

Abstract
Matrix metalloproteinases (MMPs) are proteolytic enzymes that are involved in both injury and repair mechanisms in the CNS. Pharmacological blockade of MMPs, limited to the first several days after spinal cord injury, improves locomotor recovery. This beneficial response is, however, lost when treatment is extended beyond the acutely injured cord to include wound healing and tissue remodeling. This suggests that some MMPs play a beneficial role in wound healing. To test this hypothesis, we investigated the role of MMP-2, which is actively expressed during wound healing, in white matter sparing and axonal plasticity, the formation of a glial scar, and locomotor recovery after spinal cord injury. MMP-2 increased between 7 and 14 d after injury, where it was immunolocalized in reactive astrocytes bordering the lesion epicenter. There was reduced white matter sparing and fewer serotonergic fibers, caudal to the lesion in injured MMP-2 null animals. MMP-2 deficiency also resulted in increased immunoreactivity to chondroitin sulfate proteoglycans and a more extensive astrocytic scar. Most importantly, locomotion in an open field, performance on a rotarod, and grid walking were significantly impaired in injured MMP-2 null mice. Our findings suggest that MMP-2 promotes functional recovery after injury by regulating the formation of a glial scar and white matter sparing and/or axonal plasticity. Thus, strategies exploiting MMPs as therapeutic targets must balance these beneficial effects during wound healing with their adverse interactions in the acutely injured spinal cord.
Keywords: glial fibrillary acidic protein, chondroitin sulfate proteoglycan, astrogliosis, contusion injury, gelatinase A, extracellular matrix
Introduction
Matrix metalloproteinases (MMPs), an endopeptidase family of at least 24 mammalian members, degrade a variety of substrates including extracellular matrix (ECM) proteins, proteinases and their inhibitors, chemotactic molecules, and cell surface receptors (Matrisian, 1992; Chang and Werb, 2001; Sternlicht and Werb, 2001; Yong et al., 2001). As such, they participate in both physiological and pathological events associated with cell motility, development, inflammation, and wound healing (Bjorklund and Koivunen, 2005).
In the CNS, MMPs participate in injury and disease states (Yong et al., 2001). In brain and spinal cord injury, MMPs, including MMP-9 (gelatinase B), contribute to early secondary pathogenesis by disrupting the blood–brain/spinal cord barrier and promoting inflammation (Rosenberg et al., 1994, 1995, 1996, 1998; Noble et al., 2002), oxidative stress (Gasche et al., 2001; Kim et al., 2003), and demyelination (Gijbels et al., 1992, 1993; Chandler et al., 1995; Anthony et al., 1998; Asahi et al., 2001).
MMPs also assume beneficial, specialized roles that include regulating axonal growth and guidance during development as well as promoting axonal regeneration and synaptic plasticity in the CNS (Duchossoy et al., 2001; Yong, 2005; Zhao et al., 2006). It is likely that they participate in wound healing based, in part, on their ability to degrade the ECM and release cryptic ECM molecules that regulate angiogenesis (Chang and Werb, 2001).
There is indirect evidence that MMP-2 (gelatinase A) modulates wound healing after spinal cord injury. Pharmacological blockade of MMPs, limited to the first 3 d after injury, is neuroprotective and promotes long-term recovery of hindlimb function (Noble et al., 2002). This gain of function is lost, however, if blockade of MMPs is extended to the first week after injury (Trivedi et al., 2005). One plausible explanation is that certain MMPs, expressed during wound healing, support recovery processes. A likely candidate is MMP-2, which is the dominant gelatinase in the injured cord during wound healing (Goussev et al., 2003).
Here we determine whether MMP-2 modulates the formation of a glial scar during wound healing. One of the major causes leading to regenerative failure of injured axons is the formation of a glial scar. The glial scar represents not only a physical barrier imposed by massive aggregation of reactive astrocytes (Reier, 1986) but also an inhibitory interface with aberrant expression of chondroitin sulfate proteoglycans (CSPGs), molecules that chemically arrest the regrowth of injured axons across the lesion site (McKeon et al., 1991, 1999; Levine, 1994). MMP-2 is upregulated concurrently with the emergence of reactive astrogliosis after spinal cord injury (Goussev et al., 2003) and is capable of degrading specific CSPGs (Muir et al., 2002). However, whether MMP-2 participates in the formation of a glial scar remains essentially unknown. Our findings provide the first evidence that MMP-2 is integral to wound-healing processes, including the formation of a glial scar, and is essential to motor recovery after spinal cord injury.
Materials and Methods
Breeding and surgical procedures
These studies were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco and were in accordance with the United States Department of Agriculture guidelines.
Breeding.
Homozygous MMP-2 null mice and their wild-type littermates were generated by breeding heterozygous males with heterozygous females on an FVBn background as described previously (Itoh et al., 1997). Young breeding pairs, ranging in age from 3 to 6 months, were used. MMP-2 null mice are fertile and, with the exception of a slower growth rate, are normal in appearance (Itoh et al., 1997). The genotypes of animals were identified by PCR using tail tissue and specific oligonucleotide primers (Itoh et al., 1997).
Spinal cord injury model.
Adult male mice weighing 30–35 g (∼4–6 months of age) were anesthetized with 2.5% Avertin (0.02 ml/g body weight, i.p., tribromoethanol; Sigma, St. Louis, MO) and subjected to a moderate spinal cord contusion injury as we have described previously (Noble et al., 2002). Briefly, a laminectomy was performed at the eighth thoracic vertebra, and a 2 g weight was dropped 5 cm onto the exposed dura mater. After injury, the overlying muscles were sutured, and the skin was closed with wound clips. The body temperature of the animals was maintained at 37°C with a heating pad throughout the surgery and during the recovery from anesthesia. Sham controls were subjected to laminectomy only. Postoperative care included subcutaneous administration of antibiotics and manual expression of the bladder twice per day.
Zymography
MMP-2 null and wild-type animals were killed 3, 7, or 14 d after injury (n = 3 per genotype per time point). Two additional wild-type mice received sham surgery and were killed at each of the above time points. Gelatin zymography was performed as described previously (Noble et al., 2002; Goussev et al., 2003). Briefly, segments of the spinal cord containing the lesion epicenter or matched regions of the laminectomy controls were dissected and frozen at −70°C. Each sample was weighed and homogenized in lysis buffer containing the following (in mm): 28 Tris-HCl, 22 Tris-base, pH 8, 150 NaCl, 1% Nonidet P-40 (74385; Fluka BioChemika, Ronkonkoma, NY), 0.5% sodium deoxycholate, and 0.1% SDS. The protein concentration of the homogenates was determined by the bicinchoninic acid method (BCA protein assay kit, 23225; Pierce, Rockford, IL). Equal amounts of proteins (5 μg) were loaded on a Novex 10% zymogram gel (EC61752; Invitrogen, Carlsbad, CA) and separated by electrophoresis at 4°C (125 V for 120 min). The proteins were renatured in buffer (LC2670; Invitrogen) at room temperature for 1 h to enable gelatinases to cleave the gelatin substrate copolymerized in the gel. The gel was then incubated with developing buffer (LC2676; Invitrogen) at 37°C for 72 h, stained with 0.5% Coomassie blue for 30 min, and then destained with 30% methanol containing 10% acetic acid for appropriate color contrast. Clear bands were indicative of the location of proteolytic activity. Purified human recombinant MMP-2 proenzyme was used as the positive control (PF 037; Calbiochem, La Jolla, CA). The identities of the MMPs were based on their molecular weights.
Immunohistochemistry
Preparation of tissue.
At a designated time point, animals were anesthetized and perfused with ice-cold normal saline, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The spinal cord was removed and immersed in the same fixative at 4°C for 4 h. A spinal segment, 25 mm in length and centered over the lesion epicenter, was transferred into 30% sucrose in 0.1 m PBS for cryoprotection. Frozen sections of the spinal cord were cut either transversely or horizontally at 20 μm in a cryostat, mounted onto glass slides, and stored at −20°C for additional processing.
Immunohistochemistry.
Spinal cords were selected for analysis at 7, 14, and 42 d after injury (n = 14 per genotype per time point). The following primary antibodies were used: rabbit anti-MMP-2 (1:200, AB19167; Millipore, Bedford, MA), mouse anti-glial fibrillary acidic protein (GFAP; to label astrocytes, 1: 400, clone G-A-5, G3893; Sigma), Cy3-conjugated mouse anti-GFAP (1:400, clone G-A-5, C9205; Sigma), rat anti-CD11b (to label macrophages/microglia, 1:400, clone 5C6, MCA711; Serotec, Raleigh, NC), rat anti-platelet/endothelial cell adhesion molecule-1 (PECAM-1; to label vascular endothelium, CD31, 1:400, clone MEC13.3, 550274; BD Biosciences, San Jose, CA), mouse anti-NeuN (to label neuronal nuclei, 1:1000, MAB377; Millipore), mouse anti-CSPG (1:200, clone CS-56, C8035; Sigma), and rabbit anti-serotonin (1:60,000; Immunostar, Hudson, WI). Another polyclonal anti-MMP-2 antibody (4022; Cell Signaling, Danvers, MA) was also tested on cryosections. However, this antibody yielded strong background, even in the MMP-2 null tissue, and was not adopted. Alexa Fluor 568-conjugated goat anti-rabbit IgG (1:200, A-11036; Invitrogen) was used as the secondary antibody to detect anti-MMP-2, whereas Alexa Fluor 488-conjugated goat anti-rat IgG (1:200, A-11006; Invitrogen) or Alexa Fluor 488-conjugated goat anti-mouse IgG (1:200, A-11029; Invitrogen) were used to label other primary antibodies.
Cryosections were rinsed with 0.01 m PBS and treated with blocking and permeablizing solution containing 2% normal goat serum and 0.1% bovine serum albumin in PBS at room temperature for 5 min. Incubation of anti-MMP-2 antibody diluted in the above blocking and permeablizing solution was performed in a humidified box at 4°C overnight. Primary antibodies against GFAP, PECAM-1, or NeuN were applied at 4°C overnight to colabel respective cell types with MMP-2. Some cryosections were incubated with anti-CSPG or anti-serotonin alone at 4°C overnight, and the immunolabeling was visualized by biotinylated goat anti-mouse IgM (1:200, 115-065-075; Jackson ImmunoResearch, West Grove, PA) and goat anti-rabbit IgG, respectively, using a Vectastain Elite ABC kit (PK-6100; Vector Laboratories, Burlingame, CA) in combination with 0.05% diaminobenzidine as the chromogenic substrate and 0.001% hydrogen peroxide in 0.01 m PBS.
Sections from animals examined at various time points were grouped as a set and processed simultaneously in the same experimental condition to limit variables for subsequent quantitative analyses. Immunocytochemical controls consisted of omission of the primary antibody. Fluorescently labeled sections were coverslipped with ProLong Antifade (P-7481; Invitrogen). All images were captured on an epifluorescent microscope (Optiphot EF-D3; Nikon, Tokyo, Japan) equipped with a digital camera.
Measurement of residual white matter
Transverse sections of the spinal cord 42 d after injury were stained histochemically with Luxol Fast Blue (Noble and Wrathall, 1985; Noble et al., 2002) to visualize the residual white matter in the lesion epicenter in both groups of mice (n = 8 per genotype). We have demonstrated previously that the residual white matter is the best single measurement for the severity of injury in the contused spinal cord and is, in fact, predictive of motor recovery (Noble and Wrathall, 1985). The total cross-sectional area of the injured cord and the area of residual white matter were measured using the Neurolucida imaging system (MicroBrightField, Williston, VT). Eight sections at 480 μm intervals across the lesion site were sampled from each animal. Among these eight sections, three sections with the least residual white matter were chosen, and their measurements were averaged for quantitative comparison.
Assessment of the astrocytic scar
Formation of an astrocytic scar was assessed using Cy3-conjugated anti-GFAP antibody as described above (in Immunohistochemistry). A transverse section of the spinal cord was subdivided into 12 regions. These subdivisions generally separated gray matter from white matter, the dorsal half of the cord from the ventral half, and the left side from the right side. The lines demarcating these subdivisions were superimposed onto the entire image of an anti-GFAP-immunostained section using Photoshop 7.0 (Adobe Systems, San Jose, CA).
In each subdivision, the complexity of the glial scar was evaluated according to a semiquantitative scoring scale, in which the pattern of scarring was categorized mandatorily into four levels (0–3) as follows. (0) Distribution of astrocytes, arborization of processes, pattern of GFAP immunostaining, and abundance of processes approximate that of the uninjured cord. There is no evidence of astrocytic hypertrophy. (1) There is a mild thickening of some astrocytic processes. In some cases, GFAP immunostaining appears slightly elevated. In general, there is a disorganization of astrocytes as evidenced by a tendency toward clustering. There is no evidence of astrocytic hypertrophy. (2) Astrocytic processes are organized into overlapping bundles, and GFAP immunostaining appears moderately elevated to that of uninjured tissue. Astrocytic processes are moderately arborized, and many form an entangled trabecular meshwork. Hypertrophied astrocytes are only occasionally observed. (3) Astrocytic processes are organized into dense bundles that collectively form an intricate meshwork of overlapping processes. GFAP immunostaining is pronounced relative to that of uninjured tissue. A prominent glial limitans, separating the lesion from residual cord tissue, is evident. Hypertrophied astrocytes are noted within the residual cord tissue that is separated from the lesion by the glial limitans.
Central GFAP-quiescent regions, composed of non-neuronal cells and dominated by macrophage-like cells, were excluded from the analysis. Eight transverse sections, at intervals of 480 μm, were sampled across the lesion epicenter in each animal. In each sample section, the total obtained from all scored subdivisions represented the severity and/or extent of astrogliosis in the injured cord.
Analysis of CSPG immunoreactivity
The intensity of CSPG immunoreactivity was quantified 42 d after injury (n = 6 per genotype), based on a protocol described previously with some modifications (Jones and Tuszynski, 2002; Jones et al., 2002). In each animal, a transverse section of the injured cord was selected from the middle of the lesion epicenter to measure the intensity of immunoreactivity. Another transverse section, ∼1500 μm rostral to the lesion epicenter where there was no overt tissue damage, was selected as the reference section for background staining. Digital images of each section were taken using a 4× objective under consistent illumination and exposure parameter. With the use of the ImageJ image processing program (1.33u; National Institutes of Health, Bethesda, MD), the digital image of the reference section was imported as an 8-bit grayscale image, and its empty area, including the meninges, was trimmed off to eliminate artifact. The total number of pixels in the remaining area was obtained from the histogram. The threshold value was then adjusted so that only 1% of the darkest pixels in the image remained. This threshold value, which eliminated 99% of the background staining, was then applied to the meninges-trimmed image of the injured section of the same animal. The number of pixels, with intensity higher than threshold value, was counted. The percentage of this number over the total pixel number of the meninges-trimmed image was calculated, averaged with those of the same mouse group, and reported.
In vitro CSPG synthesis assay
Preparation of cultures.
Newborn MMP-2 null and wild-type mice were generated by breeding homozygous parents of respective genotypes. Cortical astrocytes were cultured from the cerebral cortex of neonates at postnatal day 1 or 2 based on a previously described protocol (Rose et al., 1993). Briefly, the cerebral cortices were dissected from the brain stem. After the hippocampus, adherent meninges, and vessels were removed, the cerebral cortices were triturated repeatedly with a 5 ml serological pipette to dissociate single cortical cells in a mixture containing 0.05% trypsin and 0.05% DNase in Ca2+- and Mg2+- free HBSS. Cells were centrifuged at 250 rpm for 5 min, resuspended, filtered through a 70 μm nylon cell strainer, and plated in a poly-l-lysine-coated flask containing Earle's MEM supplemented with 10% fetal bovine serum, 22 mm glucose, and 2 mm GlutaMAX-1 (35050-061; Invitrogen). After 24 h of incubation in 5% CO2 at 37°C, the flask was shaken vigorously to remove debris and unattached cells before fresh medium replacement. Shaking was repeated before changing the culture medium at 3, 5, and 7 d after isolation and weekly thereafter. The purity of astrocyte cultures was ∼95% and was determined by monoclonal anti-GFAP immunostaining and nuclear staining with 0.0002% Hoechst dye.
Immunoblot analysis.
Astrocyte cultures were replated in 12-well plates. When confluent, the serum-containing medium was removed and replaced with serum-free medium for 24 h, at which time this medium was again replaced with fresh serum-free medium. Three wells of wild-type or MMP-2 mutant astrocytes were maintained in this medium without additional treatment, whereas the remaining wild-type or mutant astrocytes were treated twice over a 4 d period with TGF-β1 (10 ng/ml; R & D Systems, Minneapolis, MN). After 4 d, the conditioned medium from each well was collected into individual tubes and concentrated 10-fold by centrifugation using Microcon filters (Millipore) with a 50,000 MW cutoff according to the manufacturer's instructions. Total protein concentration of the retentate was determined by the BCA method (Pierce), and 20 μg of each sample was treated with protease-free chondroitinase ABC (0.1 U/ml; Seikagaku, Tokyo, Japan) in 100 mm Tris and 10 mm sodium acetate, pH 8.0, at 37°C for 1 h to degrade glycosaminoglycan side chains before being loaded onto 4–15% gradient mini-gels (Bio-Rad, Hercules, CA) for SDS-PAGE. The separated proteins were then electroblotted onto polyvinylidene difluoride membranes (150 mA, overnight at 4°C) using a high molecular weight transfer buffer (50 mm Tris, 380 mm glycine, 0.1% SDS, 20% methanol). The membranes were blocked with 5% dry milk in Tris-buffered saline (TBS) for 2 h and probed for 1 h at room temperature with antibodies to either the 4-sulfated (1:1000, clone 2B6; Seikagaku) or the 6-sulfated (1:1000, clone 3B3; Seikagaku) moiety of CSPGs after chondroitinase ABC digestion. The membranes were then washed three times in TBS, incubated with a peroxidase-conjugated secondary antibody for 1 h at room temperature, followed by repeated washes for 2 h, and visualized with chemiluminescence (Amersham Biosciences, Piscataway, NJ).
Quantification of serotonergic fibers
Serotonergic axons, caudal to the lesion site, were quantified 42 d after injury in horizontal sections of the spinal cord immunostained with anti-serotonin (5-HT) antibody (n = 4 per genotype) based on a design described previously (Oudega et al., 1999; Hsu and Xu, 2005). With the use of the Neurolucida imaging system, eight lines at intervals of 200 μm were superimposed onto the horizontal section perpendicularly to its longitudinal axis with the first line aligned with the center of the lesion and the remaining positioned caudally. Intersections between serotonergic axons in the gray matter and superimposed lines were counted. At a given distance, the total number of intersections obtained from six to seven sample sections, at intervals of 200 μm, of each animal was defined as the number of axons.
Assessment of recovery
Multiple behavioral tests were performed to evaluate functional improvements after spinal cord injury.
Open-field locomotion.
The 21-point Basso-Beattie-Bresnahan Locomotor Rating Scale (Basso et al., 1995) was used to examine the locomotor recovery of the injured animals. This rating scale assessed limb movement, stepping, coordination, and trunk stability in an open field (53 × 108 × 5.5 cm). Injured animals with better locomotor recovery scored higher. One trial, which lasted for 5 min, was performed at 1, 3, and 7 d after injury, followed by one trial weekly for an additional 5 weeks.
Rearing, rotarod, and grid walking.
Rearing, performance on a rotarod, and the ability to traverse a wire grid were evaluated, in sequence, at 35, 36, and 37 d after injury. There were three trials daily, with a total of nine trials for each test. In each of these tests, the best score was used to calculate the mean.
Each animal was positioned in a clear acrylic cylinder (11.5 cm in diameter and 7.5 cm in height) for 2 min per trial. A rearing event was counted each time an animal assumed an upright posture and had weight shifted to the hindlimbs. The time the animal spent on a slowly accelerating rotarod during a 2 min trial was also recorded. Performance on an inclined ladder positioned at 40° and with rungs at 9 mm intervals was evaluated by counting foot faults as the animal descended the ladder through a plastic tunnel (42 × 4.5 × 5 cm). A foot fault was defined as when either hindpaw missed a rung and extended through the space between adjacent grids.
Statistical analysis
All experiments that required quantitative evaluation, including morphometric measurements and behavioral assessments, were performed in a blinded manner. Data are expressed as means ± SD. Comparisons between MMP-2 null and wild-type groups were made by unpaired Student's t tests. ANOVA, followed by Bonferroni's post hoc test, was performed for multiple comparisons between groups. A statistically significant difference was defined at p < 0.05.
Results
MMP-2 level increases during wound healing
We first examined MMP expression during the wound-healing phase after spinal cord lesions by gelatin zymography. Consistent with our previous findings (Goussev et al., 2003), MMP-2 was not detected in the wild-type mice 3 d after injury but was strongly detected at 7 and 14 d after injury, with protease bands corresponding to the active form of MMP-2 and the inactive zymogen (proMMP-2) (Fig. 1). Wild-type mice subjected to laminectomy only (sham) showed no MMP-2 expression at all three time points examined. The MMP-2 null mouse did not display any detectable MMP-2, as expected, but did show a compensatory increase in MMP-9 at 3, 7, and 14 d after injury.
Figure 1.

Representative gelatin zymograms showing gelatinase levels in the sham-operated and injured spinal cord. A, No MMP-2 is detected in wild-type animals receiving only laminectomy or within the lesion epicenter 3 d after injury. B, However, MMP-2 expression is apparent in lesion epicenters of wild-type mice 7 and 14 d after injury. As expected, no MMP-2 is detected in MMP-2 null mice at 3, 7, or 14 d after spinal cord injury. The MMP-9 level is upregulated in the injured spinal cord of MMP-2 null mice relative to the wild-type mice at 3, 7, and 14 d after injury. Sham, Wild-type animals receiving only laminectomy; WT, wild-type mice; Null, MMP-2 null.
MMP-2 is preferentially expressed in astrocytes bordering the lesion epicenter
We then used double immunolabeling to determine which cell types express MMP-2. In contrast to MMP-9 expression in the acute phase after injury (Noble et al., 2002), MMP-2 was colocalized to a subset of GFAP-positive reactive astrocytes that bordered the lesion epicenter 7 d after injury (Fig. 2 A,C,E). These reactive astrocytes and their processes segregated the lesion core from the residual cord tissue. The intensity of both MMP-2 and GFAP immunoreactivity increased 14 d after injury (Fig. 2 B,D,F). As expected, MMP-2 was not detected in the injured spinal cord of MMP-2 null mice (Fig. 2 G,H) or in the uninjured wild-type mice (data not shown). MMP-2 did not colocalize with endothelial cells or the majority of macrophages at either 7 or 14 d after injury (Fig. 3). PECAM-1-positive microvessels were observed in the lesion epicenter at 7 and 14 d after injury (Fig. 3 A,B). These vascular structures exhibited an angiogenic phenotype, namely diameters larger than that seen in controls and tortuous morphologies. Densely packed, CD11b-positive macrophages/microglia were confined to the lesion epicenter (Fig. 3 C,D). A subset of these macrophages, adjacent to the lesion border, appeared to immunoexpress MMP-2. MMP-2 did not colocalize with NeuN-positive neurons (data not shown).
Figure 2.
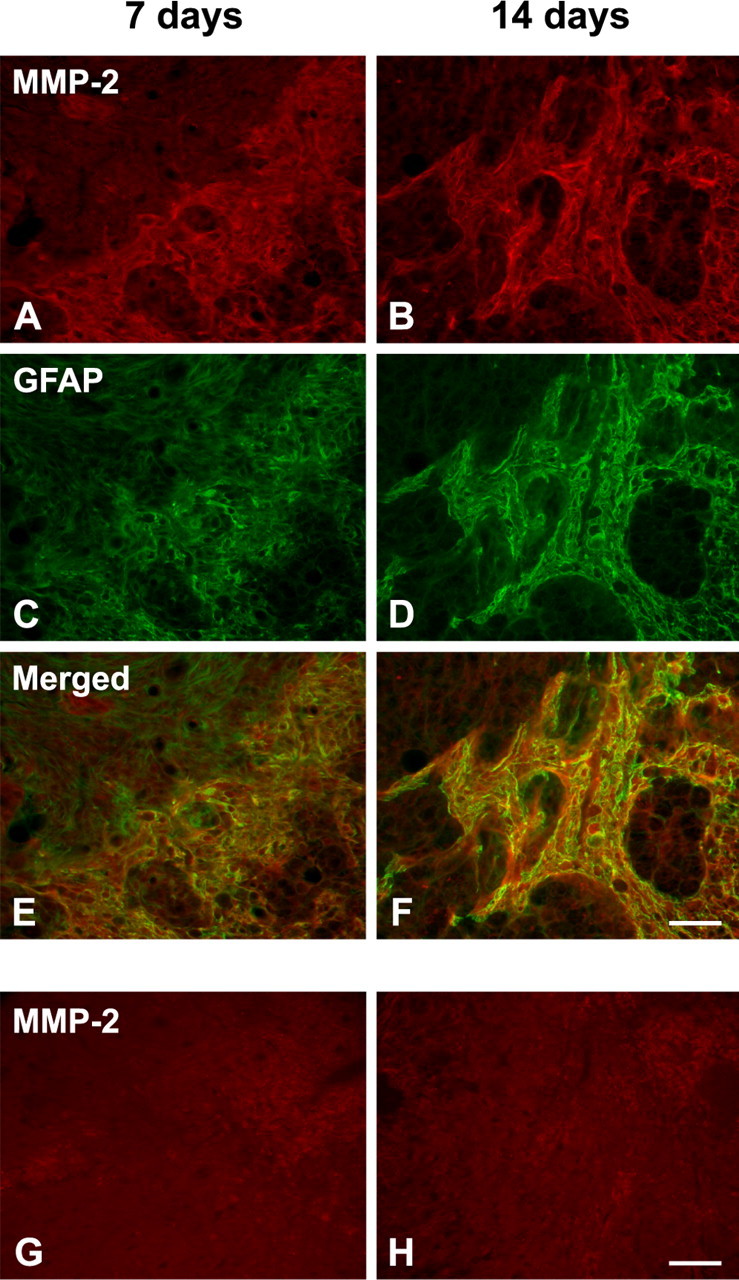
A–D, Colocalization of MMP-2 (A, B; red, Alexa 568) and GFAP (C, D; green, Alexa 488) at the lesion epicenter 7 and 14 d after injury in wild-type mice. E, F, MMP-2 is localized in reactive astrocytes that form a boundary between the lesion epicenter and the residual cord tissue (digitally merged images). G, H, No MMP-2 immunostaining is found in the injured spinal cord of MMP-2 null mice at either time point. Scale bars, 50 μm.
Figure 3.
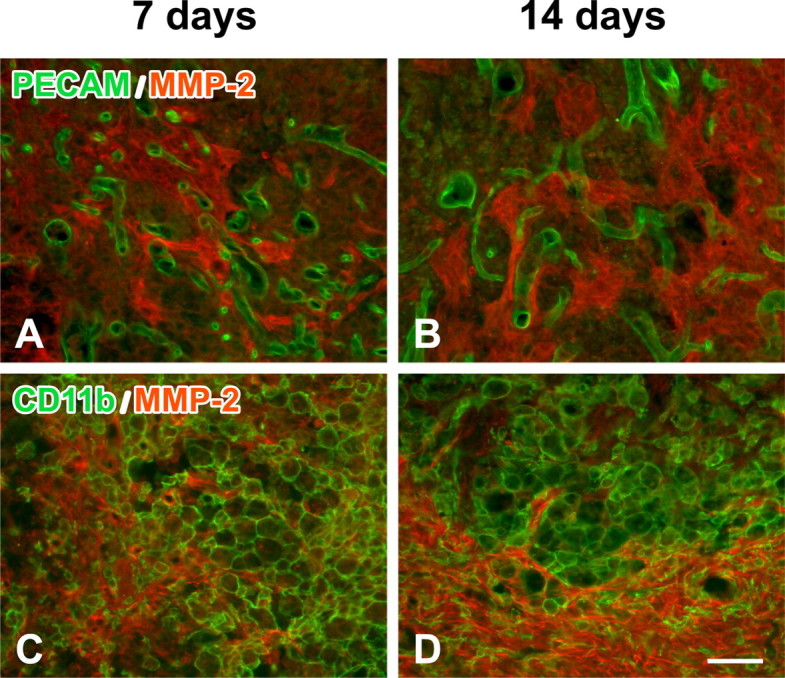
Colocalization of MMP-2 (red, Alexa 568) with either PECAM-1-positive vessels (A, B; green, Alexa 488) or CD11b-positive macrophages/microglia (C, D; green, Alexa 488) at the lesion epicenter of wild-type mice 7 and 14 d after injury. Digitally merged images reveal that neither blood vessels nor the majority of macrophages immunoexpress MMP-2. Scale bar, 50 μm.
Glial scarring is more extensive in MMP-2 deficient animals
After CNS injury, the glial scar is formed by the aggregation of reactive astrocytes that typically exhibit hypertrophic cell bodies and intense GFAP immunostaining (McKeon et al., 1999). More scarring correlates with poorer functional recovery. In the contused spinal cord of wild-type mice 42 d after injury, the glial scar mostly surrounded a central GFAP-quiescent core (Fig. 4 A). As demonstrated previously (Whetstone et al., 2003), these GFAP-quiescent areas were filled with tissue matrix and non-neuronal cells rather than cystic cavities. Mild reactive astrogliosis was found in less injured tissue around the ventral or lateral margin of the spinal cord. Conversely, the pattern of astrogliosis in MMP-2 null mice was more prominent and dispersed throughout the lesion epicenter (Fig. 4 B). Moreover, the overall intensity of the GFAP immunoreactivity within the epicenter of MMP-2 null mice appeared higher than that of the wild-type mice. At higher magnification, the glial scar in MMP-2 null mice exhibited a more intricate configuration of densely intertwined astrocytic processes relative to that of wild-type mice (Fig. 4 C,D).
Figure 4.

Localization of GFAP at the lesion epicenter of the spinal cord in wild-type (A, C) and MMP-2 null (B, D) mice 42 d after injury. GFAP-quiescent areas (asterisks) are evident within the lesion epicenter and are separated from the remaining cord tissue by reactive astrocytes. GFAP immunoreactivity appears more widespread in the null relative to the wild-type animal. At higher magnification (C, D; taken from boxed area in A and B, respectively), a more elaborate form of astrogliosis, characterized by heavily tangled astrocytic processes, is apparent in the null relative to the wild-type animal. Scale bars, 100 μm.
We next evaluated the severity of glial scarring based on its complexity and extent in eight sample sections across the entire lesion site in each animal (Fig. 5 A). The higher the total scores, the more elaborate and/or widespread the glial scar. When the section having the highest score among these eight sample sections was selected for comparison, this highest score was significantly greater in MMP-2 null mice than in wild-type controls (23 ± 4 vs 17 ± 2; p < 0.05) (Fig. 5 B). We then analyzed whether such a difference in the highest score was a result of a more extensive distribution (more sectors scored ≥1) or a more complicated pattern (higher score per sector) of glial scar formation. Among the total 12 sectors within the sample section, MMP-2 null mice had significantly more sectors scored ≥1 than wild-type mice, indicating a more extensive glial scarring in the former (Fig. 5 C). The average scores of the sectors within the sections were similar between MMP-2 null and wild-type mice (2.43 ± 0.08 vs 2.41 ± 0.09; p = 0.86), suggesting the complexity of the astrogliosis was comparable between groups. However, when the scores of all eight sample sections were combined, MMP-2 null mice showed a trend toward a higher sum total than the wild-type mice (59 ± 11 vs 45 ± 12), but this was not statistically significant. Together, these data demonstrate that the glial scar is more extensive in MMP-2 null mice than in the wild-type mice 42 d after spinal cord injury.
Figure 5.

A, A diagram illustrating the design and sampling strategy for the semiquantitative analysis of the glial scar. Twelve subdivisions (sectors) were arranged as shown in each of eight sample sections per animal (n = 8 per genotype). A semiquantitative scale, ranging from 0 to 3, was applied to each subdivision based on the criteria described in Materials and Methods. B, The section having the highest score among all eight sample sections is selected for comparison. This value is significantly greater in MMP-2 null mice than in wild-type mice. C, Within the section showing the highest score, there are significantly more subdivisions manifesting a score ≥1 in the MMP-2 null mice than in the wild-type controls. This finding suggests that the extent of the glial scar is more widespread in MMP-2 null mice. *p < 0.05. Error bars indicate SD.
MMP-2 deficiency results in elevated expression of CSPGs
MMP-2 is known to enhance neurite outgrowth by degrading CSPGs in vitro (Zuo et al., 1998). CSPGs were immunoexpressed at relatively high levels in the lesion epicenter in both groups of mice 42 d after injury compared with background immunostaining in the uninjured tissue (reference sections) (Fig. 6). Within the injured segment of both groups, the intensity of CSPG immunoreactivity was most pronounced at the interface between the lesion core and the residual cord tissue (Fig. 6 C,D). Such a staining pattern generally mirrored the distribution of astrogliosis observed with anti-GFAP immunostaining in the injured cord, suggesting a close correlation between the glial scar formation and CSPG expression. The relative intensity of CSPG immunoreactivity at the lesion epicenter was significantly greater in MMP-2 null mice than in the wild-type controls 42 d after injury (38.5 ± 4.3 vs 27.4 ± 6.1%; p < 0.05). These findings suggest that proteolytic degradation of CSPGs is reduced in the absence of MMP-2 and raises the possibility that the environment may be particularly inhibitory to axonal plasticity and/or regeneration.
Figure 6.

CSPG immunoreactivity (clone CS-56) 42 d after spinal cord injury. A, B, In the uninjured sections (reference sections) ∼1500 μm rostral to the lesion epicenter, background immunostaining is observed mostly in the gray matter. C, D, In the lesion epicenter, CSPG immunoreactivity appears enhanced in both groups compared with the staining intensity of respective reference sections. CSPGs are expressed at high levels along the penumbral zone lying between the core of the lesion and the less injured tissue. This staining pattern resembles the distribution of astrogliosis. Scale bar, 250 μm.
Both wild-type and MMP-2 null astrocytes produce CSPGs in response to TGF-β1 stimulation
Although MMPs are capable of degrading CSPGs (Muir et al., 2002), their affect on proteoglycan synthesis has not been examined in detail. Increased CSPG deposition in the MMP-2 null mice may be because of limited CSPG degradation or increased synthesis of CSPGs by mutant astrocytes. To examine whether CSPG synthesis was altered in the mutant astrocytes, we compared the ability of wild-type and MMP-2 null astrocytes to synthesize CSPGs in vitro in control conditions, and after stimulation, by TGF-β1. TGF-β is one of a number of injury-induced cytokines and has been demonstrated to stimulate CSPG synthesis in astrocytes (Asher et al., 2000; Smith and Strunz, 2005). As demonstrated in Figure 7, both wild-type and MMP-2 null astrocytes produced high-molecular-weight 2B6-immunopositive CSPGs, indicating the presence of a 4-sulfated moiety after chondroitinase ABC digestion. After TGF-β1 stimulation, both groups of astrocytes increased their production of the higher-weight CSPGs as well as a lower-weight CSPG. Similar results were obtained from immunoblots using anti-3B3 (data not shown). Individual proteoglycans, expressed by these astrocytes, were not determined because the 2B6 and 3B3 antibodies identify a 4-sulfated and a 6-sulfated epitope, respectively, on all CSPGs. However, these data demonstrate that both wild-type and mutant astrocytes synthesize CSPGs normally and in response to TGF-β1, suggesting that increased expression of CSPGs in MMP-2 null mice after injury is likely caused by a limited ability to degrade CSPGs in these animals.
Figure 7.
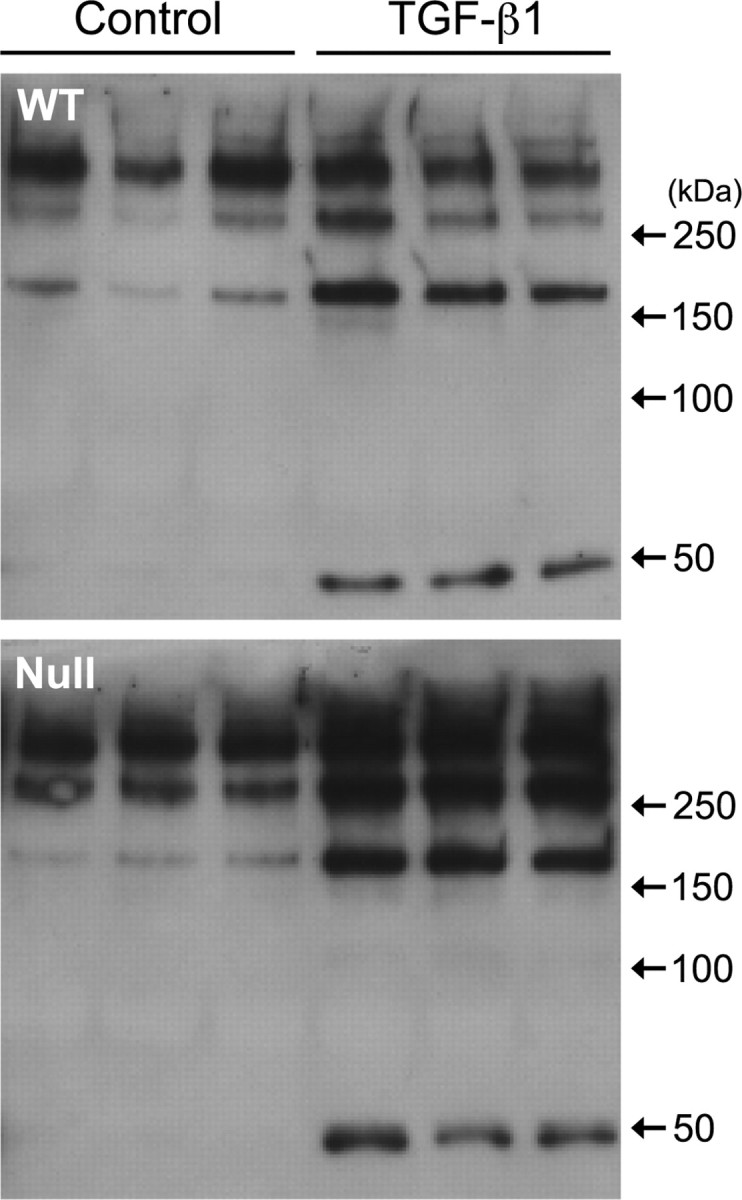
CSPG synthesis by wild-type and MMP-2 null astrocytes in vitro. Conditioned medium from untreated (Control) or TGF-β1-treated wild-type (WT) or MMP-2 null (Null) astrocytes contains a number of CSPGs with a 4-sulfated epitope demonstrated using the 2B6 anti-CSPG antibody. TGF-β1 exposure not only increases the synthesis of the higher-weight CSPGs but also stimulates the expression of a lower molecular weight species in both wild-type and MMP-2 null astrocytes.
MMP-2 deficiency results in reduced axonal plasticity and white matter sparing
The population of serotonergic fibers caudal to the lesion site has been used as an indicator of plasticity and/or sparing of these supraspinal projections after spinal cord injury (Faden et al., 1988; Wrathall et al., 1994; Teng and Wrathall, 1997). To determine whether MMP-2 deficiency and altered CSPG expression affect axonal plasticity after spinal cord injury, we quantified serotonergic axons in horizontal sections in the spinal gray matter around the lesion site (Fig. 8 A). Depending on the distance from the lesion site, serotonergic axons were distributed in the superficial dorsal horn, intermediolateral column, and around the central canal in relatively intact tissue, or in the remnant of the ventral horn in the injured area. Serotonergic axons in the remnant of the ventral horn exhibited arborized profiles with some varicosities (Fig. 8 B). In the core of the lesion, few to no serotonergic axons were found in each animal regardless of the genotype. In the ventral horn 400 and 600 μm caudal to the lesion epicenter, MMP-2 null mice showed significantly fewer serotonergic axons than wild-type mice (400 μm, 21 ± 25 vs 96 ± 24, p < 0.05; 600 μm, 88 ± 55 vs 246 ± 62, p < 0.05) (Fig. 8 C). This trend continued caudally up to 1400 μm from the lesion epicenter, which was the furthest point examined in this study; although, it was not statistically significant.
Figure 8.

A, Quantitative analysis of serotonergic axons caudal to the lesion site 42 d after injury. Horizontal sections are immunostained with anti-5-HT. B, In the spinal gray matter (arrowheads), serotonergic axons labeled by anti-5-HT are characterized by arborized profiles with varicosities . C, Axons were quantified caudal to the lesion epicenter at 200 μm consistent intervals, using the midline of the epicenter as the starting point. The numbers of serotonergic axons increase progressively with distance in both groups of animals. Compared with wild-type mice, however, MMP-2 null mice show significantly fewer serotonergic axons 400 and 600 μm caudal to the lesion epicenter (n = 4 per genotype). *p < 0.05. Scale bars: A, 500 μm; B, 50 μm. Error bars indicate SD.
We next quantified the residual white matter in the lesion epicenter. MMP-2 null and wild-type mice showed comparable total cross-sectional areas of the spinal cord at the lesion epicenter 42 d after injury (1.65 ± 0.21 vs 1.74 ± 0.19 mm2). This finding suggests that MMP-2 deficiency does not affect the gross architecture of the injured spinal cord during wound healing. However, there was significantly less residual white matter in the lesion epicenter of MMP-2 null mice than the wild-type controls 42 d after injury (0.53 ± 0.19 vs 0.67 ± 0.16 mm2; p < 0.05).
MMP-2 deficiency results in impaired motor recovery after injury
Behavioral recovery was examined by a battery of tests including locomotion in an open field, rearing, performance on a rotarod, and the ability to descend a ladder. Adult MMP-2 null mice exhibited no notable difference in their appearance and body size compared with wild-type controls. Before the injury, locomotor performance between MMP-2 null and wild-type mice was comparable. Differences in locomotion were not evident between groups within the first week after injury (Fig. 9). Thereafter, there was a clear trend toward impaired recovery of locomotor function in the MMP-2 null animals. Forty-two days after injury, the locomotor score of MMP-2 null mice was significantly lower than that of wild-type mice (13 ± 4 vs 17 ± 4; p < 0.05). At this time point, both groups showed forelimb–hindlimb coordination. However, wild-type controls typically showed consistent weight-supported plantar stepping, whereas the MMP-2 null mice were characterized by less consistent weight-supported plantar stepping.
Figure 9.

Functional recovery after spinal cord injury. Before the injury, both MMP-2 null and wild-type mice exhibited comparable activity and locomotor performance in the open field (data not shown). Based on the 21-point locomotor rating scale, MMP-2 null mice show impaired motor recovery after spinal cord injury compared with their wild-type littermates, reaching statistical significance 42 d after injury (n = 14 per genotype). *p < 0.05. Error bars indicate SD.
Spinal cord-injured MMP-2 null mice reared less frequently (3 ± 4 vs 11 ± 8 per trial; p < 0.05), performed significantly worse on a rotarod (28 ± 18 vs 71 ± 26 s/trial; p < 0.01), and exhibited a greater number of foot faults when descending an inclined ladder (12 ± 3 vs 5 ± 2 per trial; p < 0.01) compared with wild-type mice. Together, these data demonstrate that recovery of function is reduced in spinal cord-injured MMP-2-deficient animals and are consistent with the morphologic findings.
Discussion
This is the first study to assess the role of MMP-2 in the injured spinal cord. We show that MMP-2 is integral to those wound-healing events that support recovery of function in spinal cord-injured mice. Mice deficient in MMP-2 showed reduced white matter sparing and supraspinal efferents around the lesion epicenter, a widespread reactive astrogliosis, and a significant impairment in locomotor recovery compared with wild-type littermates.
CNS injury and MMPs
MMPs play complex roles in neurological diseases (Yong et al., 2001), and we are only now beginning to appreciate their involvement in spinal cord injury. A number of MMPs are upregulated in the injured spinal cord (Wells et al., 2003). Of these, MMP-9 and MMP-12 have recently been studied in some detail. MMP-9 increases in the acutely injured spinal cord (Noble et al., 2002). It degrades the CSPG NG2 and, as such, facilitates oligodendrocyte maturation and remyelination after spinal cord injury (Larsen et al., 2003). Spinal cord-injured mice with a genetic null mutation in MMP-9 exhibit stabilization of the blood–spinal cord barrier, reduced infiltration of neutrophils, and a significant improvement in locomotor recovery (Noble et al., 2002). MMP-12 (metalloelastase) mRNA markedly increases 5 d after spinal cord injury. Spinal cord-injured animals with a null mutation of this protease show greater preservation of barrier integrity and reduced macrophage/microglial activation and migration (Wells et al., 2003).
Glial scar formation and MMP-2
Although there are some beneficial effects of glial scarring (Faulkner et al., 2004), it is generally considered to be a nonpermissive environment causing regenerative failure of injured axons (Bush et al., 1999; Silver and Miller, 2004). Here we show that the astrocytic scar is more extensive in the spinal cord-injured MMP-2 null animal, suggesting a more inhibitory environment for axonal regrowth.
Astrogliosis is attributed primarily to increased migration of astrocytes toward the site of a lesion (Ridet et al., 1997; Fitch et al., 1999; McGraw et al., 2001; Matyash et al., 2002). Astrocytes, derived from ependymal stem cells, migrate into the lesion to participate in glial scar formation after spinal cord injury in the adult (Johansson et al., 1999). Proliferation of resident or migrating astrocytes is also involved in astrogliosis, but to a lesser extent (Rhodes et al., 2003; Hampton et al., 2004). Alterations in the intermediate filament cytoskeleton have been implicated in astrocyte motility. Astrocytes devoid of both GFAP and vimentin, an embryonic intermediate filament re-expressed in reactive astrocytes, display abnormal morphology and severely compromised motile behavior (Lepekhin et al., 2001). Although the role of MMPs in modulating intermediate filament components of astrocytes is not clear, MMPs facilitate the migration of bipotential oligodendrocyte-type 2 astrocyte progenitors on CNS myelin, a strongly inhibitory substrate that can be inactivated by proteolytic degradation mediated by metalloproteases (Amberger et al., 1997). Moreover, the increased migration capacity of white matter astrocytes on laminin is mediated by MMP-9, MT1 (membrane type 1)-MMP, and MMP-2 in vivo (Takenaga and Kozlova, 2006).
An enigmatic observation in our study is that MMP-2 deficiency resulted in the formation of substantially extensive astrocytic scar in vivo, implying increased migratory behavior of reactive astrocytes. These findings are counterintuitive considering the ability of MMP-2 to promote ECM remodeling and cell migration. However, it is possible that other MMPs are responsible for astrocyte migration. Several MMPs either increase or decrease during wound healing in the injured spinal cord (Wells et al., 2003). As such, there may be a feedback interaction or reciprocal regulation among these different MMPs. In this study, we show anomalous MMP-9 upregulation in the spinal cord-injured MMP-2 null animal. Similarly, MMP-9 is upregulated in a model of experimental autoimmune encephalomyelitis in MMP-2 null mice (Esparza et al., 2004). As a consequence of compensatory MMP-9 upregulation in lymphocytes and endothelial cells, MMP-2 null mice display an early onset of the disease with a worsened outcome. It is thought that increased lymphocyte transendothelial migration contributes to this poor clinical condition. In this study, a compensatory increase in MMP-9 activity could likewise contribute to the enhanced migratory behavior of MMP-2-deficient astrocytes.
It is noteworthy that MMPs can regulate cell motility dichotomously by proteolytic release of cryptic ECM fragments that either promote or inhibit cell migration (Giannelli et al., 1997; Hamano et al., 2003). A number of antiangiogenic factors, for instance, are encrypted in an extracellular microenvironment and become bioactive after MMP-mediated cleavage (Mott and Werb, 2004). In this regard, increased migration of astrocytes may be a consequence of an imbalance between inhibitory and promotive factors that regulate motility, with the latter assuming precedence in the absence of MMP-2.
Inhibitory CSPGs and MMPs
CSPGs, a group of proteoglycans that show differential inhibitory properties to neurite outgrowth, are expressed by reactive astrocytes in the glial scar (McKeon et al., 1999; Jones et al., 2003; Tang et al., 2003). Although CSPGs play a restrictive role and are essential for axonal guidance during normal development (Pindzola et al., 1993; Hsu et al., 2005), these inhibitory molecules form a chemical barrier that arrests regenerative axons in adults despite the absence of glial scarring (Davies et al., 1997). Enzymatic treatment of the injured spinal cord with chondroitinase ABC, which attenuates CSPG activity, markedly improves axonal regeneration and functional recovery (Bradbury et al., 2002; Chau et al., 2004). MMP-2 exhibits comparable proteolytic capability in decomposing certain CSPGs including neurocan (Muir et al., 2002), which is expressed at high levels after spinal cord injury (Jones et al., 2003; Tang et al., 2003). This protease enhances neurite outgrowth of dorsal root ganglionic neurons on peripheral nerve explants by degrading a still unidentified CSPG, which may otherwise inhibit laminin-promoted neurite extension (Zuo et al., 1998). Likewise, peripheral nerve segments, predegenerated by endogenous MMP-2 or neural stem cell-produced MMP-2, support axonal regeneration as a result of CSPG degradation (Krekoski et al., 2002; Heine et al., 2004).
In accordance with these studies, we show that CSPG immunoreactivity increases in spinal cord-injured MMP-2 null mice. Interestingly, this occurs despite the fact that there is a compensatory increase in MMP-9, a protease that is capable of processing CSPGs (NG2) in injured spinal cord (Larsen et al., 2003) and in degenerated peripheral nerve with an even higher effectiveness than MMP-2 (Ferguson and Muir, 2000). Similar to the pattern of glial scarring demonstrated by GFAP immunostaining, we found that the strongest CSPG immunoreactivity bordered the lesion area with relatively low immunoreactivity in the lesion core and less damaged tissue. Thus, extensive reactive astrogliosis in MMP-2 null mice may have generated CSPGs that exceeded the proteolytic activity of relevant MMPs.
Axonal plasticity, tissue sparing, and MMPs
We report a significant reduction in serotonergic axons caudal to the lesion in spinal cord-injured MMP-2 null mice, a finding consistent with a role for MMP-2 in axonal sprouting and regeneration. Extensive glial scarring, seen in MMP-2 null mice, should also be considered in this context. Indeed, mice deficient in both GFAP and vimentin, or with selective ablation of reactive astrocytes, show enhanced axonal sprouting and better functional recovery after injury as a result of reduced reactive astrogliosis (Bush et al., 1999; Pekny et al., 1999; Menet et al., 2003).
Fewer serotonergic axons caudal to the lesion may also reflect the degree of white matter sparing. There is reduced white matter in MMP-2 null mice. How MMP-2 promotes white matter sparing remains unclear. MMP-2 participates in wound-healing events such as angiogenesis that may favor sparing. The inflammatory cascade may also modulate white matter sparing. In this regard, MMP-2 can attenuate inflammation by cleaving chemokines such as monocyte chemoattractant protein-3 to terminate the chemotaxis of inflammatory cells (McQuibban et al., 2000).
MMPs are also implicated in Wallerian degeneration by proteolytic degradation of axonal components, myelin debris, and axon growth inhibitory CSPGs (Siebert et al., 2001; Hughes et al., 2002; Krekoski et al., 2002). Consistent with a beneficial role, delayed onset of Wallerian degeneration adversely affects locomotor recovery after spinal cord injury (Zhang et al., 1998). It is thus conceivable that MMP-2 deficiency results in ineffective removal of axonal debris leading to impaired functional recovery.
MMPs play diverse roles in the spinal cord. Their ability to support or inhibit normal cellular functions or participate in secondary pathogenesis likely depends on when and where they are expressed and their overall activity. We show that MMP-2 is active during wound healing, when it is preferentially expressed in reactive astrocytes. Genetic deletion of MMP-2 results in untoward wound healing, as evidenced by the formation of a more pronounced inhibitory glial scar and impaired recovery of motor function. Additional research, with the use of transgenic mice overexpressing MMPs and tissue inhibitors of MMPs, and pharmacological inhibitors to individual MMPs, should shed added light on their functions in both secondary pathogenesis and wound-healing events.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS39278 and R01 NS42717, the Roman Reed Fund from the State of California, and the Georgia Tech/Emory Center for the Engineering of Living Tissues. We are grateful to Nino Maida for his excellent technical assistance. We also thank Samuel Beckerman for his help with data analysis.
References
- Amberger VR, Avellana-Adalid V, Hensel T, Baron-van Evercooren A, Schwab ME. Oligodendrocyte-type 2 astrocyte progenitors use a metalloendoprotease to spread and migrate on CNS myelin. Eur J Neurosci. 1997;9:151–162. doi: 10.1111/j.1460-9568.1997.tb01363.x. [DOI] [PubMed] [Google Scholar]
- Anthony DC, Miller KM, Fearn S, Townsend MJ, Opdenakker G, Wells GM, Clements JM, Chandler S, Gearing AJ, Perry VH. Matrix metalloproteinase expression in an experimentally-induced DTH model of multiple sclerosis in the rat CNS. J Neuroimmunol. 1998;87:62–72. doi: 10.1016/s0165-5728(98)00046-0. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in the injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–2438. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Bjorklund M, Koivunen E. Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta. 2005;1755:37–69. doi: 10.1016/j.bbcan.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Chandler S, Coates R, Gearing A, Lury J, Wells G, Bone E. Matrix metalloproteinases degrade myelin basic protein. Neurosci Lett. 1995;201:223–226. doi: 10.1016/0304-3940(95)12173-0. [DOI] [PubMed] [Google Scholar]
- Chang C, Werb Z. The many faces of metalloproteases: cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001;11:S37–S43. doi: 10.1016/s0962-8924(01)02122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau CH, Shum DK, Li H, Pei J, Lui YY, Wirthlin L, Chan YS, Xu XM. Chondroitinase ABC enhances axonal regrowth through Schwann cell-seeded guidance channels after spinal cord injury. FASEB J. 2004;18:194–196. doi: 10.1096/fj.03-0196fje. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- Duchossoy Y, Horvat JC, Stettler O. MMP-related gelatinase activity is strongly induced in scar tissue of injured adult spinal cord and forms pathways for ingrowing neurites. Mol Cell Neurosci. 2001;17:945–956. doi: 10.1006/mcne.2001.0986. [DOI] [PubMed] [Google Scholar]
- Esparza J, Kruse M, Lee J, Michaud M, Madri JA. MMP-2 null mice exhibit an early onset and severe experimental autoimmune encephalomyelitis due to an increase in MMP-9 expression and activity. FASEB J. 2004;18:1682–1691. doi: 10.1096/fj.04-2445com. [DOI] [PubMed] [Google Scholar]
- Faden AI, Gannon A, Basbaum AI. Use of serotonin immunocytochemistry as a marker of injury severity after experimental spinal trauma in rats. Brain Res. 1988;450:94–100. doi: 10.1016/0006-8993(88)91548-x. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson TA, Muir D. MMP-2 and MMP-9 increase the neurite-promoting potential of schwann cell basal laminae and are upregulated in degenerated nerve. Mol Cell Neurosci. 2000;16:157–167. doi: 10.1006/mcne.2000.0859. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- Gijbels K, Masure S, Carton H, Opdenakker G. Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol. 1992;41:29–34. doi: 10.1016/0165-5728(92)90192-n. [DOI] [PubMed] [Google Scholar]
- Gijbels K, Proost P, Masure S, Carton H, Billiau A, Opdenakker G. Gelatinase B is present in the cerebrospinal fluid during experimental autoimmune encephalomyelitis and cleaves myelin basic protein. J Neurosci Res. 1993;36:432–440. doi: 10.1002/jnr.490360409. [DOI] [PubMed] [Google Scholar]
- Goussev S, Hsu JY, Lin Y, Tjoa T, Maida N, Werb Z, Noble-Haeusslein LJ. Differential temporal expression of matrix metalloproteinases after spinal cord injury: relationship to revascularization and wound healing. J Neurosurg Spine. 2003;99:188–197. doi: 10.3171/spi.2003.99.2.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton DW, Rhodes KE, Zhao C, Franklin RJ, Fawcett JW. The responses of oligodendrocyte precursor cells, astrocytes and microglia to a cortical stab injury, in the brain. Neuroscience. 2004;127:813–820. doi: 10.1016/j.neuroscience.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Heine W, Conant K, Griffin JW, Hoke A. Transplanted neural stem cells promote axonal regeneration through chronically denervated peripheral nerves. Exp Neurol. 2004;189:231–240. doi: 10.1016/j.expneurol.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Xu XM. Early profiles of axonal growth and astroglial response after spinal cord hemisection and implantation of Schwann cell-seeded guidance channels in adult rats. J Neurosci Res. 2005;82:472–483. doi: 10.1002/jnr.20662. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Stein SA, Xu XM. Temporal and spatial distribution of growth-associated molecules and astroglial cells in the rat corticospinal tract during development. J Neurosci Res. 2005;80:330–340. doi: 10.1002/jnr.20472. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Wells GM, Perry VH, Brown MC, Miller KM. Comparison of matrix metalloproteinase expression during Wallerian degeneration in the central and peripheral nervous systems. Neuroscience. 2002;113:273–287. doi: 10.1016/s0306-4522(02)00183-5. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25–34. doi: 10.1016/s0092-8674(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Jones LL, Tuszynski MH. Spinal cord injury elicits expression of keratan sulfate proteoglycans by macrophages, reactive microglia, and oligodendrocyte progenitors. J Neurosci. 2002;22:4611–4624. doi: 10.1523/JNEUROSCI.22-11-04611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci. 2002;22:2792–2803. doi: 10.1523/JNEUROSCI.22-07-02792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol. 2003;182:399–411. doi: 10.1016/s0014-4886(03)00087-6. [DOI] [PubMed] [Google Scholar]
- Kim GW, Gasche Y, Grzeschik S, Copin JC, Maier CM, Chan PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: role of matrix metalloproteinase-9 in early blood–brain barrier disruption? J Neurosci. 2003;23:8733–8742. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krekoski CA, Neubauer D, Graham JB, Muir D. Metalloproteinase-dependent predegeneration in vitro enhances axonal regeneration within acellular peripheral nerve grafts. J Neurosci. 2002;22:10408–10415. doi: 10.1523/JNEUROSCI.22-23-10408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NG2 proteoglycan. J Neurosci. 2003;23:11127–11135. doi: 10.1523/JNEUROSCI.23-35-11127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepekhin EA, Eliasson C, Berthold CH, Berezin V, Bock E, Pekny M. Intermediate filaments regulate astrocyte motility. J Neurochem. 2001;79:617–625. doi: 10.1046/j.1471-4159.2001.00595.x. [DOI] [PubMed] [Google Scholar]
- Levine JM. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci. 1994;14:4716–4730. doi: 10.1523/JNEUROSCI.14-08-04716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisian LM. The matrix-degrading metalloproteinases. BioEssays. 1992;14:455–463. doi: 10.1002/bies.950140705. [DOI] [PubMed] [Google Scholar]
- Matyash M, Matyash V, Nolte C, Sorrentino V, Kettenmann H. Requirement of functional ryanodine receptor type 3 for astrocyte migration. FASEB J. 2002;16:84–86. doi: 10.1096/fj.01-0380fje. [DOI] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD. Modulating astrogliosis after neurotrauma. J Neurosci Res. 2001;63:109–115. doi: 10.1002/1097-4547(20010115)63:2<109::AID-JNR1002>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- Menet V, Prieto M, Privat A, Gimenez y Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci USA. 2003;100:8999–9004. doi: 10.1073/pnas.1533187100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir EM, Adcock KH, Morgenstern DA, Clayton R, von Stillfried N, Rhodes K, Ellis C, Fawcett JW, Rogers JH. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res. 2002;100:103–117. doi: 10.1016/s0169-328x(02)00132-8. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Wrathall JR. Spinal cord contusion in the rat: morphometric analyses of alterations in the spinal cord. Exp Neurol. 1985;88:135–149. doi: 10.1016/0014-4886(85)90119-0. [DOI] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudega M, Vargas CG, Weber AB, Kleitman N, Bunge MB. Long-term effects of methylprednisolone following transection of adult rat spinal cord. Eur J Neurosci. 1999;11:2453–2464. doi: 10.1046/j.1460-9568.1999.00666.x. [DOI] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pindzola RR, Doller C, Silver J. Putative inhibitory extracellular matrix molecules at the dorsal root entry zone of the spinal cord during development and after root and sciatic nerve lesions. Dev Biol. 1993;156:34–48. doi: 10.1006/dbio.1993.1057. [DOI] [PubMed] [Google Scholar]
- Reier PJ. Gliosis following CNS injury: the anatomy of astrocytic scars and their influences on axonal elongation. In: Federoff S, Vernadakis A, editors. Astrocytes. Orlando, FL: Academic; 1986. pp. 263–324. [Google Scholar]
- Rhodes KE, Moon LD, Fawcett JW. Inhibiting cell proliferation during formation of the glial scar: effects on axon regeneration in the CNS. Neuroscience. 2003;120:41–56. doi: 10.1016/s0306-4522(03)00285-9. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine cortical cell culture. In: Tyson CA, Frazier JM, editors. In vitro biological methods. San Diego: Academic; 1993. pp. 46–60. [Google Scholar]
- Rosenberg GA, Dencoff JE, McGuire PG, Liotta LA, Stetler-Stevenson WG. Injury-induced 92-kilodalton gelatinase and urokinase expression in rat brain. Lab Invest. 1994;71:417–422. [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 1995;703:151–155. doi: 10.1016/0006-8993(95)01089-0. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Dencoff JE, Correa N, Jr, Reiners M, Ford CC. Effect of steroids on CSF matrix metalloproteinases in multiple sclerosis: relation to blood-brain barrier injury. Neurology. 1996;46:1626–1632. doi: 10.1212/wnl.46.6.1626. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Siebert H, Dippel N, Mader M, Weber F, Bruck W. Matrix metalloproteinase expression and inhibition after sciatic nerve axotomy. J Neuropathol Exp Neurol. 2001;60:85–93. doi: 10.1093/jnen/60.1.85. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. 2005;52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaga K, Kozlova EN. Role of intracellular S100A4 for migration of rat astrocytes. Glia. 2006;53:313–321. doi: 10.1002/glia.20284. [DOI] [PubMed] [Google Scholar]
- Tang X, Davies JE, Davies SJ. Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res. 2003;71:427–444. doi: 10.1002/jnr.10523. [DOI] [PubMed] [Google Scholar]
- Teng YD, Wrathall JR. Local blockade of sodium channels by tetrodotoxin ameliorates tissue loss and long-term functional deficits resulting from experimental spinal cord injury. J Neurosci. 1997;17:4359–4366. doi: 10.1523/JNEUROSCI.17-11-04359.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi A, Hsu JY, Lin Y, Goussev S, Gan J, Topp KS, Noble-Haeusslein LJ. The effects of acute and extended inhibition of matrix metalloproteinases on demyelination and functional recovery after spinal cord injury. Int J Neuroprotect Neuroregen. 2005;2:30–38. [Google Scholar]
- Wells JE, Rice TK, Nuttall RK, Edwards DR, Zekki H, Rivest S, Yong VW. An adverse role for matrix metalloproteinase 12 after spinal cord injury in mice. J Neurosci. 2003;23:10107–10115. doi: 10.1523/JNEUROSCI.23-31-10107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetstone WD, Hsu JY, Eisenberg M, Werb Z, Noble-Haeusslein LJ. Blood-spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. J Neurosci Res. 2003;74:227–239. doi: 10.1002/jnr.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrathall JR, Choiniere D, Teng YD. Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci. 1994;14:6598–6607. doi: 10.1523/JNEUROSCI.14-11-06598.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Guth L, Steward O. Mechanisms of motor recovery after subtotal spinal cord injury: insights from the study of mice carrying a mutation (WldS) that delays cellular responses to injury. Exp Neurol. 1998;149:221–229. doi: 10.1006/exnr.1997.6717. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zuo J, Ferguson TA, Hernandez YJ, Stetler-Stevenson WG, Muir D. Neuronal matrix metalloproteinase-2 degrades and inactivates a neurite-inhibiting chondroitin sulfate proteoglycan. J Neurosci. 1998;18:5203–5211. doi: 10.1523/JNEUROSCI.18-14-05203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]