present study delineates the underlying mechanism of NAC mediated attenuation of inhibition of OL development in LPS-stimulated mixed glial cultures. Factors released by LPS-stimulated mixed glial cultures inhibited the OL development as showed by decrease in both proliferation (BrdU+/NG2+) and differentiation (O4+ and MBP+) of OL-progenitors. Correspondingly, an impairment of peroxisomal proliferation was showed by decrease in the level of peroxisomal proteins in the developing OLs following exposure to LPS conditioned media (LCM). Both NAC and WY14643, a peroxisome proliferator-activated receptor (PPAR)-α agonist attenuated these LCM-induced effects in OL-progenitors. Similar to WY14643, NAC attenuated LCM-induced inhibition of PPAR-α activity in developing OLs. Studies conducted with cytokines and diamide (a thiol-depleting agent) confirmed that cytokines are active agents in LCM which may be responsible for inhibition of OL development via peroxisomal dysfunction and induction of oxidative-stress. These findings were further corroborated by similar treatment of developing OLs generated from PPAR-α (-/-) and wild-type mice or B12 oligodendroglial cells co-transfected with PPAR-α siRNAs/pTK-PPREx3-Luc plasmids. Collectively, these data provide evidence that the modulation of PPAR-α activity thus peroxisomal function by NAC attenuates LPS-induced glial factors mediated inhibition of OL development suggesting new therapeutic interventions to prevent the devastating effects of maternal infections.
Keywords: Lipopolysaccharide, oligodendrocyte, cerebral white matter injury, reactive oxygen species, peroxisome proliferator-activated receptor-α and N-acetyl cysteine
INTRODUCTION
Maternal systemic infection and inflammation are two important factors involved in the development of cerebral white matter injury (hypomyelination) in premature infants (Dammann & Leviton 1997, Back & Rivkees 2004). Systemic inflammation subsequent to maternal systemic infection gives rise to the secretion of proinflammatory mediators which are toxic to the developing fetal brain (Bell et al. 2004). This is followed by activation of resident brain glial cells and the inhibition of the differentiation of oligodendrocyte (OL) progenitors into myelin-forming OLs (Back et al. 2002, Leviton & Gilles 1996) which contributes to neonatal brain injury and later developmental disability (Dammann & Leviton 2000). Damage to the developing OLs is the hallmark of cerebral white matter injury due to an increase in production of reactive oxygen species (ROS) and nitrogen species, glutamate, cytokines, and adenosine in perinatal insults (Kinney & Back 1998). Maternal injection of lipopolysaccharide (LPS) at embryonic gestation day 18 (E18) and E19 induces an increase in expression of TNF-α and IL-1β in the maternal and fetal compartments, including the fetal brain (Wang et al. 2006, Cai et al. 2000). Clinical studies have established that higher levels of proinflammatory cytokines in the amniotic fluid observed during intrauterine infections are positively correlated with cerebral white matter injury (Yoon et al. 1997a, Hagberg et al. 2005). In line with this, immature OLs (premyelinating OLs) are reported to be more vulnerable to proinflammatory cytokines compared with myelin-forming OLs (Pang et al. 2005, Levison et al. 2001).
Animal studies showed that the administration of LPS (ip) causes peroxisomal dysfunction (β-oxidation inhibition) in the liver (Khan et al. 2000). Peroxisomes are ubiquitous, metabolically active sub-cellular organelles responsible for metabolism of myelin lipids (i.e., plasmalogen, cholesterol, and very long chain fatty acids [VLCFAs]) (Lazarow 1995) as well as the detoxification of ROS (Lazarow 2003). Peroxisomal dysfunction due to impaired β-oxidation results in the accumulation of VLCFAs which is responsible for neuroinflammation and demyelination in the human X-adrenoleukodystrophy brain, a hereditary peroxisomal disorder (Paintlia et al. 2003). Peroxisomal dysfunction has been linked with ROS generation in apoptosis (Baumgart et al. 2001), aging (Lavrovsky et al. 2000), and ischemia/reperfusion injury (Deplanque et al. 2003, Schrader & Fahimi 2006) including pro-inflammatory disease processes (Poynter & Daynes 1998). Biogenesis of peroxisomes is regulated by PPAR-α in combination with the retinoic acid receptor by acting on PPREs present in the promoter region of most of the genes of peroxisomal proteins (Qi et al. 2000). PPAR-α binds to a diverse set of ligands, namely arachidonic acid metabolites (prostaglandins and leukotrienes) and synthetic fibrate drugs (Sher et al. 1993). These PPAR-α ligands induce peroxisomal proliferation in the cell (Sher et al. 1993). An increase in ROS generation and NF-κB activation has been shown to inhibit the expression of both acyl CoA oxidase (AOX) and PPAR-α, and thereby inhibit peroxisomal proliferation in skeletal muscle cells which contributes to cardiac hypertrophy (Cabrero et al. 2003, Cabrero et al. 2002).
Since bacterial infection (LPS) induced inflammatory responses and ROS generation are involved in OL-injury/hypomyelination possibly mediated via peroxisomal dysfunction, we proposed to understand the role of LPS-induced effect on PPAR-α activity in developing OLs. The understanding of mechanisms involved in the increased vulnerability of developing OLs to various insults may be exploited for the development of strategies for neuroprotection against such insults. One of the popular therapeutic approaches for the inhibition of ROS-mediated injury is the use of glutathione-modulating agents such as NAC, a precursor of glutathione. NAC is likely one of the most widely investigated agents with both antioxidant and anti-inflammatory properties. Earlier, NAC has been shown to attenuate inflammation in various disease models such as ischemia-reperfusion injury in brain (Khan et al. 2004, Sekhon et al. 2003), lethal endotoximia (Victor et al. 2003), experimental autoimmune encephalomyelitis (Stanislaus et al. 2005), and hypoxia-ischemic brain injury in neonatal brains (Jatana et al. 2006). In addition, we and others have showed that NAC attenuates brain white mater injury in a systemic maternal infection model of periventricular leukomalacia (PVL) (Cai et al. 2000, Paintlia et al. 2004). In this study, we showed that the modulation of PPAR-α activity by NAC attenuates the inhibition of OL development by factors secreted by LPS stimulated glial cells.
MATERIALS AND METHODS
Chemicals & reagents
LPS (Escherichia coli, serotype 055:B5), NAC, bovine serum albumin (BSA) and other chemicals were purchased from Sigma-Aldrich. DMEM was purchased from Cellgro and FBS was purchased from GIBCO. Recombinant proteins of platelet-derived growth factor (PDGF-A), basic fibroblast growth factor (bFGF) and Thyroid hormone (TH) were purchased from R & D Systems. Rabbit anti-glial fibrillary acidic protein (GFAP) and mouse anti-BrdU FITC conjugated antibodies were purchased from Abcam (Cambridge, MA). Mouse and rabbit anti-NG2, -O4, -O1, -A2B5 (clone A2B5, 105), -integrin alphaM OX42 (microglia), and –anti-oligodendrocyte (clone NS-1; RIP) antibodies were purchased from Chemicon International. Anti-myelin basic protein (MBP clone 1; 129-138) was purchased from Serotec. Neutralizing anti-TNFR1 and –IL1R1 monoclonal antibodies were purchased from Santa Cruz Biotech, Inc. Polyclonal antibodies against PMP70 were purchased from Abcam (Abcam, Cambridge, UK) and polyclonal antibodies against acyl CoA: dihydroxyacetonephosphate-acyltransferase (DHAP-AT) were synthesized commercially as described earlier (Khan et al. 2005). Anti-peroxisome proliferator-proliferated receptor (PPAR)-α antibody was purchased from Santa Cruz. Goat anti-mouse or anti-rabbit IgG conjugated with FITC or Texas Red antibodies were purchased from Vector Lab.
Generation of mixed glial cultures, purification of OL progenitors, and treatments
Mixed glial cultures were generated from brains of rats (SD; Charles River, Wilmington, MA), PPAR-α (-/-) and wild-type mice at PND 1–2 as described earlier (Paintlia et al. 2005). B12 oligodendroglial cell line was kind gift from Dr. D. Schubert from the Salk Institute, La Jolla, CA. For purification of OL progenitors, mixed glial cultures were shaken for 30 min at 200 × g at 37 °C to remove microglia, followed by further shaking for 8 h to collect OL-progenitors. Then, supernatants containing OL-progenitors were centrifuged and plated in 100-mm plates after suspending them in fresh media. Cells were incubated for 30 min at 37 °C to remove remaining microglia and unattached OL-progenitors were transferred to new plates. Cultures were examined by FACS using specific markers for different cell types. Ninety-seven percent of OL-progenitors were A2B5+ containing approximately 3% of both OX42+ and GFAP+ cells. OL-progenitors were cultured in defined media i.e., DMEM containing 10% FBS and supplemented with growth factors (PDGF-A and bFGF; 10 ng/ml each) (Gregori et al. 2002), 24 h prior to treatment. Developing OLs were generated from OL-progenitors cultured initially for 96 h in defined media supplemented with 10 ng/ml of PDGF-A and TH each to enrich the O4+/GalC- population (Gregori et al. 2002) prior to treatment (confirmed by FACS analysis).
Mixed glial cells were plated in 100-mm plates at a density of 1 × 105 cells/ml. After 24 h, fresh DMEM without FBS was changed and cells were exposed to LPS (0.5 μg/ml) directly or after a 2-h pretreatment with NAC (10 mM). LPS-conditioned media (LCM) or NAC plus LPS-treated conditioned media (NLCM) were collected 48 h post-treatment. Likewise, conditioned media (CCM) from untreated mixed glial cells or those treated with NAC (10 mM, NCM) were collected after 48 h. For experimental studies, conditioned media were diluted with DMEM without FBS (50:50 v/v) to reduce the expected cell death of OL-progenitors by factors secreted by LPS stimulated glial cells. For proliferation studies, OL-progenitors (1 × 105 cells/ml) were plated on glass chamber slides (LAB TEK® II) and cultured in conditioned media generated from mixed glial cultures. For determination of peroxisomal protein levels, similarly treated OL-progenitors or developing OLs were double immunostained with anti-NG2/-PMP70, and anti-RIP/-PMP70 antibodies, respectively, 48 h post-treatment using standard protocols as described for immunocytochemistry. To determine the effect of NAC or WY14643 on the differentiation of OLs, developing OLs were treated with 20 μM of WY14643 (30 min) or 10 mM of NAC (2 h) prior to culturing in LCM or CCM. For mRNA expression analysis, OL progenitors or developing OLs were cultured in 100-mm plates and treated similar to that described above and harvested 48 h post-culturing. For nuclear translocation studies of PPAR-α, developing OLs were pretreated with 20 μM of WY14643 (30 min) and 10 mM of NAC (2 h) followed by culturing in CCM or LCM for 2 h.
Immunocytochemistry
Immunocytochemistry was performed using standard protocols. Double immunolabeling was performed by fixing cells in freshly prepared 10% (w/v) p-formaldehyde in phosphate buffered saline (PBS; pH 7.4), 15 min at room temperature (RT). Then cells were incubated with primary antibodies i.e., anti-NG2 (1: 200), -MBP (1:200), -RIP (1:5,000) overnight at 4°C. Following incubation, cells were washed three times with PBS-Tween 20 (0.01%) and incubated with Texas Red conjugated secondary anti-mouse IgG (Vector Lab) antibodies for 1 h at RT. Then cells were fixed again and permeabilized by treating with Triton X-100 (0.1%) in PBS containing 2% (v/v) fetal calf serum for 15 min, washed three times with PBS and incubated with primary antibodies for PMP70 (1:200) or PPAR-α (1:200) for 2 h at RT followed by incubation with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG for 1 h at RT. Likewise, for BrdU double labeling, prestained cells with anti-NG2 antibodies were fixed and permeabilized as described earlier (Paintlia et al. 2005). Then cells were treated with 2N HCl for 15 min at 37°C and then neutralized in sodium tetra-borate buffer (Sigma). Then cell were incubated with FITC-conjugated anti-BrdU antibodies at 1:100 for 1 h at RT. Slides were also incubated with Texas Red or FITC-conjugated IgG without primary antibody as a negative control. After thorough washings with PBS, slides were mounted and examined under immunofluorescence microscopy (Olympus BX-60) with an Olympus digital camera (Optronics, Goleta, CA) using a dual band-pass filter as described earlier (Paintlia et al. 2005). Counting of NG2+/brdU- and NG2+/BrdU+ cells was performed at 200× magnification, 5 fields/slide in duplicate in each set from three identical experiments and plotted after statistical analysis. The number and length of process of differentiating OLs was determined by using ImagePro plus4 software (Media Cybernetics, Inc. Bethesda, MD) and 10-15 cells were sampled/slide in three identical experiments.
FACS analysis
Cells (1 × 106) were washed twice and suspended in PBS containing 3% BSA followed by incubation with 10 μg/ml non-immune mouse IgG for 15 min. After washing, cells were incubated with 2 μg/ml of anti-O4 and –MBP antibodies diluted 1:100 in PBS containing 3% BSA at 4 °C for 30 min. After washing, cells were incubated with FITC-conjugated anti-mouse IgM diluted at 1:200. Cells were washed before analysis and measured in a FL-1 channel (530 ± 15 nm band pass filter) on a FACS caliber flow cytometer (BD Biosciences) operating with Cell Quest™ software (Paintlia et al. 2006). Dead cells and debris were excluded from the analysis by gating live cells from size/structure density plots, with at least 10,000 events gated.
RNA purification and quantitative real-time-PCR analysis
Total RNA from brain tissues or cells was purified using TRIZOL reagent (Gibco BRL) as described earlier (Paintlia et al. 2003). Single-stranded cDNA was synthesized from total RNA by using the superscript pre-amplification system for first-strand cDNA synthesis and performed quantitative real-time-PCR as described earlier (Paintlia et al. 2003). The primer sets (IDT, Coralville, IA, USA) used was as follows: PMP-70 (Accession No. D90038): forward, 5’-acaccacgagtactacctgcacat-3’ and reverse, 5’-ccgaactcaactgtctcttctgtg-3’, DHAP-AT (Accession No. AF218826): forward, 5′-catcatcctcacagacaaaggg-3′ and reverse, 5′-cttcatgcaagaggcatttgga-3′, acyl CoA oxidase (AOX, Accession No. BC085743): forward, 5′-gaggtccatgaatcttaccaca-3′ and reverse, 5′-gtgagtagaggaagaagttttctgtg-3′, PPAR-α (Accession Nos. NM_013196): forward, 5′-tcgggatgtcacacaatgcaatcc-3′ and reverse, 5′-cgtgttcacaggtaaggatttctgcc-3′, MBP (Accession No. AF439750): forward, 5′-aatcggctcacaagggattcaagg-3′ and reverse, 5′-gctgtctcttcctcccagcttaaa-3′, GAPDH (Accession No. DQ403053): forward, 5′-cctacccccaatgtatccgttgtg-3′ and reverse, 5′-ggaggaatgggagttgctgttgaa-3′, and 18S rRNA: forward, 5’-ccagagcgaaagcatttgccaaga-3’ and reverse, 5’-tcggcatcgtttatggtcggaact-3’. Thermal cycling conditions were as follows: activation of iTaq DNA polymerase (Bio-Rad) at 95 °C for 10 min, followed by 35 cycles of amplification at 95 °C for 30 s and 55−60 °C for 1 min. The detection of threshold was set above the mean baseline fluorescence determined by the first 20 cycles. Amplification reactions in which the fluorescence increased above the threshold were defined as positive. In addition, a dissociation or temperature-melting curve was generated each time for confirmation of specific amplification. The quantity of target gene expression was normalized to the corresponding GAPDH mRNA quantity in respective test samples. Similar results were obtained when normalized with reference genes such as GAPDH or 18S rRNA.
Plasmids and small interfering RNA (siRNA) oligonucleotides
The source of peroxisome proliferator-response element (PPRE)-containing reporter plasmids, pTK-PPREx3-Luc used in the study are the same as previously described (Paintlia et al. 2006). A pool of three siRNAs duplexes for PPAR-α i.e., nucleotide 940: ggaugucacacaaugcaau, nucleotide 1535: gaaguucaaugccuuagaa and nucleotide 2725: cagcuccuuugauaugaua, and respective scramble siRNA (sequences were not disclosed by the company) including transfection reagent and media, were purchased from Santa Cruz Biotechnology, Inc. The protocol for transient co-transfection used in these studies was described in the product manual.
Luciferase reporter assays
Luciferase reporter assays were performed with a luciferase assay reagent kit (Roche) as described earlier (Paintlia et al. 2006). The protein concentration was determined with the Bradford protein assay (Bio-Rad; Hercules, CA) and used to normalize luciferase enzyme activities in samples.
Cytotoxicity assays
Cytotoxicity was evaluated by measuring the release of lactate dehydrogenase (LDH) in the culture medium with a commercially available kit (Roche Diagnostics GmbH, Mannheim, Germany).
Preparation of Nuclear Extracts
Nuclear extracts of treated cells were prepared as described earlier (Paintlia et al. 2006). In brief, cells were treated and washed with ice-cold PBS, and homogenized in 200 μl of buffer A (10 mM HEPES, pH 7.9; 1 mM EDTA; 1 mM EGTA; 1 mM PMSF; 50 μg/ml leupeptin; 10 μg/ml aprotinin; 5 mM benzamidine; 1 mM sodium orthovandate; 2 mM NaF; and 1 mM DTT). After 10 min at 4 °C, Nonident P-40 was added to a final concentration of 0.5%. The tubes were vigorously vortexed for 10 s and the nuclei were collected by centrifugation at 13,000 × g for 30 s. The supernatants were stored at −80 °C and the nuclear pellet was re-suspended by gentle shaking for 30 min at 4 °C in 100 μL of buffer A supplemented with 20% glycerol and 0.4 M NaCl. Nuclear proteins were obtained by centrifugation at 13,000 × g for 5 min and the supernatants were stored at −80 °C. Protein concentration was quantified using Bradford protein assay (Bio-Rad; Hercules, CA).
Immunoblotting
Western blotting of protein samples was carried out as described earlier (Paintlia et al. 2005).
Determination of PPARα transcription factor activity
PPARα transcription factor activity in nuclear extracts of treated cells was determined by using ELISA based Cayman Chemical PPAR-α transcription factor assay kit (Cayman Chemicals, Ann Arbor, Michigan). 40- 50 μg of nuclear extract protein/sample was used for determination of PPARα activity using protocol described in the product manual and absorbance measured at 450 nm.
VLC fatty acid analysis
Fatty acid methyl ester (FAME) from astrocytes or primary OL progenitors was prepared and analyzed by gas chromatography (GC) as described earlier (Paintlia et al. 2005).
Measurement of ROS
ROS was determined using the membrane permeable dye 6-carboxy 2’,7’-dichlorodihydrofluorescein diacetate (DCFH2-DA) in serum-free medium as described earlier (Khan et al. 2005).
Statistical analysis
Using the Student’s unpaired t-test and ANOVA (Student-Newman-Keuls to compare all pairs of columns) p values were determined for the respective experiment from three identical experiments using GraphPad software (GraphPad Software Inc. San Diego, CA USA). The criterion for statistical significance was p<0.05.
RESULTS
Factors released by LPS stimulated glial cells block OL development via inhibition of peroxisomal proliferation which is reversed by NAC
Because LPS-induced effects on developing OLs occur by activation of microglia and astrocytes (Cai et al. 2000, Lehnardt et al. 2002), we sought to investigate the effect of factors secreted by LPS stimulated glial cells on peroxisomes in developing OLs. For this, OL-progenitors were cultured in conditioned media i.e., LCM, NLCM, NCM and CCM. Conditioned media were diluted with FBS free DMEM (50:50 v/v) to study its effect on peroxisomes in OL-progenitors without induction of their cell death. OL-progenitors cultured in LCM showed a significant decrease in BrdU+/NG2+ cell population 24 h post-incubation compared to those cultured in CCM or NLCM (Fig. 1A and C) suggesting that LCM exposure inhibits the proliferation of OL-progenitors. Interestingly, NLCM exposure induced a significant increase in the proliferation of OL-progenitors compared with CCM as showed by increase in BrdU+/NG2+ cell population (Fig. 1A). Following LCM exposure, total NG2 cell population was not significantly changed when compared with NG2+/BrdU- cell population (Fig. 1B) suggesting that dilute LCM was not able to induce cell death of OL-progenitors. Conversely, total NG2 cell populations were significantly increased when compared with NG2+/BrdU- cell populations following CCM or NLCM exposure (Fig. 1B). Furthermore, LCM exposure caused a significant decrease in late OL-progenitors (O4+) and myelin-forming OLs (MBP+) 72 h and 144 h post-treatment, respectively, compared to those cultured in CCM or NLCM or NCM (Fig. 1D and E). Similar to NG2+/BrdU+ cells, the differentiation of OL-progenitors was significantly increased when cultured in NLCM compared with CCM as showed by significant increase in O4+ and MBP+ cell populations (Fig. 1D and E). No significant cell death of OL-progenitors was observed under experimental conditions especially following LCM exposure as determined by LDH release (data not shown). These data suggest that factors secreted by LPS stimulated mixed glial cultures inhibit OL development.
Figure 1. Factors released by LPS-stimulated glial cells (LCM) inhibit OL development which is attenuated by NAC.
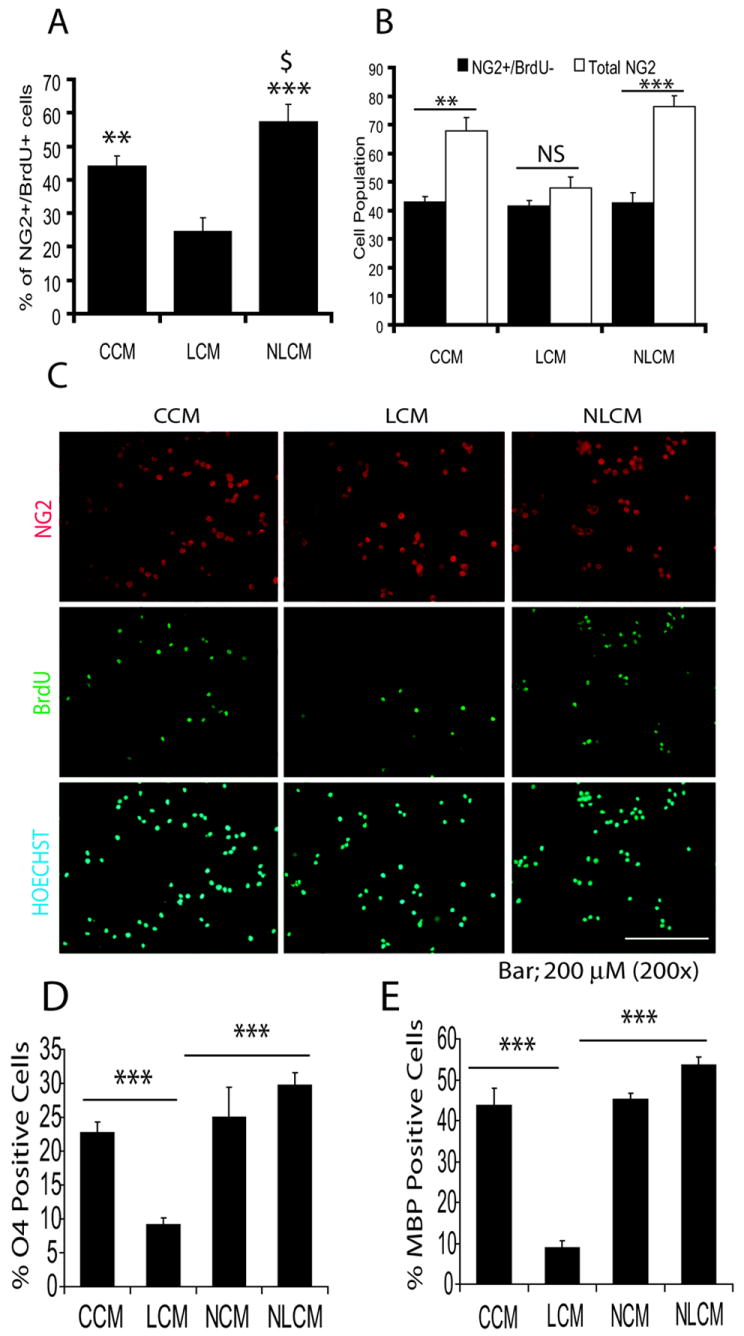
OL-progenitors generated from rat mixed glial cultures were cultured in conditioned media generated from mixed glial cultures treated with LPS (LCM), NAC plus LPS (NLCM), NAC alone (NCM) and/or untreated control (CCM) as described under ‘Materials and Methods’. Plot depicts percentage of NG2+/BrdU+ cells/10-fields/slide, 24 h post-culturing (A). Plot depicts comparison of total NG2 with NG2+/BrdU- cell population/5-fields/slide in similarly treated cells (B). Representative field of slides demonstrates NG2+ and BrdU+ cells counter stained with nuclei stain (Hoechst) at 24 h post-culturing (C). Plots depict percentage change in count of O4+ (late OL progenitors) (D) and MBP+ (differentiating OLs) (E) in cultured OL progenitors for 72 h and 144 h, respectively. Plot data are expressed as Mean ± SD of 3 independent experiments conducted in triplicate. Statistical significance is indicated as **p< 0.01 and ***p<0.001 versus LCM, and $p<0.05 versus CCM. Asterisks **<0.01 and ***p< 0.001 and NS (non-significant) (B).
Corresponding with these findings, the message for peroxisomal proteins i.e., PMP70 (Fig. 2A) and AOX (Fig. 2B) transcripts were significantly reduced in both OL-progenitors and developing OLs (O4+) when cultured in LCM for 48 h as compared to those were cultured in CCM. As expected, no difference in the level of these transcripts was observed among OL-progenitors and developing OLs when cultured in NLCM, NCM and CCM (Fig. 2A and B). Immunofluorescence studies further revealed that the level of PMP70 in both NG2+ (OL-progenitors) and RIP+ (developing OLs) cells was reduced when cultured in LCM for 48 h as compared to those were cultured in NLCM or CCM (Fig. 2C and D). Western blotting further confirmed that PMP70 level in both OL-progenitors and developing OLs was significantly decreased following LCM exposure compared with CCM or NLCM (Fig. E and F). Of note, no significant cell death was observed in cultured OL progenitors and developing OLs in LCM for 48 h as determined by LDH-release assay (data not shown). Altogether, these data suggest that factors secreted by LPS stimulated glial cells (i.e., microglia/astrocytes) inhibit peroxisomal proliferation in OL-progenitors thus their development.
Figure 2. The inhibition of OL development by LCM is associated with the reduction of peroxisomes in OL progenitors which is attenuated by NAC.
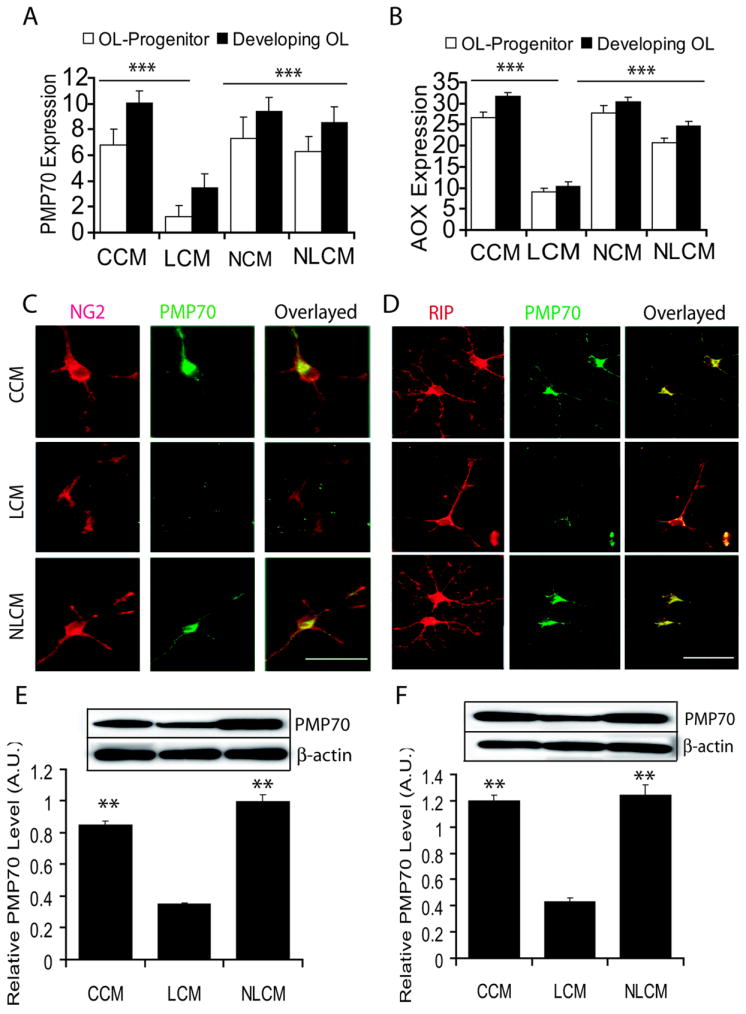
Primary OL-progenitors and developing OLs generated by rat mixed glial cultures were exposed to LCM, NLCM, NCM and CCM separately. Plots depict the levels of transcripts for PMP70 (A) and AOX (B) in both OL-progenitors and developing OLs, 48 h exposure to conditioned media. Representative field of slides depicts double-immunostaining for NG2 and PMP70 in OL-progenitors (C) and both RIP and PMP70 in developing OLs at 48 h of post-culturing (D). Plots depict relative expression of PMP70 compared with β-actin determined by western blotting in similarly treated OL-progenitors (E) and developing OLs (F) 48 h of post-culturing. Insert is representing autoradiograph of western blot. Plot data are expressed as Mean ± SD of 3 independent experiments conducted in triplicate. Statistical significance is indicated as **p< 0.01 and ***p<0.001 versus LCM. Bars, 100 μM at original magnification 600× (C) and 400× (D).
Inhibition of peroxisomal proliferation by LCM in developing OLs is via inhibition of PPAR-α trans-activation
To further elucidate the underlying mechanism of LCM mediated inhibition of peroxisomal proliferation in developing OLs and its attenuation by NAC, we proposed to determine the status of PPAR-α (a transcription factor regulates the expression of peroxisomal proteins) activity in the developing OLs. For this, developing OLs were cultured in LCM after pretreatment with NAC or WY14643 (PPAR-α agonist). Interestingly, there was a corresponding decrease in the level of PMP70 (Fig. 3A) and PPAR-α (Fig. 3B) transcripts in the developing OLs when cultured in LCM for 48 h compared with those cultured in CCM. NAC and WY14643 treatment showed the attenuation of LCM-induced effect and subsequently increased the level of PMP70 and PPAR-α transcripts in the developing OLs compared with untreated ones (Fig. 3A and B). Of note, WY14643 and NAC treatment showed a relative increase in the level of PMP70 and PPAR-α transcript in the developing OLs following LCM exposure when compared with CCM (Fig. 3A and B). Immunocytochemistry further revealed that level of both PMP70 and MBP was decreased in developing OLs when cultured in LCM, which was attenuated by WY14643 (Fig. 3C). Western blotting further supported these finding and showed a decrease in level of PMP70 in developing OLs following LCM exposure (Fig. 3D). This finding was further validated by determining the length and number of processes in the differentiating OLs. Developing OLs cultured in LCM had a significant decrease in length and number of processes in OLs compared with those cultured in CCM which was attenuated by WY14643 (Fig. 3E and F). Likewise, developing OLs cultured in LCM had a significant decrease in MBP transcripts compared with those cultured in CCM, which was attenuated by WY14643 (Fig. 3G). Developing OLs cultured in WY14643 plus LCM or WY14643 plus CCM had significant increase in level of MBP transcripts and differentiation of OLs compared to those cultured in CCM (Fig. 3E-G).
Figure 3. NAC induced effect was mimicked by WY14643 (WY) in LCM exposed OL progenitors.
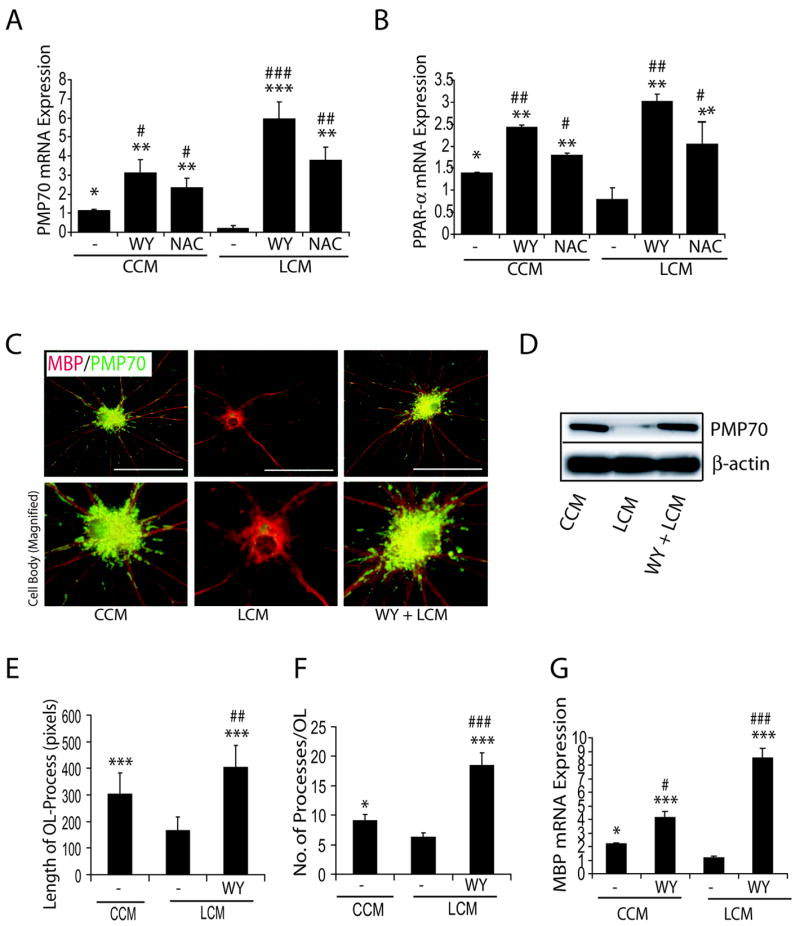
Developing OLs were pretreated with WY (20 μM) or NAC (10 mM) for 30 min and 2 h, respectively, prior to culturing in CCM or LCM. Plots depict transcript levels PMP70 (A) and PPAR-α (B) in developing OLs after 48 h culturing in CCM and LCM in the presence/absence of WY or NAC. Representative sections depict double-immunolabeled myelin-forming OLs with anti-MBP and -PMP70 antibodies (upper panel) when developing OLs were cultured in CCM or LCM in the presence of WY for 72 h (C). Magnification depicts an increase in PMP70 level in the cell body of developing OLs (lower panel) (C). Representative western blot depicts level of PMP70 in similarly treated developing OLs for 72 h (D). Plots depict the magnitude of process increase (E) and number of processes/OL (F) in WY treated developing OLs cultured in CCM or LCM for 72 h. A total of 10 cells/slide were sampled from each group. Plot depicts the level of MBP transcripts in WY-treated developing OLs when cultured in CCM or LCM for 48 h (G). Plot data are expressed as Mean ± SD of 3 identical experiments. Statistical significance is indicated * p<0.05, **p< 0.01 and ***p<0.001 versus LCM and #p<0.05, ##p< 0.01 and ###p<0.001 versus CCM. Bars 100 μM at original magnification (600 ×).
Double immunostaining revealed that LCM exposure causes the reduction of PPAR-α protein in the cytoplasm probably by its degradation and inhibits its nuclear translocation in the developing OLs, which was attenuated by WY14643 (Fig. 4A). Furthermore, NAC mimicked the WY14643 effect in LCM-exposed developing OLs (Fig. 4A). This was confirmed by nuclear translocation studies. Western blot studies showed that LCM exposure significantly inhibits the nuclear translocation of PPAR-α thus its relative activity (Fig. 4B) in the developing OLs and that was attenuated by both NAC and WY14643 pretreatment. This decrease in PPAR-α activity in LCM exposed developing OLs was further validated by ELISA based assay (Fig. 4C). Together, these data suggest that LCM exposure inhibits OL development via peroxisomal dysfunction and down-regulation of PPAR-α trans-activation.
Figure 4. Similar to WY14643 (WY), NAC attenuates LCM-induced inhibition of PPAR-α trans-activation in the developing OLs.

Developing OLs were pretreated with WY (20 μM) or NAC (10 mM) for 30 min and 2 h, respectively, prior to culturing in LCM or CCM. Field of representative slides of double-immunostained for MBP/PPAR-α depicts the nuclear translocation of PPAR-α in the developing OLs at 2 h of post-treatment (A). Likewise, representative immunoblot autoradiogram depicts the level of PPAR-α in the nuclear fraction (NF) including total cell lysates (TCL) of the developing OLs after 2 h of treatment as described above (B, insert). Plot depicts relative activity of PPAR-α determined with respect to level of nuclear PPAR-α and total PPAR-α in the cell lysate as determined by densitometric analysis of immunblot autoradiogram (n=3) and data is presented as arbitrary units (A.U.) (B). Plot depicts PPAR-α activity in the nuclear extract of similarly treated developing OLs for 2 h determined by ELISA based assay from three identical experiments (C). Statistical significance is indicated *p< 0.05 and **p<0.01 versus LCM and # p<0.05 and NS (non-significant) versus CCM.
LCM contains cytokines (IL-1β and TNF-α) which are responsible for ROS generation and peroxisomal dysfunction in developing Ols
Because maternal LPS exposure induces IL-1β and TNF-α production in the fetal brain (Cai et al. 2000, Wang et al. 2006, Paintlia et al. 2004), we next investigated the effect of these cytokines on peroxisomes in developing OLs and astrocytes. Developing OLs or astrocytes were treated separately with a cocktail of cytokines (Cyt-Mix: IL-1β and TNF-α; 10 ng/ml each) in the presence/absence of NAC. Interestingly, Cyt-Mix treatment caused the accumulation of VLCFAs (C26:0/C22:0 ratio) as an indicator of peroxisomal dysfunction in the developing OLs when compared with controls, and this was reversed by NAC pretreatment (Fig. 5A). In addition, Cyt-Mix treatment induced ROS generation in the developing OLs and that was reversed by both NAC and WY14643 (Fig. 5C). Interestingly, VLCFAs accumulation in similarly treated astrocytes for same duration was negligible or absent (Fig. 5B). Although, ROS generation in similarly treated astrocytes was increased compared with controls (Fig. 5D), but it was relatively low when compared with similarly treated developing OLs. To explore it further whether LCM consists of TNF-α and IL-1β, we cultured developing OLs and astrocytes in LCM in the presence/absence of anti-TNFR1 and -IL1R1 monoclonal antibodies (MAbs). Interestingly, an increase in the generation of ROS both in developing OLs and astrocytes following LCM exposure was significantly inhibited in the presence of anti-TNFR1 and -IL1R1 MAbs when used in combination or individually (Fig. 5E and F). Together, these data provide evidence that factors released by LPS stimulated glial cells consists of proinflammatory cytokines which are responsible for peroxisomal dysfunction and ROS generation in the developing OLs.
Figure 5. LCM consists of proinflammatory cytokines (TNF-α and IL-1β) involved in the ROS generation and peroxisomal dysfunction in developing OLs.
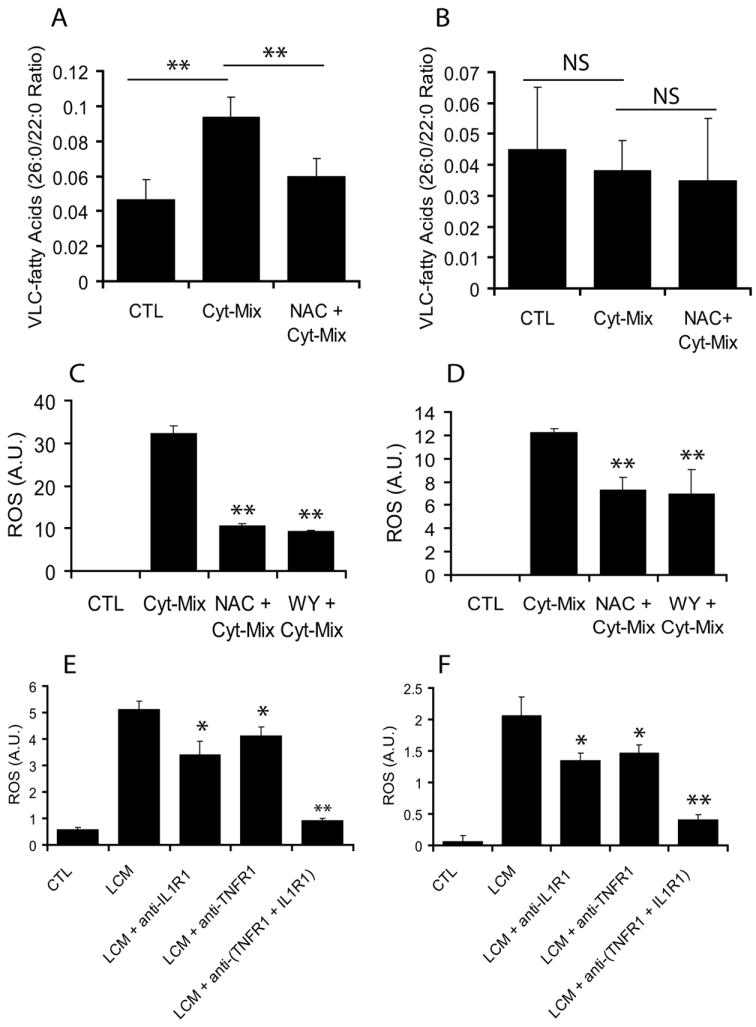
Rat developing OLs and astrocytes were treated separately with a cocktail of pro-inflammatory cytokines (Cyt-Mix: TNF-α and IL-1β; 10 ng/ml each) in the presence/absence of NAC (10 mM) or WY (20 μM) for 48 h followed by determination of VLCFAs accumulation and ROS generation. Plots depict VLCFAs (C26:0/22:0 ratio) in developing OLs (A) and astrocytes (B). Plots depict ROS generation in similarly treated developing OLs (C) and astrocytes (D). Further, rat developing OLs and astrocytes were exposed to LCM for 48 h in the presence/absence of anti-IL1R1 and –TNFR1 MAbs in combination or individually. Plots depict ROS generation in treated developing OLs (E) and astrocytes (F) for 48 h. Plot data are presented as Mean ± SD of three identical experiments. Statistical significance is indicated as **p<0.01and NS (non-significant) versus Cyt-Mix (A-D) and *p< 0.05 and **p<0.01 versus LCM (E and F).
Depletion of GSH inhibits peroxisomal proliferation in developing Ols
Previously, we documented that NAC pretreatment normalizes reduced-GSH in either diamide- (a thiol-depleting agent) or cytokine treated primary OLs (Singh et al. 1998). Cytokine-induced depletion of reduced-GSH in OLs is mediated via degradation of sphingomyelin into ceramide (Singh et al. 1998). Therefore, we next investigated whether the depletion of reduced-GSH has an impact in the inhibition of peroxisomal proliferation/function in the developing OLs. Transcripts for both peroxisomal proteins i.e., DHAP-AT (Fig. 6A) and PMP70 (Fig. 6B), and MBP (Fig. 6C) including PPAR-α (Fig. 6D) were significantly reduced in developing OLs on treatment with diamide in a dose-dependent manner (Fig. 6A-D). Of note, the expression of these proteins was significantly reduced with the dose of diamide (<0.2 mM) used to treat developing OLs 48 h post-treatment where no detectable cell death was seen (Fig. 6E). Higher dose of diamide (>0.2 mM) however was found to be toxic to the developing OLs (Fig. 6E). These data suggest that the depletion of intracellular reduced-GSH level enhances ROS generation which in turn inhibits peroxisomal proliferation and OL development.
Figure 6. Similar to LCM, diamide treatment (deplete GSH) reduces peroxisomes and inhibits the differentiation of developing OLs.

Rat primary developing OLs were treated with diamide (DIAM; 0.1–2.0 mM). Plots depict transcript levels for DHAP-AT (A), PMP70 (B), PPAR-α (C), and MBP (D) in the developing OLs 48 h post-treatment. Plot depicts LDH release in the diamide-treated developing OLs for 48 h (E). Although real-time PCR amplified products for PMP70 and MBP genes were detected in 2% agarose gel after the completion of the run, real-time PCR analysis showed a dose-dependent decrease in PMP70 and MBP transcripts in developing OLs by diamide (F). Data in plots are presented as Mean ± SD of three identical experiments. Statistical significance is indicated as *p< 0.05, **p<0.01 and ***<0.001 versus control (untreated).
NAC-induced protective effect was diminished in LCM-exposed developing OLs lacking PPAR-α or B12 oligodendroglial cells co-transfected with PPAR-α siRNAs and pTK-PPREx3-Luc
To further gain insight into the mechanism of NAC-induced protection in developing OLs via modulation of PPAR-α activity, we evaluated the protection of developing OLs lacking PPAR-α generated from PPAR-α (-/-) mice or B12 OLs co-transfected with PPAR-α siRNA and pTK-PPREx3-Luc (reporter plasmids) under similar conditions. LCM exposed wild-type developing OLs showed a significant decrease in MBP+ OL population compared to those exposed to CCM and that was attenuated by both NAC or WY14643 pretreatment (Fig. 7A). In contrast, these NAC or WY14643 induced effects in LCM exposed developing OLs lacking PPAR-α were absent (Fig. 7A) thereby confirming our observations that PPAR-α mediates NAC induced attenuation of peroxisomal dysfunction and OL development in LCM exposed OL progenitors. To further confirm this, we measured luciferase activity in NAC- or WY14643-treated B12 oligodendroglial cells transiently cotransfected with pTK-PPREx3-Luc and PPAR-α siRNAs or scramble siRNAs. Interestingly, LCM treatment decreased luciferase activity in B12 cells cotransfected with pTK-PPREx3-Luc and scramble siRNAs which was significantly reversed by NAC or WY14643 as compared to controls (Fig. 7B). This reversal of the LCM-induced decrease in luciferase activity by NAC or WY14643 did not occur in B12 cells co-transfected with pTK-PPREx3-Luc and PPAR-α siRNA (Fig. 7B). Together, these data suggest that LPS induced activation of brain glial cells inhibit OL development via generation of ROS and peroxisomal dysfunction. On the other hand, NAC reverses these LPS-induced effects by quenching of ROS and restoration of peroxisomal function via modulation of PPAR-α activity in developing OLs.
Figure 7. Knock out and antisense RNA based transfection studies established that NAC modulates PPAR-α activity in LCM exposed developing OLs.
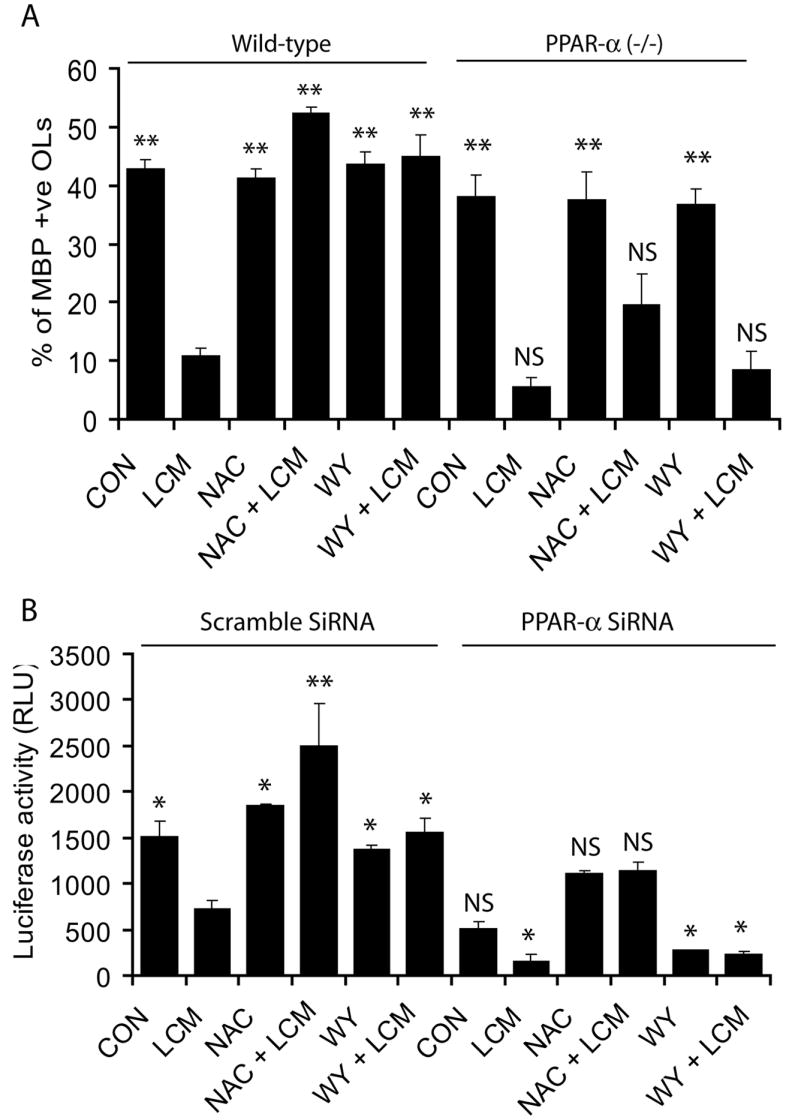
To confirm the modulation of PPAR-α activity by NAC in LCM exposed developing OLs, we used PPAR-α (-/-) and wild-type developing OLs and treated similarly with LCM, NAC and WY14643 (WY) as described under methods. Plot depicts the percentage of MBP+ OLs generated from wild-type/PPAR-α (-/-) mice 48 h post-treatment analyzed by FACS (A). Plot depicts relative luciferase activity in B12 oligodendroglial cells co-transfected with pTK-PPREx3-Luc and PPAR-α siRNAs or scramble and treated with NAC (10 mM) or WY (50 μM) prior to incubation with Cyt-Mix as described under Methods (B). Plot data are presented as Mean ± SD of three identical experiments. Statistical significance is indicated as *p<0.05, **p<0.01 and NS (non-significant) versus LCM (wild-type and scramble SiRNA).
DISCUSSION
Maternal LPS exposure has been shown to stimulate the secretion of pro-inflammatory cytokines i.e., TNF-α, IL-1α and IL-6 as detected in the maternal serum and amniotic fluid of pregnant mice (Fidel et al. 1994), mimicking maternal infection (Yoon et al. 1997b). Results presented here delineate the underlying mechanism of NAC mediated attenuation of LPS-induced inhibition of OL development during prenatal maternal infections which is responsible for cerebral white matter injury in premature infants. Factors secreted by LPS stimulated glial cells inhibited the proliferation of OL-progenitors and disturbed their differentiation into myelin-forming OLs. Correspondingly, the level of peroxisomal proteins such as PMP70 and AOX was reduced in developing OLs when exposed to LPS conditioned media generated from mixed glial cultures. NAC and WY14643 attenuated these LPS-induced effects on developing OLs in mixed glial cultures thus suggested the involvement of peroxisomes in OL development. Similar to WY14643, NAC modulated PPAR-α activity in the developing OLs. TNF-α and IL-1β are found to be active agents in the LPS conditioned media generated from mixed glial cultures, responsible for reduced-GSH depletion and ROS generation in the developing OLs. Furthermore, studies with OL-progenitors generated from PPAR-α (-/-)/wild-type mice and B12 oligodendroglial cells co-transfected with anti-PPAR-α SiRNA and PPRE-Luc plasmids confirmed our findings. It suggested that NAC modulates PPAR-α activity and peroxisomal functions in the developing OLs which are crucial for their development. Together, these findings provide evidence that NAC modulates PPAR-α activity/peroxisomal function in developing OLs against factors secreted byLPS stimulation of glial cells.
Several lines of evidence suggest that free radical injury to the developing OLs underlies (at least in part) the pathogenesis of cerebral white matter injury (hypomyelination) observed in periventricular leukomalacia long-term survivors of preterm labor (Haynes et al. 2005). In human cerebral palsy patients, the presence of free radical brain injury is supported by the existence of both oxidative and nitrative stress markers of lipid peroxidation and nitrosylation/nitration, respectively (Haynes et al. 2003). LPS has been shown to cause the depletion of intracellular GSH and an increase in malondialdehyde, a byproduct of lipid peroxidation (Topal et al. 2004, Millan-Plano et al. 2003). This LPS-induced oxidative stress and lipid peroxidation leads to production of potentially toxic aldehydes such as 4-hydroxynonenal (4-HNE) in the cell (Siems et al. 1992). 4-HNE has been reported to cause cellular damage mainly by modification of intracellular proteins (Uchida 2003, Toyokuni et al. 1994, Uchida & Stadtman 1993) and induction of inflammatory mediator expression such as COX-2, especially in macrophages (Kumagai et al. 2004). LPS-induced maternal proinflammatory cytokines or LPS itself can activate microglia in the fetal brain (Haynes et al. 2005). Therefore, activation of microglia in the fetal brain triggers events associated with reactive astrogliosis which disturbs the maturation of myelin-forming OLs, and thus hypomyelination in the postnatal brain. NAC replenishes the level of reduced-GSH thereby provides protection by quenching ROS in the glial cells following LPS exposure. In addition, anti-inflammatory effects of NAC are attributed to the suppression of pro-inflammatory cytokine expression/release (Tsuji et al. 1999), and activation of glial cells (Moynagh et al. 1994). Antioxidants have been shown to attenuate the generation of ROS and their cytotoxic effects on neurons in vivo (Bahat-Stroomza et al. 2005).
Peroxisomal dysfunction has been shown to contribute partly to the generation of oxidative stress in the cell. Defects in the targeting of catalase to peroxisomes or pharmacologic inhibition of catalase activity has been shown to promote generation of ROS and activation of NF-κB in cells (Sheikh et al. 1998, Singh 1997). In contrast, treatment of these cells with antioxidants normalizes the metabolic function of peroxisomes and stems oxidative stress (Bahat-Stroomza et al. 2005). Defects in peroxisomal functioning can exacerbate demyelination in the brain, resulting in neurological deficits in animals (Elias et al. 1998, Khan et al. 2005, Paintlia et al. 2003). Recent study revealed that peroxisomal function in OLs are vital for brain integrity and their specific loss in OLs causes axonal loss followed by demyelination and neuroinflammation in adult mice brain (Kassmann et al. 2007). Likewise, neuroinflammation has been shown to cause peroxisomal dysfunction and demyelination in an acute experimental autoimmune encephalomyelitis model (Singh et al. 2004). So far peroxisomal dysfunction has been shown to be associated with several inherited metabolic diseases due to the deficiency of one or more peroxisomal proteins, and these defects are responsible for about 18 different fatal neurological disorders (Moser 1996, Moser 1999). In addition, animal studies revealed that inhibition of VLCFAs β-oxidation by thioridazine affects the rate of myelination in the developing brain (Van den Branden et al. 1989, Van den Branden et al. 1990). Our findings suggest that LPS-induced oxidative stress, in part, is contributed by peroxisomal dysfunction which increases the vulnerability of the developing OLs to targeted injury, perhaps due to their high lipid environment and decrease in synthesis of plasmalogen. In addition, decrease GSH peroxidase and release of iron also reported to increase the vulnerability of OLs to oxidative-stress (Juurlink et al. 1998). On the other hand, LPS-induced oxidative stress causes the activation of microglia and astrocytes, albeit peroxisomal functioning remains normal, likely due to the availability of strong endogenous antioxidant systems in these glial cells (Min et al. 2006).
Major function of peroxisomes in the myelin-forming OLs is the metabolism of ROS and myelin lipids (Paintlia et al. 2003, Sztriha et al. 1997). The metabolism of ROS (H2O2) is vital for the survival of OLs against catastrophic effects of hypotension/hypoxia and/or neuroinflammation. In addition, peroxisomes support myelin biogenesis which is important for the differentiation of myelin-forming OLs (Sztriha et al. 1997). An increase in ROS generation and NF-κB activation has been shown to inhibit the expression of both AOX and PPAR-α which in turn inhibit peroxisomal proliferation in skeletal muscle cells resulting in cardiac hypertrophy (Cabrero et al. 2003, Cabrero et al. 2002). In agreement with these data, cytokine exposure causes peroxisomal dysfunction and ROS generation in the developing OLs, but not in astrocytes, which was attenuated by both NAC and WY14643. It suggests that factors secreted by LPS stimulated glial cells, inhibit peroxisomal function and enhance ROS generation via inhibition of PPAR-α activity in the developing OLs. In support of these data, WY14643 has been shown to protect hippocampal neurons against β-amyloid peptide-induced ROS generation in Alzheimer’s disease via peroxisomal proliferation (Santos et al. 2005). In addition, fibrates are shown to exert their anti-inflammatory effects by antagonizing NF-κB activity in aortic smooth muscle cells (Deplanque et al. 2003). Anti-inflammatory cytokines such as interleukin-4 has been documented to inhibit the activation of NF-κB in glial cells via trans-activation of PPAR-γ (Paintlia et al. 2006). This increase in activation of PPAR-γ by IL-4 is mediated via increase in synthesis of natural PPAR-γ ligand via modulation of 12/15 lipoxygenase activity in glial cells. In light of this, an observed decrease in PPAR-α activity and its nuclear translocation in the developing OLs when cultured in LPS-conditioned media and its reversal by NAC similar to WY14643 treatment suggesting that NAC likely increases the availability of endogenous ligands for PPAR-α in developing OLs. Secretary phospholipase A2 (sPLA2) has been shown to produce lipid mediators such as free fatty acids and prostaglandins that are endogenous ligands for PPAR-α (Corton et al. 2000). TNF-α induces an increase in sPLA2 activity and PPAR-α activation in mesangial cells via an autocrine mechanism (Beck et al. 2003). We hypothesize perhaps NAC-induced generation of a reducing environment increases the availability of endogenous PPAR-α ligands synthesized by sPLA2 in the developing OLs when exposed to LPS conditioned media generated from mixed glial cultures. However, more detailed investigations are required in this regard.
In summary, this study delineates the underlying mechanism of LPS-induced inflammation mediated inhibition of OL development in brain glial cells (microglia/astrocytes). The impaired peroxisomal proliferation/function in the developing OLs via down regulation of PPAR-α activity (Figure 8) is crucial in OL development. NAC attenuates this LPS-induced inhibition of OL development via replenishment of reduced GSH, quenching of ROS, and maintenance of peroxisomal proliferation/function via up-regulation of PPAR-α activity. Together, these findings suggest new therapeutic interventions to prevent the debilitating effects of prenatal maternal infections.
Figure 8. The underlying mechanism of NAC-induced attenuation of OL development inhibition in LPS-stimulated mixed glial cultures.
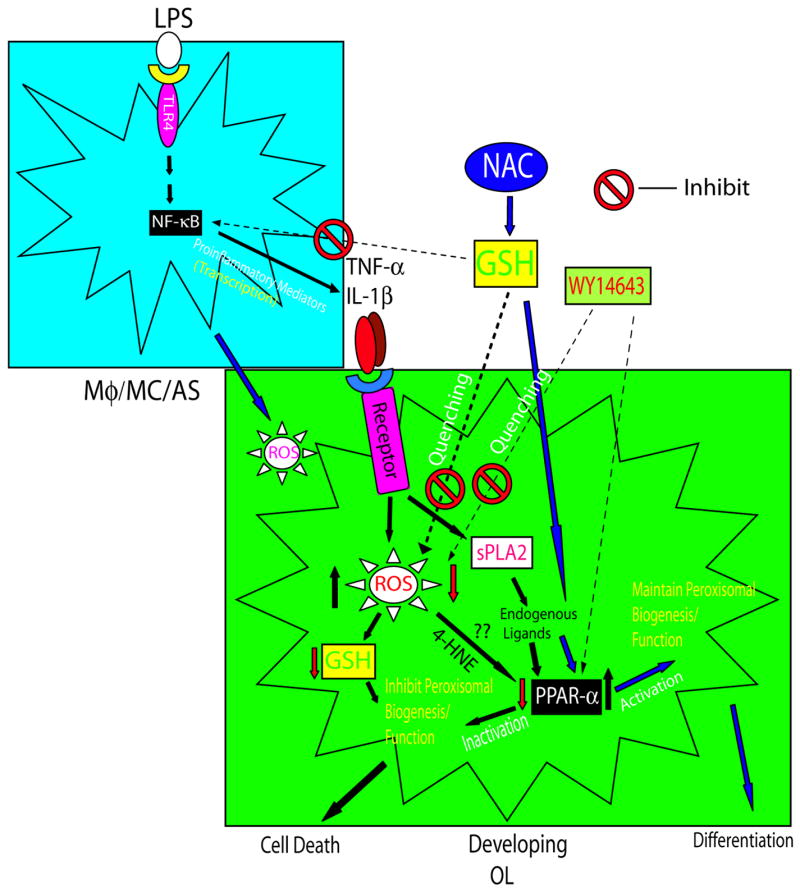
Maternal LPS exposure activates macrophages (Mφ) and microglia (MC) and, in turn, astrocytes (AS), resulting in the secretion of pro-inflammatory cytokines (i.e., TNF-α and IL-1β). This results in generation of ROS and depletion of GSH producing oxidative stress in developing OLs. Oxidative stress inhibits peroxisomal biogenesis/function by down regulation of PPAR-α activity, possibly by an increase in 4-HNE and inaccessibility of endogenous PPAR-α ligands synthesized by inflammation-induced sPLA2 (Beck et al. 2003, Corton et al. 2000) in developing OLs, leading to their targeted injury. The attenuation of LPS-induced activation of immune cells (macrophage, microglia, and astrocytes) and secretion of pro-inflammatory mediators by NAC treatment is mediated via replenishment of reduced GSH and NF-κB inactivation. NAC treatment attenuates pro-inflammatory cytokine-induced ROS generation by replenishment of depleted GSH and quenching of ROS, thereby maintaining peroxisomal biogenesis/function (i.e., β-oxidation and plasmalogen synthesis) by up regulation of PPAR-α activity in developing OLs perhaps by increasing the availability of endogenous PPAR-α ligands synthesized by sPLA2.
Acknowledgments
We thank all members of our laboratory for their valuable comments and help during the course of this study. We thank especially Dr. Jennifer G. Schnellmann for critical reading of this manuscript and Ms. Joyce Brian for her technical assistance. This study was supported in part by grants from the National Institutes of Health: NS-22576, NS-34741, NS-37766, NS-40810, C06 RR018823, and C06 RR015455.
Abbreviations
- PPAR-α
Peroxisome Proliferator Activated Receptor-α
- LPS
Lipopolysaccharide
- OL
oligodendrocyte
- ROS
reactive oxygen species
- NAC
N-acetyl cysteine
- GSH
glutathione
- AOX
acyl-CoA oxidase
- DHAP-AT
dihydroxyacetonephosphate acyltransferase
- PMP70
peroxisomal membrane protein 70
- LCM
LPS conditioned media
- NLCM
NAC plus LPS conditioned media
- NCM
NAC conditioned media
- CCM
control conditioned media
- VLCFA
very long chain fatty acids
Contributor Information
Manjeet K. Paintlia, Department of Pediatrics, Medical University of South Carolina, Charleston, South Carolina, USA.
Ajaib S. Paintlia, Department of Pediatrics, Medical University of South Carolina, Charleston, South Carolina, USA.
Mushfiquddin Khan, Department of Pediatrics, Medical University of South Carolina, Charleston, South Carolina, USA.
Inderjit Singh, Department of Pediatrics, Medical University of South Carolina, Charleston, South Carolina, USA.
Avtar K. Singh, Department of Pathology and Laboratory Medicine, Ralph H. Johnson VA Medical Center, Charleston, South Carolina, USA
References
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405–414. doi: 10.1053/j.semperi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Bahat-Stroomza M, Gilgun-Sherki Y, Offen D, Panet H, Saada A, Krool-Galron N, Barzilai A, Atlas D, Melamed E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson’s disease. Eur J Neurosci. 2005;21:637–646. doi: 10.1111/j.1460-9568.2005.03889.x. [DOI] [PubMed] [Google Scholar]
- Baumgart E, Vanhorebeek I, Grabenbauer M, Borgers M, Declercq PE, Fahimi HD, Baes M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse) Am J Pathol. 2001;159:1477–1494. doi: 10.1016/S0002-9440(10)62534-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Lambeau G, Scholz-Pedretti K, Gelb MH, Janssen MJ, Edwards SH, Wilton DC, Pfeilschifter J, Kaszkin M. Potentiation of tumor necrosis factor alpha-induced secreted phospholipase A2 (sPLA2)-IIA expression in mesangial cells by an autocrine loop involving sPLA2 and peroxisome proliferator-activated receptor alpha activation. J Biol Chem. 2003;278:29799–29812. doi: 10.1074/jbc.M211763200. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Hallenbeck JM, Gallo V. Determining the fetal inflammatory response in an experimental model of intrauterine inflammation in rats. Pediatr Res. 2004;56:541–546. doi: 10.1203/01.PDR.0000139407.89883.6B. [DOI] [PubMed] [Google Scholar]
- Cabrero A, Alegret M, Sanchez RM, Adzet T, Laguna JC, Carrera MV. Increased reactive oxygen species production down-regulates peroxisome proliferator-activated alpha pathway in C2C12 skeletal muscle cells. J Biol Chem. 2002;277:10100–10107. doi: 10.1074/jbc.M110321200. [DOI] [PubMed] [Google Scholar]
- Cabrero A, Merlos M, Laguna JC, Carrera MV. Down-regulation of acyl-CoA oxidase gene expression and increased NF-kappaB activity in etomoxir-induced cardiac hypertrophy. J Lipid Res. 2003;44:388–398. doi: 10.1194/jlr.M200294-JLR200. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res. 2000;47:64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Corton JC, Anderson SP, Stauber A. Central role of peroxisome proliferator-activated receptors in the actions of peroxisome proliferators. Annu Rev Pharmacol Toxicol. 2000;40:491–518. doi: 10.1146/annurev.pharmtox.40.1.491. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Role of the fetus in perinatal infection and neonatal brain damage. Curr Opin Pediatr. 2000;12:99–104. doi: 10.1097/00008480-200004000-00002. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, et al. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias ER, Mobassaleh M, Hajra AK, Moser AB. Developmental delay and growth failure caused by a peroxisomal disorder, dihydroxyacetonephosphate acyltransferase (DHAP-AT) deficiency. Am J Med Genet. 1998;80:223–226. [PubMed] [Google Scholar]
- Fidel PL, Jr, Romero R, Wolf N, Cutright J, Ramirez M, Araneda H, Cotton DB. Systemic and local cytokine profiles in endotoxin-induced preterm parturition in mice. Am J Obstet Gynecol. 1994;170:1467–1475. doi: 10.1016/s0002-9378(94)70180-6. [DOI] [PubMed] [Google Scholar]
- Gregori N, Proschel C, Noble M, Mayer-Proschel M. The tripotential glial-restricted precursor (GRP) cell and glial development in the spinal cord: generation of bipotential oligodendrocyte-type-2 astrocyte progenitor cells and dorsal-ventral differences in GRP cell function. J Neurosci. 2002;22:248–256. doi: 10.1523/JNEUROSCI.22-01-00248.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Jacobsson B. Role of cytokines in preterm labour and brain injury. Bjog. 2005;112(Suppl 1):16–18. doi: 10.1111/j.1471-0528.2005.00578.x. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Baud O, Li J, Kinney HC, Volpe JJ, Folkerth DR. Oxidative and nitrative injury in periventricular leukomalacia: a review. Brain Pathol. 2005;15:225–233. doi: 10.1111/j.1750-3639.2005.tb00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA, Volpe JJ, Kinney HC. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol. 2003;62:441–450. doi: 10.1093/jnen/62.5.441. [DOI] [PubMed] [Google Scholar]
- Jatana M, Singh I, Singh AK, Jenkins D. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr Res. 2006;59:684–689. doi: 10.1203/01.pdr.0000215045.91122.44. [DOI] [PubMed] [Google Scholar]
- Juurlink BH, Thorburne SK, Hertz L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia. 1998;22:371–378. doi: 10.1002/(sici)1098-1136(199804)22:4<371::aid-glia6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Kassmann CM, Lappe-Siefke C, Baes M, et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–976. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- Khan M, Contreras M, Singh I. Endotoxin-induced alterations of lipid and fatty acid compositions in rat liver peroxisomes. J Endotoxin Res. 2000;6:41–50. doi: 10.1177/09680519000060010601. [DOI] [PubMed] [Google Scholar]
- Khan M, Haq E, Giri S, Singh I, Singh AK. Peroxisomal participation in psychosine-mediated toxicity: implications for Krabbe’s disease. J Neurosci Res. 2005;80:845–854. doi: 10.1002/jnr.20529. [DOI] [PubMed] [Google Scholar]
- Khan M, Sekhon B, Jatana M, Giri S, Gilg AG, Sekhon C, Singh I, Singh AK. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J Neurosci Res. 2004;76:519–527. doi: 10.1002/jnr.20087. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Back SA. Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, Uchida K. A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. J Biol Chem. 2004;279:48389–48396. doi: 10.1074/jbc.M409935200. [DOI] [PubMed] [Google Scholar]
- Lavrovsky Y, Chatterjee B, Clark RA, Roy AK. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol. 2000;35:521–532. doi: 10.1016/s0531-5565(00)00118-2. [DOI] [PubMed] [Google Scholar]
- Lazarow PB. Peroxisome structure, function, and biogenesis--human patients and yeast mutants show strikingly similar defects in peroxisome biogenesis. J Neuropathol Exp Neurol. 1995;54:720–725. doi: 10.1097/00005072-199509000-00015. [DOI] [PubMed] [Google Scholar]
- Lazarow PB. Peroxisome biogenesis: advances and conundrums. Curr Opin Cell Biol. 2003;15:489–497. doi: 10.1016/s0955-0674(03)00082-6. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ. Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev Neurosci. 2001;23:234–247. doi: 10.1159/000046149. [DOI] [PubMed] [Google Scholar]
- Leviton A, Gilles F. Ventriculomegaly, delayed myelination, white matter hypoplasia, and “periventricular” leukomalacia: how are they related? Pediatr Neurol. 1996;15:127–136. doi: 10.1016/0887-8994(96)00157-9. [DOI] [PubMed] [Google Scholar]
- Millan-Plano S, Garcia JJ, Martinez-Ballarin E, Reiter RJ, Ortega-Gutierrez S, Lazaro RM, Escanero JF. Melatonin and pinoline prevent aluminium-induced lipid peroxidation in rat synaptosomes. J Trace Elem Med Biol. 2003;17:39–44. doi: 10.1016/S0946-672X(03)80044-5. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser HW. Peroxisomal disorders. Semin Pediatr Neurol. 1996;3:298–304. doi: 10.1016/s1071-9091(96)80033-7. [DOI] [PubMed] [Google Scholar]
- Moser HW. Peroxisomal disorders: classification and overview of biochemical abnormalities. Rev Neurol. 1999;28(Suppl 1):S45–48. [PubMed] [Google Scholar]
- Moynagh PN, Williams DC, O’Neill LA. Activation of NF-kappa B and induction of vascular cell adhesion molecule-1 and intracellular adhesion molecule-1 expression in human glial cells by IL-1. Modulation by antioxidants. J Immunol. 1994;153:2681–2690. [PubMed] [Google Scholar]
- Paintlia AS, Gilg AG, Khan M, Singh AK, Barbosa E, Singh I. Correlation of very long chain fatty acid accumulation and inflammatory disease progression in childhood X-ALD: implications for potential therapies. Neurobiol Dis. 2003;14:425–439. doi: 10.1016/j.nbd.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, Singh I. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. Faseb J. 2005;19:1407–1421. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. IL-4-induced peroxisome proliferator-activated receptor gamma activation inhibits NF-kappaB trans activation in central nervous system (CNS) glial cells and protects oligodendrocyte progenitors under neuroinflammatory disease conditions: implication for CNS-demyelinating diseases. J Immunol. 2006;176:4385–4398. doi: 10.4049/jimmunol.176.7.4385. [DOI] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Barbosa E, Singh I, Singh AK. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J Neurosci Res. 2004;78:347–361. doi: 10.1002/jnr.20261. [DOI] [PubMed] [Google Scholar]
- Pang Y, Cai Z, Rhodes PG. Effect of tumor necrosis factor-alpha on developing optic nerve oligodendrocytes in culture. J Neurosci Res. 2005;80:226–234. doi: 10.1002/jnr.20450. [DOI] [PubMed] [Google Scholar]
- Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- Qi C, Zhu Y, Reddy JK. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem Biophys. 2000 Spring;32:187–204. doi: 10.1385/cbb:32:1-3:187. [DOI] [PubMed] [Google Scholar]
- Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC. Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem. 2005;280:41057–41068. doi: 10.1074/jbc.M505160200. [DOI] [PubMed] [Google Scholar]
- Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763:1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Sekhon B, Sekhon C, Khan M, Patel SJ, Singh I, Singh AK. N-Acetyl cysteine protects against injury in a rat model of focal cerebral ischemia. Brain Res. 2003;971:1–8. doi: 10.1016/s0006-8993(03)02244-3. [DOI] [PubMed] [Google Scholar]
- Sheikh FG, Pahan K, Khan M, Barbosa E, Singh I. Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proc Natl Acad Sci U S A. 1998;95:2961–2966. doi: 10.1073/pnas.95.6.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher T, Yi HF, McBride OW, Gonzalez FJ. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32:5598–5604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- Siems WG, Grune T, Beierl B, Zollner H, Esterbauer H. The metabolism of 4-hydroxynonenal, a lipid peroxidation product, is dependent on tumor age in Ehrlich mouse ascites cells. Exs. 1992;62:124–135. doi: 10.1007/978-3-0348-7460-1_13. [DOI] [PubMed] [Google Scholar]
- Singh I. Biochemistry of peroxisomes in health and disease. Mol Cell Biochem. 1997;167:1–29. doi: 10.1023/a:1006883229684. [DOI] [PubMed] [Google Scholar]
- Singh I, Pahan K, Khan M, Singh AK. Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem. 1998;273:20354–20362. doi: 10.1074/jbc.273.32.20354. [DOI] [PubMed] [Google Scholar]
- Singh I, Paintlia AS, Khan M, Stanislaus R, Paintlia MK, Haq E, Singh AK, Contreras MA. Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res. 2004;1022:1–11. doi: 10.1016/j.brainres.2004.06.059. [DOI] [PubMed] [Google Scholar]
- Stanislaus R, Gilg AG, Singh AK, Singh I. N-acetyl-L-cysteine ameliorates the inflammatory disease process in experimental autoimmune encephalomyelitis in Lewis rats. J Autoimmune Dis. 2005;2:4. doi: 10.1186/1740-2557-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztriha LS, Nork MP, Abdulrazzaq YM, al-Gazali LI, Bakalinova DB. Abnormal myelination in peroxisomal isolated dihydroxyacetonephosphate acyltransferase deficiency. Pediatr Neurol. 1997;16:232–236. doi: 10.1016/s0887-8994(97)00014-3. [DOI] [PubMed] [Google Scholar]
- Topal T, Oter S, Korkmaz A, Sadir S, Metinyurt G, Korkmazhan ET, Serdar MA, Bilgic H, Reiter RJ. Exogenously administered and endogenously produced melatonin reduce hyperbaric oxygen-induced oxidative stress in rat lung. Life Sci. 2004;75:461–467. doi: 10.1016/j.lfs.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Toyokuni S, Uchida K, Okamoto K, Hattori-Nakakuki Y, Hiai H, Stadtman ER. Formation of 4-hydroxy-2-nonenal-modified proteins in the renal proximal tubules of rats treated with a renal carcinogen, ferric nitrilotriacetate. Proc Natl Acad Sci U S A. 1994;91:2616–2620. doi: 10.1073/pnas.91.7.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji F, Miyake Y, Aono H, Kawashima Y, Mita S. Effects of bucillamine and N-acetyl-L-cysteine on cytokine production and collagen-induced arthritis (CIA) Clin Exp Immunol. 1999;115:26–31. doi: 10.1046/j.1365-2249.1999.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- Van den Branden C, Dacremont G, Hooghe R, Roels F. Inhibition of peroxisomal beta-oxidation and brain development in rats. Glia. 1989;2:260–265. doi: 10.1002/glia.440020407. [DOI] [PubMed] [Google Scholar]
- Van den Branden C, Leeman J, Dacremont G, Collumbien R, Roels F. Experimental inhibition of peroxisomal beta-oxidation in rats: influence on brain myelination. Glia. 1990;3:458–463. doi: 10.1002/glia.440030604. [DOI] [PubMed] [Google Scholar]
- Victor VM, Rocha M, De la Fuente M. N-acetylcysteine protects mice from lethal endotoxemia by regulating the redox state of immune cells. Free Radic Res. 2003;37:919–929. doi: 10.1080/1071576031000148727. [DOI] [PubMed] [Google Scholar]
- Wang X, Rousset CI, Hagberg H, Mallard C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Semin Fetal Neonatal Med. 2006;11:343–353. doi: 10.1016/j.siny.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Kim CJ, Romero R, Jun JK, Park KH, Choi ST, Chi JG. Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. Am J Obstet Gynecol. 1997a;177:797–802. doi: 10.1016/s0002-9378(97)70271-0. [DOI] [PubMed] [Google Scholar]
- Yoon BH, Romero R, Kim CJ, Koo JN, Choe G, Syn HC, Chi JG. High expression of tumor necrosis factor-alpha and interleukin-6 in periventricular leukomalacia. Am J Obstet Gynecol. 1997b;177:406–411. doi: 10.1016/s0002-9378(97)70206-0. [DOI] [PubMed] [Google Scholar]