

Summary
BubR1 is an essential mitotic checkpoint protein with multiple functional domains. It has been implicated in mitotic checkpoint control -as an active kinase at unattached kinetochores and as a cytosolic inhibitor of APC/CCdc20 activity- as well as in mitotic timing and stable chromosome-spindle attachment. Using BubR1-conditional knockout cells and BubR1 domain mutants, we demonstrate that the N-terminal Cdc20 binding domain of BubR1 is essential for all of these functions, whereas its C-terminal Cdc20-binding domain, Bub3-binding domain, and kinase domain are not. We find that the BubR1 N terminus binds to Cdc20 in a KEN box-dependent manner to inhibit APC/C activity in interphase, thereby allowing accumulation of cyclin B in G2 phase prior to mitosis onset. Together, our results suggest that kinetochore-bound BubR1 is non-essential and that soluble BubR1 functions as a pseudosubstrate inhibitor of APC/CCdc20 during interphase to prevent unscheduled degradation of specific APC/C substrates.
Keywords: BubR1, Cdc20, APC/C, kinetochore, mitotic checkpoint, chromosome missegregation, aneuploidy
Introduction
The mitotic checkpoint (or spindle assembly checkpoint) is a surveillance mechanism that inhibits sister chromatid separation until all chromosomes are correctly bioriented on the mitotic spindle (Musacchio and Salmon, 2007). Core components of this checkpoint, including Mad1, Mad2, Bub1, BubR1 and Bub3, accumulate at unattached kinetochores shortly after onset of mitosis. There, they generate a negative signal (called “wait anaphase” signal) that prevents Cdc20 from activating the anaphase-promoting complex/cyclosome (APC/C). This signal quenches after the last sister chromatid pair becomes attached to the mitotic spindle, allowing the activation of APC/C by Cdc20. Once active, APC/C initiates the separation of sister chromosomes by targeting two key substrates, cyclin B and securin, for destruction by the 26S proteosome through polyubiquitination.
The molecular basis of the Cdc20-APC/C inhibitory signals emanated by mitotic checkpoint proteins at unattached kinetochores remains unclear. One hypothesis is that kinetochore-bound mitotic checkpoint proteins provide a catalytic surface for the structural activation of soluble mitotic checkpoint molecules so that they can efficiently bind and inhibit Cdc20. This idea is largely based on elegant structural studies showing that Mad2 exists in two distinct conformations (reviewed by: Musacchio and Salmon, 2007; Yu, 2006). In one conformation, known as the closed confirmation (C-Mad2), Mad2 locks Mad1 into a binding pocket via a mobile structure referred to as the “safety belt”. This mechanism ensures that Mad1 and Mad2 form a stable complex at unattached kinetochores. In the mitotic cytosol, Mad2 exists in an open confirmation (O-Mad2) that does not allow it to bind Mad1 or Cdc20. Recruitment of cytosolic O-Mad2 to C-Mad2-Mad1 at unattached kinetochores via C-Mad2-O-Mad2 dimerization, converts O-Mad2 into a conformer (I-Mad2) that can capture Cdc20, and thus inhibit APC/C activity.
A less well understood Cdc20 inhibitor is the BubR1 kinase. BubR1, like Mad2, is also present at unattached kinetochores and in the mitotic cytosol. A “GLEBS-like” motif that mediates binding to Bub3 is necessary for BubR1 accumulation at kinetochores in early mitosis (Wang et al., 2001). At kinetochores, BubR1 kinase activity is turned on by microtubule motor protein CENP-E (Mao et al., 2003; Mao et al., 2005). Inhibition of BubR1 kinase activity in astrocytoma cells causes massive chromosome missegregation and cell death (Kops et al., 2004), suggesting that the enzymatic functions of kinetochore-bound BubR1 are essential for cell viability.
Like Mad2, BubR1 binds directly to Cdc20 and inhibits APC/C activity in vitro (Tang et al., 2001). Combined, however, BubR1 and Mad2 are much more potent APC/CCdc20 inhibitors than the individual proteins (Fang, 2002). This, together with the discovery of an in vivo mitotic checkpoint complex (MCC) consisting of Mad2, BubR1, Bub3 and Cdc20 and the demonstration that this complex strongly inhibits APC/C activity in vitro (Sudakin et al., 2001) led to speculation that Mad2-Cdc20 complexes generated at unattached kinetochores and released into the cytosol assemble with BubR1-Bub3 complexes to form the ultimate cytosolic APC/C inhibitor. Yet, how BubR1-Bub3 binding to Cdc20 or Cdc20-Mad2 is regulated is unknown. One theory is that kinetochore-bound BubR1 or other kinetochore-bound proteins recruit BubR1 or BubR1-Bub3 from the mitotic cytosol and prime these complexes for binding to Cdc20 or Cdc20-Mad2. Alternatively, kinase-active BubR1 at kinetochores might activate a signaling pathway that alters the binding ability of cytosolic BubR1-Bub3 for Cdc20 or Cdc20-Mad2.
In addition to functioning in the mitotic checkpoint, BubR1 plays a role in the timing of mitosis (Meraldi et al., 2004). This BubR1 function involves cooperation with Mad2 and is kinetochore independent, but is otherwise not understood at the molecular level. BubR1 is further required for stable kinetochore-microtubule attachments (Ditchfield et al., 2003; Lampson and Kapoor, 2005), yet how it does so remains unknown.
Inactivation of BubR1 in mice causes early embryonic lethality, as do null mutations in other mitotic checkpoint proteins (Ricke et al., 2008). However, although gene knockout studies have firmly established the essential nature of mammalian mitotic checkpoint proteins, it remains to be determined how individual components of this surveillance mechanism control cell viability. Using BubR1 conditional knockout cells and a series of BubR1 mutants with one or more defective functional domains, we set out to identify the critical functional domain(s) of BubR1 and the mechanisms by which the kinetochore-bound and the cytosolic BubR1 fractions regulate the mitotic checkpoint, mitotic timing and kinetochore-microtubule attachment. We demonstrate that the N-terminal Cdc20 binding domain of BubR1 is essential for all of these functions and acts as a pseudosubstrate for APC/CCdc20 to prevent cyclin B destruction in interphase.
Results
The N Terminus of BubR1 Is Required for Cell Viability
To dissect the essential BubR1 domains, we utilized mouse embryonic fibroblasts (MEFs) that contain BubR1 hypomorphic (BubR1H) alleles in which exon 5 is flanked by loxP sites (Baker et al., 2004). Our approach was to stably introduce mutant BubR1 expression constructs into these mutant MEFs, inactivate the BubR1H alleles by expression of Cre recombinase, and then monitor cells for growth and survival in the absence of endogenously produced full-length BubR1 protein. Initially, we created a set of four Flag (F)-tagged BubR1 mutants, each lacking a distinct functional domain (Fig. 1A). F-BubR1(KD2) has two point mutations (K784>R in the ATP binding pocket and K802>R in the catalytic domain of the kinase domain) that inactivate BubR1 kinase activity (Harris et al., 2005; Mao et al., 2003; Tang et al., 2001). F-BubR1(E406K) has a point mutation in the GLEBS motif that interrupts Bub3 binding (Harris et al., 2005, see also Fig. 2E). F-BubR1(357-1052) lacks the N-terminal Cdc20-binding domain (Cdc20-BD1), while F-BubR1Δ(525-700) lacks the C-terminal Cdc20-binding domain (Cdc20-BD2) (Davenport et al., 2006; Tang et al., 2001). All four mutants and full-length BubR1 were cloned into the pMSCV-GFP retroviral vector and expressed in BubR1H/H MEF cells, which were immortalized by homozygous p53 gene disruption (BubR1H/H/p53−/− MEFs). GFP-positive cells were sorted by flow cytometry. There were no notable changes in the rates of cell death and cell proliferation in BubR1H/H MEFs expressing any of our mutant BubR1 proteins (Fig. S1), excluding the possibility that these mutants interfere with cell viability in a dominant-negative fashion. Cultures of GFP-positive cells were then infected with a Cre encoding retrovirus (pMSCV-Cre/Puro) to abort expression of wild-type BubR1 protein from the endogenous BubR1 hypomorphic allele. Following infection, Cre-expressing cells were selected by addition of puromycin. As expected, cells infected with empty vector failed to proliferate after disruption of endogenous BubR1 expression and typically died within 4-6 days after Cre-induction (Fig. 1B and 1C). When wild-type Flag-tagged BubR1 (F-BubR1(1-1052)) was ectopically expressed prior to disruption of endogenous BubR1, the cells survived and continued to proliferate (Fig. 1B and 1C). Cells expressing F-BubR1(KD2), F-BubR1(E406K) or F-BubR1Δ(525-700) also continued to proliferate after Cre expression, but cells expressing F-BubR1(357-1052) died (Fig. 1B and 1C). Southern blot analysis confirmed that the hypomorphic alleles of the surviving cells had indeed been converted into null alleles (Fig. 1D). Western blot analysis using anti-Flag antibody validated that each mutant was accurately expressed (Fig. 1E). Most flag-tagged BubR1 proteins were quite overexpressed compared to endogenous BubR1. However, we note that no dominant-negative effects were observed when these same proteins were overexpressed in BubR1+/+/p53−/− MEFs (Fig. S2). Furthermore, when the cell viability analysis was repeated using non-immortalized BubR1H/H MEFs, once again, the only mutant that was unable to rescue viability was F-BubR1(357-1052) (Fig. S3). Together, these data demonstrate that only the BubR1 N terminus is essential for cell viability.
Figure 1. Cell Growth and Survival Requires the N Terminus of BubR1.

(A) Schematic overview of wild-type and mutant BubR1 proteins.
(B) Images of BubR1H/H/p53−/− MEFs containing indicated expression constructs taken 6 days after infection with Cre retrovirus.
(C) Viability of BubR1H/H/p53−/− MEFs ectopically expressing the indicated mutant BubR1 proteins in the presence (− Cre) or absence (+ Cre) of endogenous BubR1.
(D) Southern blot of BubR1H/H/p53−/−MEFs expressing the indicated mutant BubR1 proteins subjected to Cre-mediated disruption of endogenous BubR1. DNA was digested with BamH1 and blots were probed with a 3’ BubR1 genomic probe (Baker et al., 2004).
(E) Western blot analysis of MEFs carrying the indicated BubR1 expression constructs. Blots were probed with the indicated antibodies. Panels labeled αBubR1: lanes 1-6 were probed with antibodies against hBubR1(1-350) and lanes 7 and 8 with antibodies against mBubR1(382-420). Actin was used as loading control. The genotype of MEFs in lanes 3-6 and 8 was BubR1−/−/p53−/−, that of MEFs in lane 7 was BubR1H/H/p53−/−.
Figure 2. Cdc20 Binding to BubR1 N Terminus Is Sufficient for Cell Viability.

(A) Schematic overview of wild-type and mutant BubR1 proteins. K, KEN box.
(B and C) Same as legends to Fig. 1B and 1C, respectively.
(D) Western blot analysis of MEFs carrying the indicated BubR1 expression constructs. Blots were probed with the indicated antibodies. Actin was used as loading control. The genotype of cells in lanes 1 and 5 was BubR1−/−/p53−/−, whereas that of cells in lanes 2-4 and 6-8 was BubR1H/H/p53−/−.
(E) Immunoblots of mitotic extracts of MEFs carrying the indicated BubR1 expression constructs subjected to immunoprecipitation with Cdc20 or Flag antibody, and probed with the indicated antibodies.
The Critical Function of the BubR1 N Terminus Is Cdc20 Binding
To determine whether the N-terminal BubR1 segment containing the Cdc20-BD1 would be sufficient for cell growth and survival, we expressed F-BubR1(1-363) in BubR1H/H/p53−/−and BubR1H/H MEFs (Fig. 2A). These cells were viable in the absence of wild-type BubR1 and able to proliferate (Fig. 2B and 2C, and Fig. S3). In contrast, F-BubR1(1-218) was unable to mediate cell growth and survival (Fig. 2A-D, and Fig. S3). To more precisely map the BubR1 region sufficient for cell viability, we generated F-BubR1(1-322) and F-BubR1(47-363) (Fig. 2A). Both these mutants were also unable to mediate cell growth and survival (Fig. 2B-D), indicating that the entire 1-363 BubR1 segment is essential.
Next, we tested whether Cdc20 binding to the BubR1 N terminus would be a requirement for survival. As shown in Fig. 2E, F-BubR1(1-363) coprecipitated with Cdc20 from mitotic MEF extracts, and vice versa, Cdc20 coprecipitated with F-BubR1(1-363). In contrast, F-BubR1(1-218), F-BubR1(1-322), F-BubR1(47-363) failed to coprecipitate Cdc20 (Fig. 2E). These data demonstrate that the first 363 residues define the boundaries of the BubR1 N-terminal Cdc20-binding domain and that this domain is both necessary and sufficient for cell viability. F-BubR1(357-1052), which contains the C-terminal Cdc20 binding domain, readily precipitated with Cdc20, and vice versa (Fig. 2E). Thus, while both domains are capable of binding Cdc20, the fact that only the N-terminal domain is required for viability, demonstrates that they are not functionally redundant.
ScMad3p and SpMad3p, the homologues of BubR1 in S. cerevisiae and S. pombe, contain two evolutionarily conserved KEN boxes, one of which is known to be required for binding to Cdc20. Both of these KEN boxes are conserved in mouse BubR1 and located within BubR1(1-363), one spanning amino acids 19-21 (designated KEN19), the other 298-300 (KEN298). To further investigate the role of Cdc20 binding in cell survival, we produced three F-BubR1(1-363) mutants in which these KEN boxes were mutated to AAA either individually or in combination, and expressed them in BubR1H/H/p53−/− MEFs (Fig. 2A and 2D). None of the three mutants coprecipitated with Cdc20 (Fig. 2E), indicating that both KEN boxes are required for Cdc20 binding in vivo. Consistent with this, F-BubR1(1-363)KEN19AAA, F-BubR1(1-363)KEN298AAA and F-BubR1(1-363)KEN19+298AAA were unable to pull down Cdc20 in the reverse experiment (Fig. 2E). Importantly, none of these mutants was able to sustain cell growth and survival following Cre expression (Fig. 2B and 2C). Thus, Cdc20 binding by the BubR1 N terminus appears to be necessary for cell viability.
Furthermore, F-BubR1(1-363) not only pulled down Cdc20, but also Mad2 and the APC component APC6 (Fig. 2E). As expected, Bub3 did not coprecipitate because of the absence of the GLEBS motif. F-BubR1(1-363) KEN box mutants, F-BubR1(1-218), F-BubR1(1-322) and F-BubR1(47-363), all of which did not interact with Cdc20, also failed to coprecipitate Mad2 and APC6 (Fig. 2E). These data suggest that the BubR1 N-terminus, Mad2 and Cdc20 form a protein complex that can associate with APC/C.
BubR1 Kinase Activity Sustains the Mitotic Checkpoint
Earlier work has shown that the targeting of BubR1 to kinetochores of unattached mitotic chromosomes is dependent on Bub3 binding (Wang et al., 2001). Consistent with this, mutant F-BubR1(E406K), which lacks the ability to bind Bub3 (Fig. 2E), failed to accumulate at kinetochores (Fig. 3A). This result, combined with our observation that F-BubR1(E406K) rescues cell growth and survival in the absence of endogenous BubR1, indicates that kinetochore-associated BubR1 functions are non-essential. Two additional results support this conclusion. First, F-BubR1(1-363), which does not bind Bub3 (Fig. 2E) but is sufficient for cell viability, failed to accumulate at unattached kinetochores (Fig. 3A). Second, the complementary segment, BubR1(357-1052), did concentrate at kinetochores (Fig. 3A), yet failed to rescue cells lacking endogenous BubR1.
Figure 3. The Mitotic Checkpoint Has Different Requirements for Cytosolic and Kinetochore-Bound BubR1.
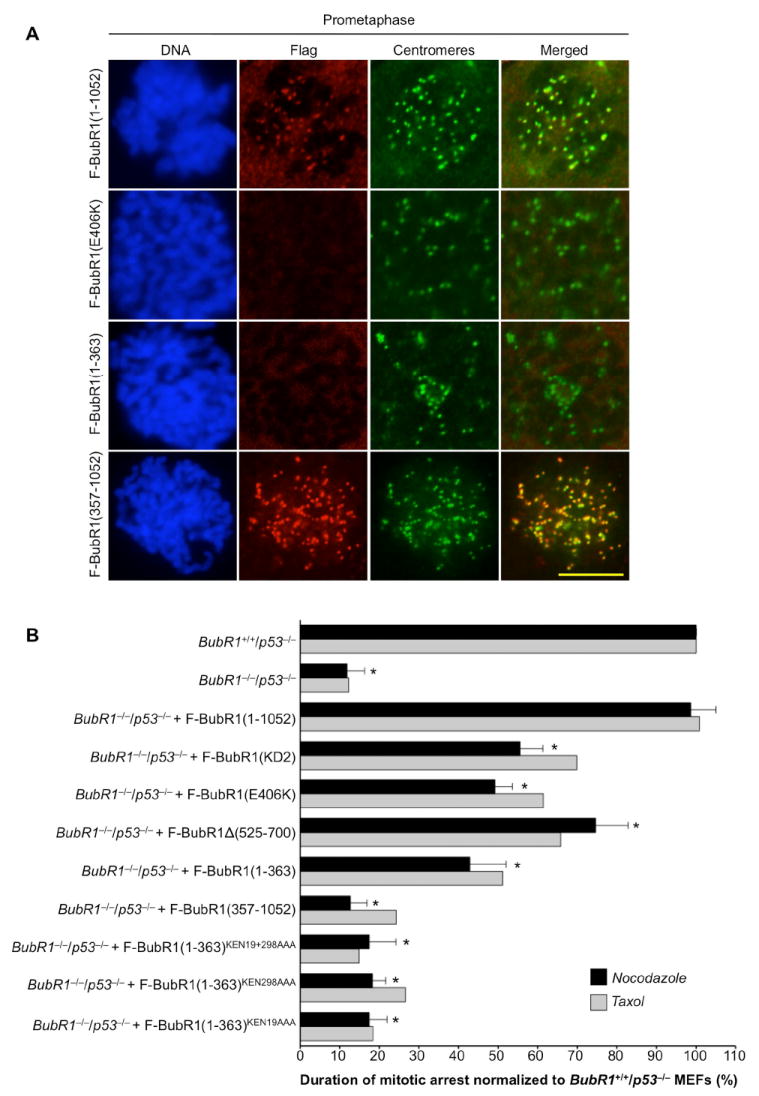
(A) Immunolocalization of Flag-tagged BubR1 proteins in MEFs during prometaphase. F-BubR1(1-1052), F-BubR1(E406K) and F-BubR1(1-363) were expressed in BubR1−/−/p53−/− MEFs and F-BubR1(357-1052) in BubR1H/H/p53−/− MEFs. Flag-tagged proteins were visualized with mouse anti-FLAG antibody and centromeres with ACA antibody. DNA was stained with Hoechst. Bar = 10 μm.
(B) Analysis of mitotic checkpoint activity of MEFs challenged with nocodazole or taxol. Three independent MEF lines per genotype were challenged with nocodazole. Error bars represent the SD. *p < 0.0001 versus BubR1+/+/p53−/− MEFs (Log rank test). One line per genotype was challenged with taxol.
Both Bub1 and Mad2 properly accumulated at unattached kinetochores of BubR1−/−/p53−/− MEFs expressing F-BubR1(1-363) (Fig. S4A and S4B), suggesting that loss of endogenous BubR1 has no major impact on kinetochore assembly. CENP-E levels at kinetochores were reduced in BubR1−/−/p53−/− MEFs expressing F-BubR1(1-363) or F-BubR1(E406K), but not in those containing F-BubR1(KD2) or F-BubR1Δ(525-700) (Fig. S4C), indicating that kinetochore-bound BubR1 modulates CENP-E binding to unattached kinetochores.
To further explore the functional relevance of kinetochore-bound BubR1, we prepared immortalized MEFs expressing various BubR1 mutants in the absence of endogenous BubR1 and measured their mitotic checkpoint activity using a standardized nocodazole-challenge assay. BubR1−/−/p53−/− MEFs generated by transducing BubR1H/H/p53−/− MEFs with Cre retrovirus 72 h prior to analysis (Fig. S5) had only 12% of normal mitotic checkpoint activity (Fig. 3B), indicating that there is virtually no checkpoint activity in the absence of BubR1. F-BubR1(1-1052) fully restored the mitotic checkpoint in these MEFs. However, F-BubR1(KD2), which binds to kinetochores (Fig. S6), only partially restored it to 56% of normal, indicating that BubR1 kinase activity contributes to mitotic checkpoint signaling. We expected F-BubR1(E406K) to have a similar corrective effect as BubR1(KD2) because activation of BubR1 kinase activity is mediated by CENP-E at kinetochores. Indeed, with 49% of normal checkpoint activity, F-BubR1(E406K) was in the same range as F-BubR1(KD2). Failure of F-BubR1(E406K) to bind Bub3 might impair its ability to fully inhibit APC/CCdc20 in the cytoplasm. It is conceivable that this potential defect contributes to F-BubR1(E406K)’s inability to fully correct checkpoint activity. However, the observation that BubR1 inhibits APC/CCdc20 equally well in vitro in the presence or absence of Bub3 (Tang et al., 2001) argues against this possibility. As shown in Fig. 3B, mitotic checkpoint activities in response to taxol were very similar to those seen in response to nocodazole. Collectively, the above data suggest that kinetochore-associated BubR1 kinase activity is important for prolonged mitotic checkpoint activation.
The BubR1 N Terminus Has a Critical Role in the Mitotic Checkpoint
To further examine the essential function of the N-terminal Cdc20-binding domain of BubR1, we analyzed the mitotic checkpoint status of BubR1−/−/p53−/− MEFs expressing F-BubR1(357-1052). Like BubR1−/−/p53−/− MEFs, these MEFs had extremely low mitotic checkpoint activity (Fig. 3B; 13% of normal). In contrast, F-BubR1(1-363) partially restored mitotic checkpoint function in BubR1−/−/p53−/− MEFs to 43% of normal. Expression of BubR1(1-363)KEN19AAA, F-BubR1(1-363)KEN298AAA or F-BubR1(1-363)KEN19+298AAA in BubR1−/−/p53−/− MEFs had no such corrective effect (Fig. 3B). These data indicate that the binding of Cdc20 to the BubR1 N terminus in the cytosol is crucial for mitotic checkpoint function.
The BubR1 N Terminus Is Required for Chromosome-Spindle Attachment
RNA interference (RNAi) studies in human cancer cell lines have implicated BubR1 in stable attachment of chromosomes to spindle microtubules (Ditchfield et al., 2003; Lampson and Kapoor, 2005). To determine the domain requirements for this function, we used three different assays. First, we treated MEFs with the proteosome inhibitor MG132 to induce metaphase arrest. Spindles, centromeres and chromosomes were then stained to examine proper chromosome alignment at the metaphase plate by confocal microscopy. Chromosome misalignment was observed in 7% of BubR1+/+/p53−/− metaphases (Fig. 4A and 4B). In all instances, only one or a few chromosomes were misaligned (minor misalignment). In contrast, 72% of BubR1−/−/p53−/− metaphases had misaligned chromosomes (Fig. 4B), half of which had minor chromosome misalignments, and half of which had major chromosome misalignments involving numerous chromosomes (Fig. 4A). Expression of F-BubR1(1-1052) in BubR1−/−/p53−/− MEFs almost completely restored proper chromosome alignment (13% of metaphases remained defective). F-BubR1(1-363) also had a major corrective effect, with only 22% of metaphases expressing this mutant exhibiting chromosome misalignment. Importantly, all misalignments were minor. Conversely, F-BubR1(1-363) proteins carrying KEN box mutations had absolutely no corrective ability. These data suggest that Cdc20 binding to the BubR1 N terminus is critical for proper chromosome-microtubule attachment.
Figure 4. The BubR1 N Terminus Regulates Chromosome-Spindle Attachment and Mitotic Timing.

(A) Cdc20 binding to the BubR1 N terminus is essential for chromosome alignment. MEFs were treated with 10 μM MG132 for 2 h, fixed and immunostained for α–tubulin and centromeres. The second and third image from the left represent BubR1−/−/p53−/− metaphases with minor and major misalignment phenotypes, respectively. Bar = 10 μm.
(B) Quantification of chromosome misalignment defects of MEFs shown in (A). MEF genotypes and BubR1 expression constructs are as indicated in (C). Shaded and non-shaded areas represent major and minor misalignments, respectively.
(C) Quantification of misalignment defects in MG132-treated MEFs by live-cell imaging. Shaded and non-shaded areas represent major and minor misalignments, respectively.
(D) Examples of metaphase-arrested MEFs displaying proper chromosome alignment (top), minor misalignment (middle) or major misalignment (bottom). Cells were MG132 treated. Arrows designate misaligned chromosomes. Bar = 10 μm.
(E) Analysis of chromosome misalignment and cell death by live-cell imaging. We note that nearly all cells dying within 8 h after mitosis had major alignment defects.
(F) Timing of anaphase onset. Three MEF lines were used per genotype. Error bars represent the SD. *p < 0.0001 versus BubR1+/+/p53−/− MEFs (Mann-Whitney test).
In the second assay, we monitored chromosome alignment in the presence of MG132 by live cell imaging. Results obtained by this method were essentially the same as those obtained by immunostaining of fixed cells (Fig. 4C and 4D, and Movie S1-S2). Furthermore, analysis of BubR1−/−/p53−/− MEFs expressing F-BubR1(357-1052) confirmed the essential role of the N-terminal Cdc20 binding domain in chromosome-spindle attachment (Fig. 4C). F-BubR1(KD2) and F-BubR1(E406K) both had a major corrective effect on chromosome misalignment when expressed in BubR1−/−/p53−/− MEFs (Fig. 4C). However, these mutants were not as effective as F-BubR1(1-1052), suggesting that BubR1 kinase activity at kinetochores plays a contributing role in chromosome-microtubule attachment.
Third, we monitored MEFs as they progressed through mitosis in the absence of MG132. As expected, high rates of chromosome misalignment were observed in BubR1−/−/p53−/− MEFs and BubR1−/−/p53−/− MEFs expressing F-BubR1(357-1052) or any of the F-BubR1(1-363) KEN box mutants (Fig. 4E). Percentages of cells with major alignment problems were similar to those seen in the presence of MG132. Most of these cells underwent mitotic catastrophe and died within 8 h (Fig. 4E, and Movie S3). However, percentages of cells with minor alignments were considerably lower in the absence than in the presence of MG132, suggesting that metaphase arrest facilitated the detection of these defects. Misalignments were rare and minor when F-BubR1(1-1052) was present. F-BubR1(1-363), F-BubR1(KD2), F-BubR1(E406K) also showed no major misalignment problems but slightly increased rates of misalignment of one or a few chromosomes (Fig. 4E), underscoring that kinetochore-bound BubR1 has a contributing rather than a critical role in chromosome-spindle attachment. No cell death was observed among cells with minor defects (Fig. 4E). We found that Cre recombinase expression per se did not cause chromosome misalignment (Fig. S7).
F-BubR1(KD2) can only partially correct the chromosome alignment and mitotic checkpoint defects of BubR1−/−/p53−/− MEFs. It cannot be excluded that this is due to the relatively low levels of F-BubR1(KD2) expression (Fig. 1E). However, this is unlikely because both defects were corrected when BubR1−/−/p53−/− MEFs expressed F-BubR1(1-1052) at F-BubR1(KD2) levels (Fig. S8).
The BubR1 N Terminus Regulates Mitotic Timing
Depletion of BubR1 in HeLa cells shortens the NEBD-anaphase time interval from about 25 to 14 min (Meraldi et al., 2004), suggesting that BubR1 is a regulator of mitotic timing. This BubR1 function is conserved in MEFs as we found the time from NEBD to anaphase onset to be significantly reduced in BubR1−/−/p53−/− MEFs (Fig. 4F). Notably, expression of F-BubR1(1-363), but not F-BubR1(357-1052) or F-BubR1(1-363) fragments containing KEN box mutations, fully restored proper timing of mitosis in BubR1−/−/p53−/− MEFs, indicating that BubR1 regulates mitotic timing through direct Cdc20 binding in the mitotic cytosol (Fig. 4F). F-BubR1(KD2), F-BubR1(E406K) and F-BubR1Δ(525-700) all restored normal NEBD-to-anaphase duration in BubR1−/−/p53−/− MEFs, confirming that the N-terminal Cdc20 binding domain is the only BubR1 domain relevant to mitotic timing.
The BubR1 N Terminus Prevents Premature Degradation of Cyclin B by APC/CCdc20
The demonstration that KEN boxes in the BubR1 N terminus are necessary for Cdc20 binding suggests that BubR1 acts as a pseudosubstrate inhibitor of APC/CCdc20 in early mitosis. To test this further, BubR1−/−/p53−/− MEFs expressing various BubR1 mutants were immunostained for cyclin B, a substrate that is normally targeted for degradation by APC/CCdc20 in late metaphase. As shown in Fig. 5A and 5B, cyclin B levels in prophase were 3- to 4-fold lower in BubR1−/−/p53−/− MEFs than in BubR1+/+/p53−/− MEFs. This decline is due to increased proteosomal degradation rather than reduced gene transcription as MG132 treatment restored normal cyclin B protein levels in BubR1−/−/p53−/− prophases (Fig. 5C and 5D). Expression of F-BubR1(1-363), but not of F-BubR1(357-1052) or F-BubR1(1-363)KEN19+298AAA, fully restored cyclin B levels in prophase (Fig. 5A and 5B), indicating that the N terminal Cdc20-binding domain indeed functions as a potent inhibitor of cyclin B polyubiquitination in early mitosis.
Figure 5. Binding of BubR1 N Terminus to Cdc20 Inhibits APC/C activity.

(A) Prophases stained for cyclin B, phosphohistone H3Ser10 (P-H3) and DNA (Hoechst).
(B) Quantification of cyclin B levels in prophase.
(C) Representative prophases treated with 10 μM MG132 1 h prior to staining for cyclin B, P-H3 and DNA (Hoechst).
(D) Quantification of cyclin B levels in prophase after MG132 treatment.
(E) Incidence of PMSCS in MEFs expressing the indicated BubR1 mutants.
Premature sister chromatid separation (PMSCS), which is an indicator of premature APC/C activity (Baker et al., 2005), was extremely high in BubR1−/−/p53−/− MEFs, with 72% of metaphase spreads showing the defect (Fig. 5E). When cells were briefly treated with MG132, this percentage reduced to 48%, confirming that the observed separation defect was, at least in part, due to premature APC/C activity. PMSCS was only 9% in BubR1−/−/p53−/− MEFs when F-BubR1(1-363) was present, and 1% when full-length BubR1 was expressed (Fig. 5E). F-BubR1(357-1052) and F-BubR1(1-363)KEN19+298AAA were unable to rescue the PMSCS phenotype. Thus, the above data support our conclusion that the BubR1 N terminus binds to Cdc20 and inhibits APC/C activity.
BubR1 N Terminus Prevents Cyclin B Degradation by APC/CCdc20 in Interphase
To further examine the degradation of cyclin B in the absence of BubR1, we transiently transfected cyclin B-GFP (Hagting et al., 2002) into BubR1−/−/p53−/− and BubR1+/+/p53−/− MEFs and tracked its level of expression by live-cell imaging (Fig. 6A). Cyclin B associates with centrosomes (Bailly et al., 1992), a feature that we utilized to screen for cells in G2 phase. BubR1+/+/p53−/− MEFs in G2 typically had high cytoplasmic levels of cyclin B-GFP (Fig. 6A, and Movie S4). At mitosis onset, cyclin B-GFP entered the nucleus and then diffused into the mitotic cytosol after NEBD. Following proper chromosome alignment in metaphase, cyclin B-GFP was promptly degraded, allowing anaphase onset. BubR1−/−/p53−/− MEFs, on the other hand, failed to accumulate high levels of cyclin B-GFP in G2 phase (Fig. 6A, and Movie S5). This was due to proteosomal degradation rather than impaired synthesis, as cyclin B-GFP increased to normal levels after MG132 treatment (Fig. 6B, and Movie S6-S7), suggesting that, in G2 phase, cyclin B-GFP is marked for destruction by APC/C when BubR1 is lacking. Consistent with this, expression of non-degradable cyclin B-GFP containing a R42A substitution in the destruction box of cyclin B (Clute and Pines, 1999) was high in both BubR1+/+/p53−/− and BubR1−/−/p53−/− G2 MEFs (Fig. 6C). Cyclin BR42A-GFP levels remained high as these cells entered mitosis and arrested in metaphase. Furthermore, F-BubR1(1-363), but not F-BubR1(1-363)KEN19+298AAA, restored normal G2 cyclin B-cerulean levels in BubR1−/−/p53−/− MEFs (Fig. 6D, and Movie S8), confirming that Cdc20 binding by the N terminus of BubR1 prevents APC/C-mediated destruction of this cyclin. Securin and cyclin A were not prematurely degraded in G2 or M phase in the absence of BubR1 (Fig. S9, and Movie S9-S10), suggesting that BubR1 is a rather specific pseudosubstrate inhibitor for cyclin B, at least in MEFs. Earlier biochemical studies showing that securin is not prematurely degraded in BubR1 hypomorphic cells support this notion (Jeganathan et al., 2005). Live-cell imaging revealed that the median time interval from G1/S to mitosis was substantially longer in BubR1−/−/p53−/− MEFs than in BubR1+/+/p53−/− MEFs (12.3 h versus 9.5 h), indicating that cyclin B hypomorphism may delay progression to mitosis (Fig. S10).
Figure 6. BubR1 Stabilizes Cyclin B in Interphase by Inhibiting APC/CCdc20.

(A) BubR1−/−/p53−/− MEFs have low cyclin B-GFP levels in G2 and mitosis. Cyclin B-GFP fluorescence of MEF cells of the indicated genotypes was measured and the average fluorescence intensity in G2, prophase (P), prometaphase (PM), metaphase (M) and anaphase (A) plotted (n > 10 cells per genotype). Error bars represent the SD. *p < 0.0001 versus BubR1+/+/p53−/− MEFs at the same cell cycle stage (Mann-Whitney test). Bar = 10 μm.
(B) MG132 treatment of BubR1−/−/p53−/− MEFs restores normal cyclin B-GFP expression during G2 and mitosis. Error bars represent the SD. *p < 0.0001 versus BubR1+/+/p53−/− MEFs at the same cell cycle stage (Mann-Whitney test). Bar = 10 μm.
(C) BubR1−/−/p53−/− MEFs express normal levels of non-degradable cyclin BR42A-GFP G2 and mitosis. Error bars represent the SD.
(D) Expression of F-BubR1(1-363) in BubR1−/−/p53−/− MEFs restores normal cyclin B-cerulean levels in G2 and mitosis, whereas F-BubR1(1-363) lacking its KEN boxes does not. Error bars represent the SD. *p < 0.0001 versus BubR1+/+/p53−/− MEFs at the same cell cycle stage (Mann-Whitney test). Bar = 10 μm.
Endogenous BubR1 Interacts with Cdc20 in Interphase
The above experiments suggested that the BubR1 N terminus stabilizes cyclin B in G2 by acting as a pseudosubstrate for APC/CCdc20. This model predicts that BubR1 forms a complex with Cdc20 not only in mitosis, but also in interphase. To test this, we prepared extracts from BubR1+/+ MEFs in G2 or prometaphase for coimmunoprecipitation experiments. To ensure that G2 extracts were devoid of mitotic cells, any such cells were removed from the cultures by shake-off prior to harvest. Analysis of Hoechst stained cells revealed that the G2 and prometaphase samples contained 0% and 95% mitotic cells, respectively (Fig. 7A). G2 and prometaphase extracts were prepared from equal amounts of cells (Fig. 7B). As shown in Fig. 7C, Cdc20 precipitated with BubR1 from both MEF extracts. Bub3 and Mad2 also precipitated with BubR1 from both G2 and mitotic extracts, as did the APC/C component APC6 (Fig. 7C). More Cdc20, Bub3 and Mad2 precipitated from mitotic extracts, which may be due to the fact that more BubR1 precipitated from these extracts then from G2 extracts. Furthermore, F-BubR1(1-363) precipitated Cdc20, Mad2 and APC6, not only from mitotic MEF extracts (Fig. 2E), but also from G2 extracts containing 0% mitotic cells (Fig. 7D). Together, these results indicate that BubR1 binds to Cdc20 and other MCC components not only in mitosis when the mitotic checkpoint is active, but also in interphase. We found that ~80-90% reduction of Mad2 levels in BubR1+/+/p53−/− MEFs by shRNAmir (Fig. 7E and Fig. S11) had no impact on cyclin B-GFP accumulation in G2 phase (Fig. 7F), indicating that BubR1 is the primary inhibitor of APC/CCdc20-mediated cyclin B in interphase.
Figure 7. BubR1 Interacts with APC/CCdc20 in Interphase Cells.

(A) Percentage of mitotic cells present in G2 and mitotic BubR1+/+/p53−/− MEF extracts used in coimmunoprecipitation experiments shown in (C).
(B) Western blot analysis of G2 and mitotic BubR1+/+/p53−/− MEF extracts for actin. 5 μl of extracts used in (C) was loaded in each lane.
(C) Cdc20 binds to BubR1 in both interphase and mitosis. G2 and mitotic MEF extracts prepared from equal amounts of cells were subjected to immunoprecipitation with anti-BubR1 antibody and probed with the indicated antibodies.
(D) As (C) but with G2 extracts from BubR1−/−/p53−/− MEFs expressing BubR1(1-363).
(E) Western blot analysis showing that BubR1+/+/p53−/− MEFs transduced with pTRIPZ lentivirus expressing mMad2-shRNAmir clone V2MM_6980 contain ~10-20% of normal Mad2 levels after 3 days of culture in medium with 1 μg/ml DOX.
(F) Mad2 depletion in BubR1+/+/p53−/− MEFs has no impact on cyclin B-GFP levels in G2 and mitosis. DOX was added 72 h prior to analysis of cyclin B-GFP stability. Error bars represent the SD. Bar = 10 μm.
(G) Model for BubR1 function in G2 phase and mitosis. BubR1 binds to Cdc20 or Cdc20 already bound to APC/C (probably in combination with Mad2 and Bub3) in a KEN-box-dependent manner in G2, thereby blocking access of cyclin B to the APC/C, allowing this cyclin to accumulate and perform its mitotic functions. In mitosis, mitotic checkpoint proteins accumulating at unattached kinetochores catalyze binding of Mad2 and BubR1-Bub3 to Cdc20 that might emerge due to degradation of APC/CCdc20 inhibitors, such as Emi1 (Miller et al., 2006), and/or activation of mechanisms that displace Mad2 and BubR1 from Cdc20 in mitosis, such as UbcH10-mediated ubiquitination of Cdc20. BubR1 kinase activity is critical for APC/C inhibition under circumstances where spindle damage persist for prolonged periods of time.
Discussion
Inhibition of APC/CCdc20 in Interphase by BubR1
In mitosis, BubR1 is believed to bind and inhibit APC/CCdc20 in a mitotic checkpoint-dependent fashion (Musacchio and Salmon, 2007). Here we provide several lines of evidence to suggest that BubR1 functions as a pseudosubstrate inhibitor of APC/CCdc20 in interphase to allow for cyclin B accumulation prior to mitosis onset (Fig. 7G). First, cells lacking BubR1 have strongly reduced cyclin B levels in G2, a defect that can be restored by inhibition of the proteosome or by mutation of the APC/CCdc20 recognition site of cyclin B. Second, the N-terminal Cdc20 binding domain is necessary and sufficient to prevent cyclin B degradation by APC/C in G2 phase. Third, the BubR1 N-terminal Cdc20 binding domain harbors two KEN boxes, mutation of which perturbs Cdc20 binding to the BubR1 N terminus and causes premature cyclin B destruction in G2. These KEN boxes are conserved in fission and budding yeast Mad3p, where they are known to serve as Cdc20p binding sites that compete with APC/C substrates for Cdc20p binding (Burton and Solomon, 2007; King et al., 2007; Sczaniecka et al., 2008). Fourth, we demonstrate that endogenous BubR1 and ectopically expressed F-BubR1(1-363) interact with Cdc20 not only in mitosis, but also in G2 phase.
Our binding studies in MEFs suggest that Cdc20, BubR1, Bub3 and Mad2 already exist as a complex during interphase, which is consistent with an earlier study in HeLa cells (Sudakin et al., 2001). These findings imply that the MCC complex can assemble in a checkpoint independent manner. Further evidence for this notion is provided in the accompanying paper, where Kulukian et al. demonstrate that functional MCC complexes can assemble in vitro from recombinant Mad2, BubR1, Bub3 and Cdc20 proteins in the absence of active kinetochores, although at much lower rates than in their presence. One possibility is that, unlike interphase, mitosis requires catalysis of MCC formation by unattached kinetochores to offset mechanisms such as Cdc20 ubiquitination by UbcH10 that drive cells into anaphase by displacing Mad2 and BubR1-Bub3 from Cdc20 (see Fig. 7G; Reddy et al., 2007; Stegmeier et al., 2007).
Although Bub3 forms a complex with BubR1 in interphase, our finding that BubR1 mutants that cannot interact with Bub3 have normal cyclin B stability and Cdc20 binding indicates that Bub3 is not essential for BubR1’s role in inhibiting APC/CCdc20 in interphase and mitosis. The same may hold true for Mad2 as we find that its depletion from MEFs by RNAi has no impact on cyclin B stability in interphase. A recent study showing that cyclin B levels are normal as fly cells lacking Mad2 enter mitosis (Buffin et al., 2007) supports this notion.
An earlier study demonstrated that Emi1 functions as a substrate inhibitor of APC/CCdh1 during interphase, allowing for cyclin A accumulation from G1/S through early mitosis (Miller et al., 2006). Thus, pseudosubstrate inhibition of APC/C activity seems to be a more general mechanism through which cyclins accumulate in interphase, with Emi1 and BubR1 inhibiting APC/CCdh1 and APC/CCdc20 activity, respectively. Cyclin B is rather selectively targeted for destruction by the APC/C when BubR1 is lacking, as cyclin A and securin are stable under these circumstances. This result is consistent with previous work on Rae1/Nup98 double haplo-insufficient MEFs showing that APC/CCdh1, rather than APC/CCdc20, targets securin for destruction in early mitosis (Jeganathan et al., 2005). It will be interesting to test in future experiments to what extent cyclin B instability drives the mitotic defects associated with BubR1 deficiency.
Critical BubR1 Function Is to Bind and Inhibit Cdc20
In contrast to BubR1, ScMad3p and SpMad3p are not essential for cell viability (Hardwick et al., 2000; Millband and Hardwick, 2002). Structurally, BubR1 differs quite extensively from the yeast Mad3p proteins (Musacchio and Salmon, 2007). The Mad3p proteins are much smaller than BubR1, lacking a BubR1 kinase domain and C-terminal Cdc20 binding domain. ScMad3p contains a GLEBS-like motif required for Bub3 binding, but SpMad3p lacks this motif. One hypothesis for why BubR1 is essential and Mad3p proteins are not is that by gaining additional domains, BubR1 acquired additional functions, some of which might be required for viability. In this light, it is surprising that the only domain that is completely conserved among Mad3p and BubR1 proteins, the N-terminal Cdc20 binding domain, turns out to be the essential BubR1 functional domain. One plausible explanation for this apparent paradox might be that the BubR1 N terminus is implicated in a wider variety of mitotic processes than ScMad3p and SpMad3p. Indeed, the BubR1 N terminus is critical for mitotic checkpoint function, mitotic timing and proper alignment of chromosomes at the metaphase plate, while ScMad3p and SpMad3p have only been implicated in the mitotic checkpoint. Interestingly, our live-cell imaging experiments revealed a strong link between severe chromosome misalignment and cell death in BubR1-null cells, indicating that the function of BubR1 in chromosome-spindle attachment is perhaps the most critical BubR1 function with respect to cell viability.
How does the N-terminal Cdc20 binding domain regulate microtubule-kinetochore attachment? As this domain functions to inhibit APC/CCdc20 away from kinetochores, it is reasonable to assume that it does so indirectly by preventing ubiquitination of specific APC/C substrates that might control this process. This seems inconsistent with the observation that attachment defects persist when cells lacking BubR1 are treated with MG132. However, proteosomal inhibition does not exclude a scenario in which polyubiquitination per se is sufficient to alter substrate activity.
Mad2 and BubR1 both regulate the timing of mitosis in a kinetochore-independent fashion (Meraldi et al., 2004). It has been proposed that this timing function requires Mad2 and BubR1 binding to Cdc20 at the onset of mitosis when checkpoint proteins have yet to assemble at kinetochores and the checkpoint is unable to protect APC/C substrates from degradation by APC/C activity (Meraldi et al., 2004; Musacchio and Salmon, 2007). That Mad2 and BubR1 bind to Cdc20 in a kinetochore-independent manner and that the N-terminal Cdc20 binding domain can restore proper mitotic timing when endogenous BubR1 is lacking, as reported here, is in support of this model.
Role of BubR1 Kinase Activity at Mitotic Kinetochores
The discovery that kinetochore-bound Mad2 converts cytosolic Mad2 to a conformer that is able to interact with Cdc20 led to speculation that kinetochore-bound BubR1 might likewise prime soluble BubR1 for binding to Cdc20 or Cdc20-Mad2 (Kops et al., 2005; Mao et al., 2003). However, our finding that BubR1 mutants that fail to localize to kinetochores can bind to Cdc20 and efficiently inhibit APC/C activity, strongly suggests that cytoplasmic BubR1 is functionally independent of kinetochore-associated BubR1.
What then is the role of BubR1 at kinetochores? Although cells expressing mutant BubR1 proteins that cannot bind to kinetochores are capable of activating the mitotic checkpoint in the presence of nocodazole, the time interval of their arrest is considerably shortened. This suggests that kinetochore-bound BubR1 plays a role in sustaining mitotic checkpoint activity under conditions where chromosome-spindle attachment problems persist for extended periods of time (Fig. 7G). Importantly, cells expressing kinase-dead BubR1 showed the exact same phenotype, implying that prolonged checkpoint signaling requires BubR1 kinase activity at kinetochores. The observation that combined loss of both kinetochore binding ability and kinase activity compromises checkpoint maintenance to a similar degree as the corresponding single mutations, further supports this notion. It will be important in future experiments to identify the substrate(s) through which kinetochore-bound BubR1 acts to maintain the mitotic checkpoint.
Although BubR1 mutants lacking kinase activity or Bub3 binding ability (or both), corrected nearly all major chromosome alignment defects of BubR1-null cells, none of these mutants was capable of reducing the minor alignment defects to the same extent as full-length BubR1, indicating that BubR1 kinase activity at kinetochores contributes to stable kinetochore-microtubule attachment during metaphase.
Experimental Procedures
Generation and Culture of MEFs
BubR1H/H MEFs and BubR1H/H/p53−/− MEFs were generated and cultured as previously described (Baker et al., 2004). For studies reported here, at least three independently generated MEF lines per genotype were used, unless noted otherwise. G2 phase MEFs were prepared by arresting them at the G1/S border (using a double thymidine block) and then releasing them in DMEM/10% FCS for 8 h. The “shake-off” method was used to remove any mitotic cells from culture flasks prior to cell harvest. Mitotic MEFs were prepared by culturing cells for 5 h in medium containing nocodazole (100 ng/ml; Sigma-Aldrich) and harvesting cells by shake-off.
Expression plasmids
A full-length cDNA clone of mouse BubR1 was used as a template for PCR to create mutant BubR1 cDNAs (Davenport et al., 2006; Harris et al., 2005). Each mutant was provided with an amino-terminal FLAG epitope tag and cloned into the MluI site of the retroviral expression plasmid pMSCV-IRES-GFP. pMC-Cre was used as a template for PCR to create pMSCV-Cre/Puro. Procedures for viral transduction and Mad2 silencing by TRIPZ lentiviral inducible shRNAmir (Open Biosystems) are described in detail in the Supplemental Experimental Procedures.
Western Blotting, Coimmunoprecipitation and Indirect Immunofluorescence
Western blot analyses, coimmunoprecipitations and indirect immunofluorescence were performed as previously described (Kasper et al., 1999). For α-tubulin stainings, cells were incubated in PHEM buffer (25 mM HEPES, 10 mM EGTA, 60 mM PIPES, 2 mM MgCl2, pH6.9) and fixed in ice-cold methanol for 10 min. A Zeiss LSM 510 laser scanning microscope with a c-Apochromat 100x oil immersion objective was used for image acquisition. For quantification of cyclin A and B levels, at least 10 cells per mitotic stage were analyzed per MEF line. The mean fluorescence intensity was determined after background subtraction of images transformed to 8 bits grayscale, using NIH Image J software. A list of antibodies used is provided in Supplemental Experimental Procedures.
Live Cell Imaging
Nocodazole-challenge assays were performed as follows. MEFs were first transduced with a retrovirus encoding a monomeric red fluorescent protein (mRFP)-tagged H2B to allow visualization of chromosomes by fluorescence microscopy. Cells were then seeded onto 35 mm glass-bottomed culture dishes (MatTek Corporation) and cultured in DMEM/10% FBS. Approximately 24 h later, experiments were performed using a Zeiss Axio Observer Z1 system with: CO2 Module S, TempModule S, Heating Unit XL S, Pln Apo 63X/1.4 oil DICIII objective, AxioCam MRm camera and AxioVision 4.6 software. The imaging medium was DMEM/10% FBS. The temperature of the imaging medium was kept at 37°C. The exposure time in nocodazole-challenge experiments were identical among experiments. Nocodazole was added to a final concentration of 100 ng/ml. Cells undergoing nuclear envelope breakdown (NEBD) were marked and monitored at 15 min intervals to determine when they decondensed their chromosomes. The duration of arrest in mitosis, which is defined as the interval between NEBD (onset of mitosis) and chromatin decondensation (exit from mitosis without cytokinesis), was then calculated and plotted. The time at which 50% of the cells had exited mitosis was used for comparison. Mitotic checkpoint activities of BubR1−/−/p53−/− MEFs and BubR1−/−/p53−/− MEFs expressing BubR1 mutants were normalized to BubR1+/+/p53−/− MEFs for comparison. Taxol-challenge experiments were essentially the same except that, instead of nocodazole, taxol (Sigma Aldrich, T1912) was added at a concentration of 1 μM.
In mitotic timing experiments, the time interval between NEBD and anaphase onset was measured as H2B-mRFP positive cells progressed through an unchallenged mitosis. Interframe intervals were 3 min. To determine the time interval between G1/S and mitosis onset, H2B-mRFP-positive cells were seeded onto 6-well plates and infected with pMSCV-Cre retrovirus. Cells were then synchronized at the G1/S border by adding 1 μg/ml aphidicolin (Sigma-Aldrich, A0781) to the culture medium. After 15 h, cells were washed 3 times with PBS, released in fresh pre-warmed culture medium and monitored at 15 min intervals for mitosis entry (chromosome condensation). Cumulative frequency plots for G1/S-to-mitosis times were generated for each cell line. Median values were calculated and used for comparison.
To measure chromosome alignment, MEFs were treated with 10 μM MG132 (Sigma Aldrich, C2211). After 30 min, 20-30 cells entering prophase were marked and, 30 min later, analyzed for chromosome alignment defects. Cells exhibiting 1 to 3 misaligned chromosomes were classified as cells with minor misalignment defects, whereas those with 4 or more misaligned chromosomes were classified as cells with major misalignment defects.
Analyses of fluorescent protein-tagged cyclin B and securin levels were as follows: H2B-mRFP expressing MEFs were infected with pMSCV-Cre/Puro for 48 h and Cre expressing MEFs were selected in culture medium containing 3 μg/ml puromycin for 36 h. Cells were harvested and nucleofected with 5 μg pCMX-Cyclin B1-GFP, 3 μg pCMX-Cyclin B1R42A-GFP, 5 μg pCerulean-N1-Cyclin B1 or 2 μg pEYFP-N1-Securin plasmid DNA (kindly provided by Dr. Jonathon Pines, Gurdon Institute, Cambridge, United Kingdom) using an Amaxa Nucleofector II. Cyclin B-cerulean was used instead of cyclin B-GFP in instances where MEFs were already GFP positive, such as BubR1−/−/p53−/− MEFs expressing BubR1(1-363) or BubR1(1-363)KEN19+298AAA.Two ×106 MEFs were used per nucleofection. Nucleofections were done in MEF2 buffer (Amaxa, VPD-1005). MEFs were immediately seeded into 35-mm dishes and analyzed 4 h later. G2 phase cells were marked and images acquired every 6 min. In specified experiments, we inhibited the proteosome by adding 10 μM MG132 to the imaging medium. Exposure times were identical among experiments for each fluorochrome/filters set. For quantification of fluorescence levels, at least 10 cells were analyzed per MEF line. The mean fluorescence intensity at each cell stage was determined, after background subtraction of images transformed to 8 bits grayscale, using NIH Image J software (http://rsb.info.nih.gov/ij/). Mean fluorescence intensities were expressed in arbitrary units. For each of the aforementioned experiments, at least three independent MEF lines were used per genotype unless otherwise noted. GraphPad Prism software was used for statistical analyses. To study the impact of Mad2 depletion on cyclin B-GFP stability, BubR1+/+/p53−/− MEFs containing mMad2-shRNAmir clone V2MM_6980 were grown in the presence or absence of 1 μg/ml doxycycline for 72 h prior to nucleofection with pCMX-Cyclin B1-GFP.
Supplementary Material
Acknowledgments
We thank members of the van Deursen lab for helpful discussions, Rick Bram, Meelad Dawlaty, Robin Ricke, Limin Liu, Darren Baker and Paul Galardy for critically reading the manuscript, Dr. Jonathon Pines for plasmids and Dr. Don Cleveland for antibodies. This work was supported by NIH grant CA96985 and the Ellison Medical Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailly E, Pines J, Hunter T, Bornens M. Cytoplasmic accumulation of cyclin B1 in human cells: association with a detergent-resistant compartment and with the centrosome. J Cell Sci. 1992;101(Pt 3):529–545. doi: 10.1242/jcs.101.3.529. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Chen J, van Deursen JM. The mitotic checkpoint in cancer and aging: what have mice taught us? Curr Opin Cell Biol. 2005;17:583–589. doi: 10.1016/j.ceb.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Buffin E, Emre D, Karess RE. Flies without a spindle checkpoint. Nat Cell Biol. 2007;9:565–572. doi: 10.1038/ncb1570. [DOI] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes & development. 2007;21:655–667. doi: 10.1101/gad.1511107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- Davenport J, Harris LD, Goorha R. Spindle checkpoint function requires Mad2-dependent Cdc20 binding to the Mad3 homology domain of BubR1. Exp Cell Res. 2006;312:1831–1842. doi: 10.1016/j.yexcr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G. Checkpoint Protein BubR1 Acts Synergistically with Mad2 to Inhibit Anaphase-promoting Complex. Mol Biol Cell. 2002;13:755–766. doi: 10.1091/mbc.01-09-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Den Elzen N, Vodermaier HC, Waizenegger IC, Peters JM, Pines J. Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J Cell Biol. 2002;157:1125–1137. doi: 10.1083/jcb.200111001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Johnston RC, Smith DL, Murray AW. MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol. 2000;148:871–882. doi: 10.1083/jcb.148.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris L, Davenport J, Neale G, Goorha R. The mitotic checkpoint gene BubR1 has two distinct functions in mitosis. Exp Cell Res. 2005;308:85–100. doi: 10.1016/j.yexcr.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007;179:255–267. doi: 10.1083/jcb.200706015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 2005;438:1036–1039. doi: 10.1038/nature04221. [DOI] [PubMed] [Google Scholar]
- Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19:764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EM, van der Sar SJ, Hardwick KG. Mad3 KEN boxes mediate both Cdc20 and Mad3 turnover, and are critical for the spindle checkpoint. PLoS ONE. 2007;2:e342. doi: 10.1371/journal.pone.0000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol. 2005;7:93–98. doi: 10.1038/ncb1208. [DOI] [PubMed] [Google Scholar]
- Mao Y, Abrieu A, Cleveland DW. Activating and Silencing the Mitotic Checkpoint through CENP-E-Dependent Activation/Inactivation of BubR1. Cell. 2003;114:87–98. doi: 10.1016/s0092-8674(03)00475-6. [DOI] [PubMed] [Google Scholar]
- Mao Y, Desai A, Cleveland DW. Microtubule capture by CENP-E silences BubR1-dependent mitotic checkpoint signaling. J Cell Biol. 2005;170:873–880. doi: 10.1083/jcb.200505040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Draviam VM, Sorger PK. Timing and checkpoints in the regulation of mitotic progression. Dev Cell. 2004;7:45–60. doi: 10.1016/j.devcel.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Millband DN, Hardwick KG. Fission yeast Mad3p is required for Mad2p to inhibit the anaphase-promoting complex and localizes to kinetochores in a Bub1p-, Bub3p-, and Mph1p-dependent manner. Mol Cell Biol. 2002;22:2728–2742. doi: 10.1128/MCB.22.8.2728-2742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, Jackson PK. Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes & development. 2006;20:2410–2420. doi: 10.1101/gad.1454006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Reddy SK, Rape M, Margansky WA, Kirschner MW. Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature. 2007;446:921–925. doi: 10.1038/nature05734. [DOI] [PubMed] [Google Scholar]
- Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008 doi: 10.1016/j.tig.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczaniecka M, Feoktistova A, May KM, Chen JS, Blyth J, Gould KL, Hardwick KG. The spindle checkpoint functions of Mad3 and Mad2 depend on a Mad3 KEN box-mediated interaction with Cdc20-anaphase-promoting complex (APC/C) J Biol Chem. 2008;283:23039–23047. doi: 10.1074/jbc.M803594200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, McDonald ER, 3rd, Li MZ, Hannon GJ, Sorger PK, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature. 2007;446:876–881. doi: 10.1038/nature05694. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–936. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, Yu H. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell. 2001;1:227–237. doi: 10.1016/s1534-5807(01)00019-3. [DOI] [PubMed] [Google Scholar]
- Wang X, Babu JR, Harden JM, Jablonski SA, Gazi MH, Lingle WL, de Groen PC, Yen TJ, van Deursen JM. The mitotic checkpoint protein hBUB3 and the mRNA export factor hRAE1 interact with GLE2p-binding sequence (GLEBS)-containing proteins. J Biol Chem. 2001;276:26559–26567. doi: 10.1074/jbc.M101083200. [DOI] [PubMed] [Google Scholar]
- Yu H. Structural activation of Mad2 in the mitotic spindle checkpoint: the two-state Mad2 model versus the Mad2 template model. J Cell Biol. 2006;173:153–157. doi: 10.1083/jcb.200601172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.