

Abstract
X-ray scattering and diffraction from non-crystalline systems have gained renewed interest in recent years, as focus shifts from the structural chemistry information gained by high-resolution studies to the context of structural physiology at larger length scales. Such techniques permit the study of isolated macromolecules as well as highly organized macromolecular assemblies as a whole under near-physiological conditions. Time-resolved approaches, made possible by advanced synchrotron instrumentation, add a critical dimension to many of these investigations. This article reviews experimental approaches in non-crystalline x-ray scattering and diffraction that may be used to illuminate important scientific questions such as protein/nucleic acid folding and structure-function relationships in large macromolecular assemblies.
Introduction
Here we review recent research trends and advances in experimental and computational techniques in structural studies of biological macromolecules in non-crystalline forms. We focus on macromolecular solution x-ray scattering (small angle x-ray scattering or SAXS) and fiber diffraction studies (non-crystalline diffraction or NCD) that are playing increasingly important roles in interpreting high-resolution structural information deduced by crystallography, NMR and electron microscopy in the context of molecular physiology. The advent of synchrotron radiation has contributed immensely to technological advances in this field in much the same way as in macromolecular crystallography. Due to space limitations we omit studies of other important biological systems including lipid membranes and refer those interested to a recent review. [1]
Instrumental requirements and experimental considerations
Randomly oriented or partially ordered systems provide only weak and generally more diffuse X-ray scattering primarily at small scattering angles, requiring synchrotron-based instruments for these studies in contrast to strong single crystal x-ray diffraction where diffraction spots can be measured at high scattering angles up to atomic resolution. Undulator radiation at third generation storage rings such as the Advanced Photon Source (APS), European Synchrotron Radiation Facility (ESRF), Super Photon Ring-8 (SPring-8) and other newer sources provide near ideal sources for NCD studies because of their high flux and low beam divergence necessary for high time-resolution and superior accessibility to long characteristic lengths. There are about two dozen experimental facilities for NCD studies at synchrotrons worldwide some of which are devoted to structural biological studies (http://biosync.rcsb.org). Such studies require the highest level of overall instrumental stability from x-ray source to detector since generally weak signals can be extracted only after subtracting the contribution of background scattering, especially in solution scattering. Detectors with high photon sensitivity, electronic stability and low noise are essential. High-performance CCD detectors are currently in widespread use in these experiments while silicon pixel array detectors, converting x-ray photons directly to electrons, have recently emerged as the next generation detector. Micro-fluidic chips[2] as well as other sample handling devices have been developed to minimize sample consumption in solution studies. A number of methods have been developed to mitigate radiation damage, particularly acute at high flux third generation facilities, including the use of capillary flow cells in solution scattering and flash-cooling or sample oscillation in fiber diffraction.
X-ray scattering from macromolecules in solution
Small angle x-ray scattering samples contain many copies of a macromolecule in random orientations as a solute. The resulting circularly symmetric scattering profile is usually azimuthally averaged into a one-dimensional profile to improve data statistics. The contribution of solvent x-ray scattering is subtracted to obtain the x-ray scattering from the solute, often called as the form factor, reflecting the three dimensional shape of the macromolecule of interest, including proteins, nucleic acids and biological assemblies Synchrotron instruments allow solution scattering studies on a variety of macromolecules ranging from long peptides or oligo-nucleotides of a few thousand Da to multi-megadalton assemblies. Most of these instruments can obtain useful data from dilute solutions in the sub mg/ml range and above. Intermolecular interactions, which can complicate interpretation, may be minimized by adjusting solution conditions, e.g., using a moderately high ionic strength. The information content of the form factor and advanced computational methods used to obtain three-dimensional biological structures, at near nanometer resolutions in favorable cases, have recently been reviewed.[3] An increasing number of structural biologists adopt these methods to build 3D models of macromolecular complexes that are unsuitable for crystallographic studies.[4] Here, we emphasize novel experimental approaches that complement this class of studies.
Macromolecular assemblies
biological assemblies of various sizes are formed in response to external factors. The forward scattered intensity provides a direct measure of molecular weight, thus state of assembly. When multiple molecular species exist, the overall scattering signal is weighed by the square of molecular weight. The elimination of aggregated species is thus crucial in obtaining the form factor of a single molecular species. Complementary techniques such as light scattering and gel filtration are useful to identify and purify distinct molecular species. Sokolova et al. combined analytical ultracentrifugation and negative stain electron microscopy to identify solution conditions in which intermediate filament vimentin exists as a distinctively monodispersed assembly species.[5] They built 3D structure models of vimentin tetramer, octamer, and unit-length filaments (32-mer) by rigid body modeling of the dimeric crystal structure. A similar approach was used to study discrete 25mer and 100mer assemblies of human prion protein caused by metal-induced oxidation.[6] Inline size exclusion chromatography is another approach to obtain accurate form factor of a single molecular species.[7] A new method based on singular value decomposition and an assumption of simple association pathways has been developed to obtain both component scattering curves and the equilibrium constants for individual oligomerization steps.[8]
Flexible structures
A number of biological systems are in a dynamical conformational equilibrium. Since all molecules within the volume irradiated by an x-ray beam contribute to x-ray scattering, solution x-ray scattering is both ensemble- and time-averaged. Unlike proteins in a crystal, flexible and mobile portions of a biological structure also contributes to the observed signal. This enables the study of ensembles of flexible protein structures that may take multiple conformations under given conditions. Bernado et al. have developed the ensemble optimization method, which fits the experimental scattering curve by an ensemble of 50 to 100 different conformers made up of rigidly folded domains connected by a flexible self-avoiding linker.[9] This approach revealed the dynamical equilibrium between the open active state (15% population) and the closed active conformation (85%) of Src kinase, only the latter of which had been previously identified as active conformation by crystallography.[10] Protein/RNA folding studies present another important example of the effective use of solution scattering [11] on flexible structures which are not suitable for crystallographic studies. Solution x-ray scattering complements local structural probes such as circular dichroism and NMR residual dipole coupling.[12]
Time-resolved studies
Kinetic studies of conformational changes, due to ligand binding or thermodynamic perturbation, at the tertiary and quaternary structural levels are essential for interpreting atomic resolution structures in the physiological context. Such studies require the use of a high-flux synchrotron beam, as well as a fast-readout detector and a method to trigger a conformational change. A number of different experimental procedures are available depending on triggering method and the rate of conformational change. Slow molecular processes of the order of minutes to hours can be studied by manual mixing followed by a series of successive scattering measurements. Large-scale conformational changes in virus and phage particles, which could not be contained in crystallographic lattice structures, have been studied this way. (Figure 1) HK97 bacteriophage maturation kinetics have been found to be remarkably cooperative from the initial to late stage maturation processes despite the involvement of 420 unique protein subunits in the icosahedral shell. [13] Another recent example highlights the direct observation of a 24h period oscillation between two different Kai protein binary complexes in circadian clocks of a cyanobacterium.[14] Remote-controlled rapid mixing is almost universally used for time-resolved solution x-ray scattering studies at faster time scales.[15] Stopped-flow mixers, requiring only as little as a few milligrams of macromolecule, have been successfully used for studying kinetics whose time constants are in the range of milliseconds to minutes and have been particularly valuable in the studies of the folding of proteins and nucleic acids.[11] In favorable cases, only a single mixing event is required to observe structural kinetics at a few ms time resolution.[16] Micro-machined continuous-flow rapid mixers have been recently adopted for solution x-ray scattering to reach sub-millisecond time resolution primarily in fast folding studies. [17,18] Recently, Arai et al. monitored a rapid compaction of E. coli dihydrofolate reductase within first 0.3 ms after refolding by a urea dilution jump.[19] The use of high brilliance third generation sources is mandatory for such experiments.
Figure 1.
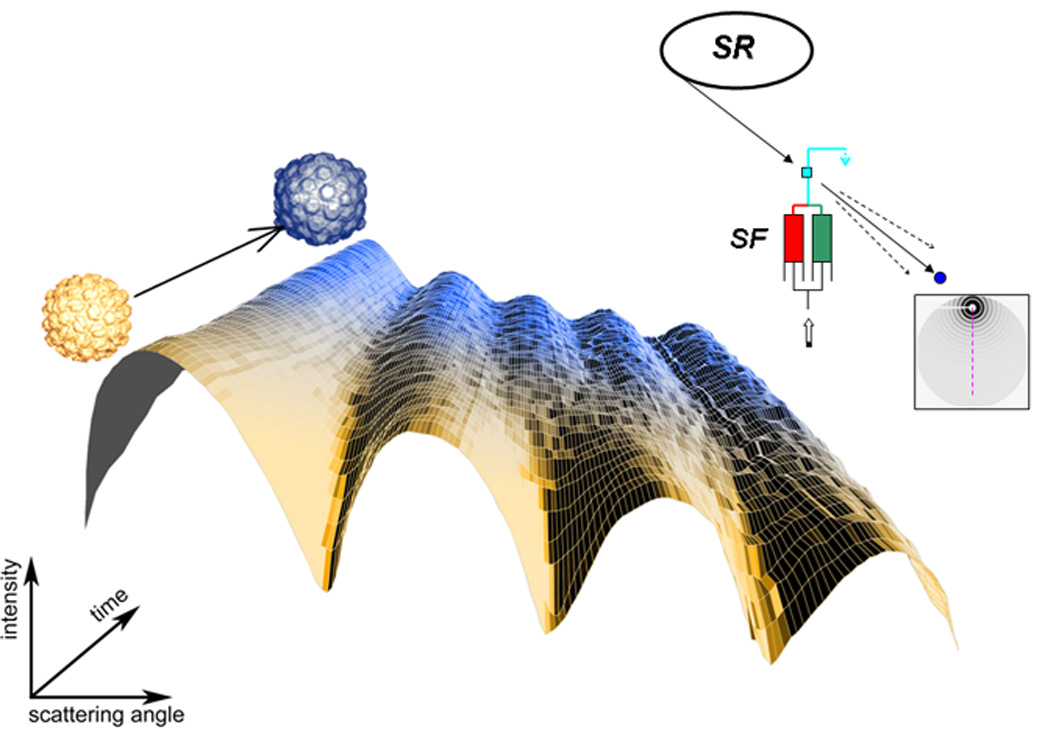
HK97 bacterophage maturation kinetics studied by time-resolved solution x-ray scattering. A stopped-flow mixer (SF) initiates the maturation by a pH-jump, followed by a series of successive x-ray scattering pattern measurements. A synchrotron storage ring (SR) provides an intense x-ray beam. The direct beam is intercepted by a beam stop (blue disk) and surrounding circularly symmetric x-ray scattering is recorded as a function of time, and azimuthally averaged into one-dimensional patter as shown in the time course illustration. The peak position shifts toward small angle reflect the larger particle size in the final stage of the maturation. Courtesy of Dr. Kelly Lee, the Scripps Research Institute.
High angle scattering studies
Protein/nucleic acid studies in solution at high scattering angles, corresponding to Bragg spacings of 1 nm or smaller, have been thought to bring structural information in this resolution range, but have been hampered by the extremely weak signals. Third generation synchrotron sources have made this class of studies feasible in recent years. The use of high angle data shows potential for drug screening [20] and most recently as a means of monitoring altered protein quaternary structure due to molecular crowding.[21] High angle data have been shown to be very useful in evaluating various nucleic acid sugar and phosphate backbone structures deduced by NMR and crystallography, in particular, as regards the base pair stacking of DNA. This approach allowed selection of the most realistic solution structure of DNA oligomers among conflicting crystallographic and NMR structures.[22] A similar approach was taken to experimentally assess the accuracy of DNA oligmer structures obtained by molecular dynamics simulation.[23]
Intermolecular interactions
The intermolecular spatial correlation or structure factor can also be observed in solution x-ray scattering even at relatively low concentrations. For instance, a solution of mildly charged molecules exhibits a broad distribution of weakly correlated intermolecular distances in the range of hundreds of Ångstroms. The structure factor offers a unique opportunity to study intermolecular interactions, which are central to many biological functions. The physiological importance of intermolecular interactions is exemplified by a series of studies performed on the eye lens protein crystalline.[24] A direct measurement of like-charge attractive interaction of oligonucleotides, essential in nucleic acid packaging in virus particles, has recently been performed.[25] Sato et al. demonstrated that the heme binding can induce strong long-range attractive potential of human serum albumin, suggesting the importance of the attractive potential in molecular recognition in protein-protein interaction.[26]
Interplay with other techniques
Neutron solution scattering has the advantage of being able to adjust solvent contrast to visualize a selected component of a heterogeneous system made up of different electron densities such as protein-nucleic acid complexes and protein-lipid complexes.[27] Simultaneous fitting of x-ray and neutron scattering data results in more reliable structural models.[28] The long range structural information contained in x-ray scattering has also been used to aid NMR structural solutions of large proteins.[29] The use of a low-resolution macromolecular envelope, derived from solution x-ray scattering, for determining initial diffraction phases in crystallography has been in development.[30] The combined use of molecular dynamics simulation has been beneficial to an increasing number of x-ray scattering studies. Human complement factor fragments have been analyzed on the basis of x-ray solution scattering and thousands of simulated structures by molecular dynamics.[31] The best probable models can be selected among the simulated structures that not only fit x-ray scattering data well, but also fulfill other constrains such as inter-domain interactions. A new computational method to utilize spatial restraints to model protein structures using solution scattering data has been developed as a part of the Integrative Modeling Platform.[32] Even though the application of these computational methods is presently limited to relatively small biological systems, we expect them to be more widely applicable as computation costs continue to decline.
X-ray fiber diffraction
Many biopolymers are composed of long helical structures. These macromolecules are typically very difficult to crystallize but have a natural tendency to line up parallel to each other to form fibers. The key difference between fibers and crystals is that in fibers, the fundamental structural motifs are randomly oriented about the fiber axis so that the resulting diffraction pattern is cylindrically averaged resulting in loss of information. Despite this limitation, fiber diffraction has been a powerful technique for the study of a wide variety of biopolymers, ranging from simple polypeptides, polynucleotides, and polysaccharides to filamentous viruses, cytoskeletal filaments, and larger assemblies such as collagen microfibrils and muscle fibers. The availability of specialized beamlines on third generation synchrotron radiation sources in combination with modern high sensitivity, high spatial resolution CCD-based detectors, have been a boon to fiber diffraction studies since these technologies allow resolution of closely spaced diffraction peaks above the high backgrounds typical of these systems to yield superior, more easily interpreted, diffraction patterns. The ability to focus beams from these sources to micron-scale spots provide new scientific opportunities that are only beginning to be realized.
Wide-angle diffraction of oriented fibers
As in conventional macromolecular crystallography, the ability to produce well-ordered systems is crucial to success. Combinations of centrifugation, controlled drying, and exposure to very high magnetic fields have been shown to produce well-oriented specimens. Even with such approaches, the sizes of well-ordered domains within these samples may be quite small (≪ 100 microns) requiring small synchrotron x-ray beams to interrogate them. All these approaches were crucial to success in the determination of the first useful structural parameters from a filamentous virus of the potyvirus family, a previously completely intractable system for structural analysis.[33] While diffraction patterns from large macromolecular systems seldom diffract past ca. 3Å, many simpler systems diffract to atomic resolution allowing application of refinement techniques similar to those used in conventional macromolecular crystallography. For instance, Nishiyama et al. used a combination of neutron and synchrotron fiber diffraction to determine the crystal and molecular structure, together with the hydrogen-bonding system of cellulose I(α) to atomic resolution (~1Å ).[34]
Small-angle x-ray diffraction of muscle
X-ray diffraction is the only technique that can provide molecular level information in muscle tissue under hydrated, physiological conditions at the physiologically relevant millisecond time scale.[35] Such experiments demand the high intensity and superior collimation of synchrotron radiation and have historically been a driving force in the development of synchrotron-based technologies. The availability of small angle instruments on large third generation synchrotrons have enabled new classes of experiments. One technique exploits the interference between the diffraction from the arrays of crossbridges in the two halves of each thick filament to measure the motions of myosin heads to ca. 1Å resolution.[35] This approach was used to provide a new explanation for the so-called “force velocity curve” of muscle whereby numbers of myosin crossbridges generating similar step sizes and unitary forces are adjusted in the muscle to match the load.[36] The new sources have also permitted other challenging studies including those of the indirect flight muscles in living Drosophila during tethered flight.[37] (Figure 2) This system is particularly promising for future studies because of its high degree of structural order and the ease of creating transgenic organisms. A major recent technical development for studies of cardiac muscle has been the development of in vivo measurements from beating hearts in living animals. These have allowed relating structural changes to questions of direct clinical relevance.[38] One of the most interesting uses of microbeam diffraction so far was an examination of the filament lattice ordering in cryogenically frozen myofibrils of insect flight muscle from a wide range of species giving insights into how insect flight muscle has evolved.[39]
Figure 2.

X-ray diffraction pattern from living Drosophila during tethered flight. The high intensity of the 19.3 nm first row line reflections (circles) indicates a high occupancy of actin binding sites by myosin heads poised for active contraction during the downstroke of the wingbeat.
In situ studies of connective tissue
Connective tissues are particularly complex biological materials, being composed of various combinations of collagen fibers, complex glycoproteins and mineralized tissues. A recent advance was the publication of a MIR structure for rat-tail collagen showing the packing of the collagen molecules within the 67 nm long unit cell providing essential structural information for revealing likely mechanisms of proteolysis.[40] For this work, techniques from macromolecular crystallography were successfully adapted to the study of natural fibers in situ. In most studies of connective tissue, however, the emphasis is on the hierarchical arrangement of molecules within the tissues, rather than on the structure of the molecules themselves, with the aim of explaining their physical properties. Diffraction techniques have been used for the study of bone[41], cartilage[42], and cornea[43]. All of these kinds of studies could benefit from using microbeams in order to interrogate the different ordered regions that make up the substructure of these complex materials.
Fiber diffraction and protein misfolding diseases
Amyloid fibers are formed when normally soluble proteins change conformation to form insoluble filaments that may aggregate and deposit in tissues, contributing to pathologies such as Alzheimer’s and Huntington’s disease among many others. Because of their complexity and insolubility, amyloid fibers have largely resisted characterization by protein crystallography and NMR, so fiber diffraction has been the method of choice for structural studies.[44] Although most amyloid proteins share a cross-β structure it is becoming apparent that there is no reason to assume that all amyloids have the same tertiary or quaternary structure. For instance fibril-forming peptides with a cross- α structure were recently reported.[45] Mammalian prions also cause disorders due to protein misfolding but unlike other amyloid systems, they are infectious. The central event appears to be a change from a predominantly α-helical, non-infectious form of the protein PrP to a predominantly β-sheet form that is then infectious. Illnesses caused by prions include Creutzfeldt-Jakob disease, bovine spongiform encephalopathy (“mad cow disease”), and scrapie. Structural analysis of prions using diffraction and other techniques has recently been reviewed.[46] An example of how microbeams could bring these kinds of studies into a new era is given by Yagi et al.[47] who used a 6 µm beam to interrogate different parts of the structure of individual insulin spherulites. In the future, similar techniques could be envisioned to obtain structural information from amyloid structures in situ using sectioned tissue samples.
Conclusions
Third generation sources have finally fulfilled the promise of synchrotron radiation for non-crystalline diffraction. Flux and beam quality, for most studies, are no longer limiting. New sources on the horizon such as the NSLS II (Brookhaven) and Petra III (Hamburg) and proposed energy-recovery linacs and ultimate storage rings such as PEP-X (Stanford) can provide extremely small source sizes and beam divergences, much more suitable for nano-beam studies than current 3rd generation sources. This will enable whole new classes of NCD studies of complex materials. It is also fair to say that the ability to obtain high quality data for SAXS and NCD has outstripped our ability to analyze it fully. The challenge for developing suitable analytical techniques has always been the diversity of the systems studied and the approaches needed. Solution studies have great potential, as yet not realized, for high-throughput structural characterization. The availability of fully coherent 4th generation sources such as XFEL (Hamburg), SPring-8 XFEL (Harima) and the LCLS (Stanford,) in the near future raises the possibility of collecting continuous diffraction patterns from single molecules and using various phase retrieval algorithms to recover their structure. One approach proposes to use laser alignment to orient a stream of macromolecules that passes through the x-ray beam as a means of mitigating radiation damage to allow reconstruction of secondary protein structures to ~7Å resolution with relatively short exposure times [48].
Acknowledgements
The authors acknowledge the National Institutes of Health, National Center for Research Resources, Biomedical Technology Grants P41 RR001209 and RR08630 for the support of the SSRL Structural Molecular Biology Program and the IIT Biophysics Collaborative Access Team at the APS, respectively. The SSRL SMB Program is additionally supported by the US Department of Energy, Office of Biological and Environmental Research. The SSRL and APS are supported by DOE, Basic Energy Sciences. The content is solely the responsibility of the authors and does not necessarily reflect the official views of NCRR or NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* of special interest
** of outstanding interest
- 1.Koch MH, Bras W. Synchrotron radiation studies of non-crystalline systems. Ann. Rep. Prog. Chem., Sect. C: Phys. Chem. 2008;104:35–80. [Google Scholar]
- 2.Toft KN, Vestergaard B, Nielsen SS, Snakenborg D, Jeppesen MG, Jacobsen JK, Arleth L, Kutter JP. High-throughput small angle X-ray scattering from proteins in solution using a microfluidic front-end. Anal Chem. 2008;80:3648–3654. doi: 10.1021/ac800011y.A 200 nl microfluidic chamber has been developed for high-throughput protein characterization using solution x-ray scattering. A fully automated stream of data collection to low-resolution 3D structure modeling is proposed based on machine learning techniques.
- 3.Petoukhov MV, Svergun DI. Analysis of X-ray and neutron scattering from biomacromolecular solutions. Curr Opin Struct Biol. 2007;17:562–571. doi: 10.1016/j.sbi.2007.06.009.This article reviews computational approaches developed by the authors’ group to obtain ab initio three dimensional models from solution x-ray or neutron scattering data as well as to construct multi-component structures based on known constituent structures. The programs from this group are highly user friendly and currently most widely used by structural biologists. Many of the programs can incorporate complementary information from other structural techniques.
- 4.Roll-Mecak A, Vale RD. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451:363–367. doi: 10.1038/nature06482.The hexameric structure model deduced from solution x-ray scattering data allowed the authors to interpret the monomeric crystal structure of the ATPase domain of the AAA ATPase spastin in the context of physiologically relevant hexamer assembly, a common oligomeric state in many AAA ATPases. A molecular mechanism is proposed for sepatin-mediated severing of microtubules, based on the 3D structure model and structure-guided mutagenesis.
- 5.Sokolova AV, Kreplak L, Wedig T, Mucke N, Svergun DI, Herrmann H, Aebi U, Strelkov SV. Monitoring intermediate filament assembly by small-angle x-ray scattering reveals the molecular architecture of assembly intermediates. Proc Natl Acad Sci U S A. 2006;103:16206–16211. doi: 10.1073/pnas.0603629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Redecke L, von Bergen M, Clos J, Konarev PV, Svergun DI, Fittschen UE, Broekaert JA, Bruns O, Georgieva D, Mandelkow E, et al. Structural characterization of beta-sheeted oligomers formed on the pathway of oxidative prion protein aggregation in vitro. J Struct Biol. 2007;157:308–320. doi: 10.1016/j.jsb.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Mathew E, Mirza A, Menhart N. Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. J Synchrotron Radiat. 2004;11:314–318. doi: 10.1107/S0909049504014086. [DOI] [PubMed] [Google Scholar]
- 8.Williamson TE, Craig BA, Kondrashkina E, Bailey-Kellogg C, Friedman AM. Analysis of self-associating proteins by singular value decomposition of solution scattering data. Biophys J. 2008 doi: 10.1529/biophysj.107.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n.It describes the ensemble optimization method, which uses genetic algorithm to optimize the fit of an ensemble of protein conformations produced by the Monte Carlo method to an experimental scattering curve. The resulting ensemble represents a set of multiple protein conformations that are likely to co-exist.
- 10.Bernado P, Perez Y, Svergun DI, Pons M. Structural characterization of the active and inactive states of Src kinase in solution by small-angle X-ray scattering. J Mol Biol. 2008;376:492–505. doi: 10.1016/j.jmb.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 11.Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu Rev Biophys Biomol Struct. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655.This article reviews the use of solution x-ray scattering in characterizing nucleic acid folding intermediates and unfolded proteins. It also discusses limitations in 3-D reconstruction techniques as well as challenges in dealing with protein-detergent complexes.
- 12.Ohnishi S, Kamikubo H, Onitsuka M, Kataoka M, Shortle D. Conformational preference of polyglycine in solution to elongated structure. J Am Chem Soc. 2006;128:16338–16344. doi: 10.1021/ja066008b. [DOI] [PubMed] [Google Scholar]
- 13.Lee KK, Gan L, Tsuruta H, Moyer C, Conway JF, Duda RL, Hendrix RW, Steven AC, Johnson JE. Global capsid expansion during virus maturation is driven by the local capture of dynamic surface loops. Structure. 2008 doi: 10.1016/j.str.2008.06.014. accepted for publication.The combination of time-resolved x-ray scattering and the kinetics of auto-catalytic inter-subunit crosslinking revealed the molecular mechanism of bacterophage HK97 maturation, based on the cryo-electron microscopy and crystallographic structures of the initial and final maturation states of HK97. The remarkably cooperative structural transition points to the crucial role of E-loop which transverse from an initial “up” position to a crosslink ready “down” position while maintaining the global icosahedral subunit symmetry.
- 14.Akiyama S, Nohara A, Ito K, Maeda Y. Assembly and Disassembly Dynamics of the Cyanobacterial Periodosome. Mol Cell. 2008 doi: 10.1016/j.molcel.2008.01.015.The key protein complexes in circadian clocks of a cyanobacterium have recently been studied in time-resolved fashion. Mixing of two or three Kai protein components exhibit oscillations between two binary complexes of different sizes in exact 24 hour periodicity. The observed phase shift between the assembly oscillation and protein phosphorylation is ascribed to the oscillation between a kinetically favored binary complex and a thermodynamically favored one. X-ray scattering profiles of the binary protein complexes have been numerically extracted from x-ray scattering of mixed populations and binary complex structure models are proposed, which are consistent with component crystal structures as well as biochemical data.
- 15.Panine P, Finet S, Weiss TM, Narayanan T. Probing fast kinetics in complex fluids by combined rapid mixing and small-angle X-ray scattering. Adv Colloid Interface Sci. 2006;127:9–18. doi: 10.1016/j.cis.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 16.West JM, Xia J, Tsuruta H, Guo W, E.M. OD, Kantrowitz ER. Time Evolution of the Quaternary Structure of Escherichia coli Aspartate Transcarbamoylase upon Reaction with the Natural Substrates and a Slow Tight Bindign Inhibitor. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.09.022. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bilsel O, Matthews CR. Molecular dimensions and their distributions in early folding intermediates. Curr Opin Struct Biol. 2006;16:86–93. doi: 10.1016/j.sbi.2006.01.007.This is a comprehensive review of the use of solution x-ray scattering, in particular time-resolved experiments, and other complementary physical methods in the studies on early protein folding intermediates.
- 18.Kwok LW, Shcherbakova I, Lamb JS, Park HY, Andresen K, Smith H, Brenowitz M, Pollack L. Concordant exploration of the kinetics of RNA folding from global and local perspectives. J Mol Biol. 2006;355:282–293. doi: 10.1016/j.jmb.2005.10.070.A continuous-flow and stopped-flow mixers were used to study the Mg2+-mediated folding of a Tetrahymena ribozyme in the time scale of a millisecond to hundreds of seconds. Two-stage compaction was observed. Time-resolved hydroxyl radical foot-printing analysis was used to examine the role of long-range tertiary contacts among different structural domains.
- 19.Arai M, Kondrashkina E, Kayatekin C, Matthews CR, Iwakura M, Bilsel O. Microsecond hydrophobic collapse in the folding of Escherichia coli dihydrofolate reductase, an alpha/beta-type protein. J Mol Biol. 2007;368:219–229. doi: 10.1016/j.jmb.2007.01.085. [DOI] [PubMed] [Google Scholar]
- 20.Rodi DJ, Mandava S, Gore DB, Makowski L, Fischetti RF. Detection of functional ligand-binding events using synchrotron x-ray scattering. J Biomol Screen. 2007;12:994–998. doi: 10.1177/1087057107306104. [DOI] [PubMed] [Google Scholar]
- 21.Makowski L, Rodi DJ, Mandava S, Minh DD, Gore DB, Fischetti RF. Molecular crowding inhibits intramolecular breathing motions in proteins. J Mol Biol. 2008;375:529–546. doi: 10.1016/j.jmb.2007.07.075.This article examines the spatial scale of protein conformational fluctuations using solution x-ray scattering data recorded up to the Bragg spacing of 2.5Å. Their computational modeling shows that rigid-body motions of secondary structures are consistent with the degree of fluctuations. The authors provide evidence for suppressed conformational fluctuations at high protein concentration, depending on tertiary or quaternary structures of the protein.
- 22.Zuo X, Tiede DM. Resolving conflicting crystallographic and NMR models for solution-state DNA with solution X-ray diffraction. J Am Chem Soc. 2005;127:16–17. doi: 10.1021/ja044533+. [DOI] [PubMed] [Google Scholar]
- 23.Zuo X, Cui G, Merz KM, Jr, Zhang L, Lewis FD, Tiede DM. X-ray diffraction "fingerprinting" of DNA structure in solution for quantitative evaluation of molecular dynamics simulation. Proc Natl Acad Sci U S A. 2006;103:3534–3539. doi: 10.1073/pnas.0600022103.DNA oligomer structures generated by molecular dynamics simulations were examined on the basis of experimental solution x-ray scattering profiles. Scattering maxima in the Bragg spacing range between 3 and 15Å are shown highly sensitive to base pair stacking as well as sugar-phosphate backbone structures. Discrepancies between experimental and theoretical scattering profiles suggest that the molecular dynamics force field is biased by short range interactions while x-ray scattering reflects global structure as well.
- 24.Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A. Ageing and vision: structure, stability and function of lens crystallins. Prog Biophys Mol Biol. 2004;86:407–485. doi: 10.1016/j.pbiomolbio.2003.11.012.This is a highly comprehensive review of properties and structures of crystallines. Chapter 4 is dedicated to the crystalline intermolecular interactions that lead to eye transparency, which was first demonstrated by an x-ray scattering study by Delaye and Tardieu in 1983.
- 25.Qiu X, Andresen K, Kwok LW, Lamb JS, Park HY, Pollack L. Inter-DNA attraction mediated by divalent counterions. Phys Rev Lett. 2007;99:038104. doi: 10.1103/PhysRevLett.99.038104.The second virial coefficient A2 was obtained by conducting a series of solution x-ray scattering measurements on oligomer DNA in the presence of various cations, which screen the net negative charge of DNA. It was demonstrated that non-specifically bound Mg2+ ions mediate attractive intermolecular forces as seen by negative A2 values in certain conditions in contrast to Na+ which does not invert the sign of A2.
- 26.Sato T, Komatsu T, Nakagawa A, Tsuchida E. Induced long-range attractive potentials of human serum albumin by ligand binding. Phys Rev Lett. 2007;98:208101. doi: 10.1103/PhysRevLett.98.208101. [DOI] [PubMed] [Google Scholar]
- 27.Grossmann JG, Callaghan AJ, Marcaida MJ, Luisi BF, Alcock FH, Tokatlidis K. Complementing structural information of modular proteins with small angle neutron scattering and contrast variation. Eur Biophys J. 2008 doi: 10.1007/s00249-008-0278-z. [DOI] [PubMed] [Google Scholar]
- 28.Petoukhov MV, Svergun DI. Joint use of small-angle X-ray and neutron scattering to study biological macromolecules in solution. Eur Biophys J. 2006;35:567–576. doi: 10.1007/s00249-006-0063-9. [DOI] [PubMed] [Google Scholar]
- 29.Grishaev A, Tugarinov V, Kay LE, Trewhella J, Bax A. Refined solution structure of the 82-kDa enzyme malate synthase G from joint NMR and synchrotron SAXS restraints. J Biomol NMR. 2008;40:95–106. doi: 10.1007/s10858-007-9211-5.The χ2SAXS value was included in the NMR refinement process as a pseudo energy term in CNS, following initial RDC and RCSA refinements. The resulting solution NMR structure of the 82 kDa enzyme malate synthase G demonstrated an improved accuracy by a smaller backbone rmsd value between the derived model and crystal structure: 3.3 vs. 4.5Å without the SAXS refinement.
- 30.Hao Q. Macromolecular envelope determination and envelope-based phasing. Acta Crystallogr D Biol Crystallogr. 2006;62:909–914. doi: 10.1107/S0907444906014089. [DOI] [PubMed] [Google Scholar]
- 31.Okemefuna AI, Gilbert HE, Griggs KM, Ormsby RJ, Gordon DL, Perkins SJ. The regulatory SCR-1/5 and cell surface-binding SCR-16/20 fragments of factor H reveal partially folded-back solution structures and different self-associative properties. J Mol Biol. 2008;375:80–101. doi: 10.1016/j.jmb.2007.09.026. [DOI] [PubMed] [Google Scholar]
- 32.Foerster F, Webb B, Krukenberg KA, Tsuruta H, Agard DA, Sali A. Integration of small angle X-ray scattering data into structural modeling of proteins and assemblies. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.07.074. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker L, Kendall A, Berger PH, Shiel PJ, Stubbs G. Wheat streak mosaic virus--structural parameters for a Potyvirus. Virology. 2005;340:64–69. doi: 10.1016/j.virol.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 34.Nishiyama Y, Sugiyama J, Chanzy H, Langan P. Crystal structure and hydrogen bonding system in cellulose I(alpha) from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc. 2003;125:14300–14306. doi: 10.1021/ja037055w. [DOI] [PubMed] [Google Scholar]
- 35.Reconditi M. Recent improvements in small angle x-ray diffraction for the study of muscle physiology. Reports on Progress in Physics. 2006;69:2709–2759. doi: 10.1088/0034-4885/69/10/R01.A review of recent applications of x-ray diffraction for understanding muscle physiology including a thorough explanation of the interferometric technique for measuring axial motions in crossbridges in working muscle.
- 36.Piazzesi G, Reconditi M, Linari M, Lucii L, Bianco P, Brunello E, Decostre V, Stewart A, Gore DB, Irving TC, et al. Skeletal muscle performance determined by modulation of number of myosin motors rather than motor force or stroke size. Cell. 2007;131:784–795. doi: 10.1016/j.cell.2007.09.045.This paper provides a molecular level explanation for one of the most fundamental phenomena in muscle physiology, the force/velocity curve requiring a rethinking of conventional thinking that had been in place for over 30 years.
- 37.Dickinson M, Farman G, Frye M, Bekyarova T, Gore D, Maughan D, Irving T. Molecular dynamics of cyclically contracting insect flight muscle in vivo. Nature. 2005;433:330–334. doi: 10.1038/nature03230. [DOI] [PubMed] [Google Scholar]
- 38.Pearson JT, Shirai M, Ito H, Tokunaga N, Tsuchimochi H, Nishiura N, Schwenke DO, Ishibashi-Ueda H, Akiyama R, Mori H, et al. In situ measurements of crossbridge dynamics and lattice spacing in rat hearts by x-ray diffraction: sensitivity to regional ischemia. Circulation. 2004;109:2976–2979. doi: 10.1161/01.CIR.0000133322.19340.EF.This is one of a series of papers where the high flux, high beam quality from third generation sources have allowed measurements of muscle function in vivo in beating hearts. Together with the Dickinson et al. [37], they show that x-ray diffraction now can be used to obtain useful structural parameters, that could not be obtained any other way, under truly physiological conditions.
- 39.Iwamoto H, Inoue K, Yagi N. Evolution of long-range myofibrillar crystallinity in insect flight muscle as examined by X-ray cryomicrodiffraction. Proc Biol Sci. 2006;273:677–685. doi: 10.1098/rspb.2005.3389.A technically impressive paper that required ground-breaking work in techniques of cryo-preservation, sample manipulation and micro-beam diffraction to obtain "end-on" diffraction patterns from a range of insect muscles.
- 40.Orgel JP, Irving TC, Miller A, Wess TJ. Microfibrillar structure of type I collagen in situ. Proc Natl Acad Sci U S A. 2006;103:9001–9005. doi: 10.1073/pnas.0502718103.The authors used multiple isomorphous replacement to solve crystallographically the supermolecular packing structure of collagen type I in its natural fibrous form at a level where the molecular conformation of each collagen segment is defined in the very large unit cell. This structure paves the way to a molecular understanding of the action of collagen binding proteins such as decorin and the Matrix Metallo-Proteinase (MMP) collagenase.
- 41.Gupta HS, Seto J, Wagermaier W, Zaslansky P, Boesecke P, Fratzl P. Cooperative deformation of mineral and collagen in bone at the nanoscale. Proc Natl Acad Sci U S A. 2006;103:17741–17746. doi: 10.1073/pnas.0604237103.This paper is an excellent example of how x-ray diffraction on third generation sources can be used to provide a structural explanation for the physical properties of a complex biological material, in this case bone. We can expect that the study of naturally occurring and synthetic biomaterials to become ever more important in the future.
- 42.Mollenhauer J, Aurich M, Muehleman C, Khelashvilli G, Irving TC. X-ray diffraction of the molecular substructure of human articular cartilage. Connect Tissue Res. 2003;44:201–207. [PubMed] [Google Scholar]
- 43.Boote C, Hayes S, Jones S, Quantock AJ, Hocking PM, Inglehearn CF, Ali M, Meek KM. Collagen organization in the chicken cornea and structural alterations in the retinopathy, globe enlarged (rge) phenotype--an X-ray diffraction study. J Struct Biol. 2008;161:1–8. doi: 10.1016/j.jsb.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 44.Makin OS, Sikorski P, Serpell LC. Diffraction to study protein and peptide assemblies. Curr Opin Chem Biol. 2006;10:417–422. doi: 10.1016/j.cbpa.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 45.Lazar KL, Miller-Auer H, Getz GS, Orgel JP, Meredith SC. Helix-turn-helix peptides that form alpha-helical fibrils: turn sequences drive fibril structure. Biochemistry. 2005;44:12681–12689. doi: 10.1021/bi0509705.This paper shows that a very short amino acid sequence in a strategic part of the peptide could induce the fibrilogenesis of α-helical based fibers including a previously not observed 'cross-α' fibril structure i.e. fibrilogenesis is not exclusively due to cross-β structures as is widely assumed.
- 46.Inouye H, Kirschner DA. X-Ray fiber and powder diffraction of PrP prion peptides. Adv Protein Chem. 2006;73:181–215. doi: 10.1016/S0065-3233(06)73006-6.This comprehensive review provides a good overview of prion biology and how diffraction techniques has contributed to our knowledge of prion structure.
- 47.Yagi N, Ohta N, Iida T, Inoue K. A microbeam X-ray diffraction study of insulin spherulites. J Mol Biol. 2006;362:327–333. doi: 10.1016/j.jmb.2006.07.041.This paper provides an excellent demonstration of the utility of microbeam diffraction to obtain structural information from complex biological structures.
- 48.Starodub D, Rez P, Hembree G, Howells M, Shapiro D, Chapman HN, Fromme P, Schmidt K, Weierstall U, Doak RB, et al. Dose, exposure time and resolution in serial X-ray crystallography. J Synchrotron Radiat. 2008;15:62–73. doi: 10.1107/S0909049507048893.This paper makes the technical case for using a stream of oriented macromolecules in a high intensity, coherent x-ray beam from either specialized beamlines on third generation sources or upcoming fourth generation sources.