

Abstract
Apoptosis plays an important role in eliminating UV damaged keratinocytes but its role in UV-induced immune suppression is not clear. Langerhans cells (LCs) may function as inducers of immune suppression. We have shown that LCs derived from mice deficient in the pro-apoptotic Bid (BH3-interacting death domain protein) gene (Bid KO) resist apoptosis and induce amplified immune responses. In this report, we examined responses in Bid KO mice to UVB exposure. Acute UV exposure led Bid KO mice to develop fewer apoptotic cells and retain a greater fraction of LCs in the epidermal layer of skin in comparison to wild type (WT) mice. Bid KO mice were also markedly resistant to local and systemic UV-tolerance induction to hapten sensitization and contact hypersensitivity (CHS) responses. Elicitation responses and inflammation at skin sensitization sites in UV-treated Bid KO mice were equal to or greater than non-suppressed control responses. In Bid KO mice, LCs accumulated in lymph nodes to greater numbers, demonstrated longer lifespans and contained fewer DNA damaged cells. These studies provide evidence that Bid activation is a critical upstream mediator in UV-induced keratinocyte and LC apoptosis and its absence abrogates UV-induced immune tolerance.
Keywords: Rodent, Skin, Dendritic Cells, Apoptosis, Immunosuppression
Introduction
The epidermis is our first line of defense against environmental damage, including highly mutagenic UVB irradiation (290 – 320 nm). Epidermal cells have evolved a highly regulated set of UV-damage responses that initially serve to both protect against the carcinogenic effect of UVB, by eliminating highly damaged cells through apoptosis, but at the same time retain and repair damaged cells to maintain skin barrier function (1). In addition to eliminating damaged cells, some molecular pathways of apoptosis are necessary for normal keratinocyte differentiation (2). Those that are not essential for homeostatic keratinocyte differentiation may be critical for certain stress-induced skin responses. One such molecule is the Bcl-2 family member Bid. While Bid protein expression is reportedly high in differentiated epidermis (3), Bid knockout (KO)3 mice manifest normal keratinocyte differentiation (4). However, since Bid is a potent proapoptotic molecule, it may be an important regulator of certain photodamage-induced responses.
As part of the “BH3-only” domain subset of proteins, Bid activates the intrinsic mitochondrial pathway of apoptosis (5). However, Bid is unique in that it links “extrinsic” death receptor signals to the “intrinsic” mitochondrial pathway. Bid activation results from death receptor induced caspase-8 mediated cleavage, producing a truncated form (tBid) that translocates to mitochondria where it efficiently activates the downstream pathways of apoptosis (6-8). Since death receptor activation plays a prominent role in developing ‘sunburn’ apoptotic cells (9, 10), and since Bid is highly expressed in differentiated keratinocytes, we reasoned that Bid may be a key mediator in this process. It is known that UV-induced sunburn cell formation is dependent on the expression of tumor suppressor p53 (11, 12), and p53 upregulates Bid gene transcription (13). Thus, Bid-dependent UV-induced responses may be upregulated in cells that normally do not express high levels Bid, such as normal primary basal cells. Indeed, Bid cleavage has been detected in human primary keratinocyte cultures in response to low doses of UV that potentiates further cleavage in the presence of TRAIL (14). While Bid cleavage has been detected in epidermal cell lines and keratinocyte cultures after UV exposure in vitro (15) (14), Bid’s role as a critical mediator of cutaneous cell apoptosis remains undefined.
Interestingly, besides the keratinocytes, epidermal Langerhans cells are also important targets of UV-induced damage. It has been observed that within 24 - 48 hours after UV exposure of epidermis, their cell numbers and dendritic morphology diminish (16-18). Further, the number of LCs able to reach the dLNs is also diminished. The loss of LCs may be due to apoptosis and/or inhibited migration to draining LNs (17, 19). It is also likely that LCs that do make it to the draining LNs have altered presentation function that promotes the development of regulatory T (Treg) cells (20-22). The altered LC function may be due to direct DNA damage by UVB, (which can be identified by antibodies to pyrimidine dimers), or due to indirect immune modulation in response to keratinocyte-derived signals, such as soluble mediators like platelet activating factor, cis-urocanic acid, IL-10 or expression of TNF-related activation-induced cytokine (TRANCE), the ligand for receptor activator of NFκB (RANK), that is produced by UV-damaged keratinocytes (23-26). Direct DNA damage has been shown to correlate with UV immune suppression. This was shown in a series of experiments in which topical liposomal delivery of bacterial restriction enzymes induced IL-10 expression and inhibited CHS responses (27, 28), while application of photolyase (which efficiently repairs pyrimidine dimers) inhibited local immunosuppression and blocked induction of Treg cells by UV-treated cutaneous APCs in vitro (29). However, these experiments do not directly assess the role of LC apoptosis in regulating UV-induced immune suppression.
In our previous studies, we showed that Bid KO mice could promote enhanced contact hypersensitivity to hapten, and isolated LCs were resistant to apoptosis and able to induce enhanced immunity when transferred to naive recipients (30). Because LC function figures prominently in the mechanism of UV-induced immune suppression, we investigated whether Bid KO mice demonstrated altered responses to UV-induced damage by keratinocytes and LCs. Our results indicate that epidermal keratinocytes and LCs depend on Bid to mediate UV-induced apoptosis. Furthermore, UV-induced immune suppression and tolerance are blocked in Bid KO mice and this correlated with increased LC accumulation within draining LNs. Our data strongly support that Bid-dependent LC apoptosis is a critical response in the mechanisms of immune downregulation following exposure to UV radiation (UVR).
Materials and Methods
Mice
Bid KO mice were bred on a C57BL/6 genetic background and generously provided by Dr Stanley L. Korsmeyer (Harvard, Boston MA). Aged and sex-matched (8 - 12 weeks) C57BL/6 mice, termed wild type (WT), were purchased from Jackson Laboratories (Bar Harbor, Maine USA). The institutional review board approved all animal protocols.
Chemicals and monoclonal antibodies
Ammonium thiocyanate, 2,4 Dinitrofluorobenzene (DNFB), olive oil, BSA, acetone reagent grade, sodium hydroxide, sodium chloride, were purchased from Sigma-Aldrich (St. Louis, MO). Monoclonal anti-cyclobutane pyrimidine dimers or an isotype control antibody (IgG1) was purchased from Kamiya Biomedical Company (Seattle, WA). Monoclonal antibody specificities: I-A (2G9), CD11c (HL3), CD8a (53-6.7), Streptavidin-PerCP from BD-Biosciences Pharmingen (San Jose, CA), CD207 (Langerin) (clones 205C1, 929F3) from Dendritics (Abcys Biologie, Paris, France) or eBiosciences (eBioRMUL.2) (San Diego, CA). Peroxidase-conjugated steptavidin, DAB kit (Vector Laboratories Inc., CA). DeadEnd Colorimetric TUNEL System from Promega Corp. (Madison, WI). Dispase II and Collagenase D were purchased from Roche Applied Sciences (Indianapolis, IN). Fluromount G (Southern Biotech, AL). Attane isoflurane (supplied through Animal Resource Program UAB, AL).
UVB irradiation
The dorsal sides of mice were shaved with electric clippers one day prior to UV exposure. Control groups of “mock” treated mice were not exposed to UVB but were also shaved. UVB irradiation was done on anesthetized mice, and their eyes and ears were covered to protect from UVB exposure. A Daavlin, UVA/UVB Research Irradiation Unit (Bryan, OH), equipped with an electronic controller to regulate UV dosage was used to emit UV radiation from a band of four FS20 UVB lamps. The UV lamps emit UVB (280–320 nm; 80% of total energy) and UVA (320–375 nm; 20% of total energy), with very little UVC emission (<1%). The majority of the resulting wavelengths of UV radiation are in the UVB range (290–320 nm) with peak emission at 314 nm. The UVB emission was also monitored with an IL-1700 research radiometer (International Light, Newburyport, MA).
Histology
Midline dorsal skin, or ears were excised promptly after euthanasia and placed immediately in 70% alcohol buffered formalin. The tissues were fixed for 24 hours in formalin and then embedded in paraffin. Sections of 6-μm thickness were cut and processed (UAB Skin Diseases Research Core Facility University of Alabama at Birmingham) for hematoxylin and eosin (H&E) staining to identify sunburn cells, TUNEL staining for apoptosis, or CPD staining (data not shown) on serial sections. Stained slide sections were examined by bright-field microscopy and images captured with an Olympus DP70 digital camera and further analyzed using ImagePro Plus software v6.0 (Media Cybernetics, Inc., Silver Springs, MD).
Detection of apoptotic cells in epidermis
Paraffin embedded tissue sections on the slides were immersed in the xylene (Sigma) twice for 5 minute each, to remove paraffin and rinsed by 100% ethyl alcohol for 5 minute. Tissue sections on slides were rehydrated through ethanol gradient in decreasing concentration (100%,95%,85%,70%, &50%) and washed by 0.85% of NaCl solution for 5 minutes. Tissues were fixed in 4% paraformaldhyde followed by two washes in PBS for 5 minutes each, then treated for 20 minutes in proteinase K solution (Promega Kit) with 20 μg/ml 100 μl /tissue to permeabilize the tissue and permeablized further in Triton-X buffer. Tissue sections were incubated in equilibration buffer for 10 minutes at room temperature, then labeled with rTDT enzyme and biotin-dUTP (Promega Kit) reaction mix 100 μl/sample during 60 minute incubation at 37°C in the humidified chamber (negative control was treated with reaction mix except rTdT enzyme). After incubation, slides were immersed for 15 minutes in 2X SSC solution and washed with PBS for 3 times for 5-minutes. Endogenous hydrogen peroxide was blocked by immersing the slide in a 3% H2O2 (Sigma) solution for 5-10 minutes and was then washed with PBS twice for 5 minutes each. Tissue sections were incubated for 30 minutes at RT in 100 μl per sample of streptavidin HRP (1:500) in PBS. Slides were rinsed with PBS 3 times for 5 minutes each and stained with 100 μl/sample DAB freshly prepared solution for 3 minutes until a brown color developed. Sections were rinsed in deionized water 3 times. Tissue sections were mounted with Crystal/Mount (Biomeda Corp. Foster City, CA). Images were captured at 20× and 40× magnification, and 10 fields per section, 3 sections per animal, and 3 animals per treatment were analyzed in a blinded fashion for quantification of TUNEL positive cells.
Staining of Langerhans cells in epidermis
Aged match females of WT and BID KO were anesthetized and exposed to varying doses of UVB as indicated. After 24 hours, ears were harvested from euthanized mice and separated into dorsal and ventral halves. Ear halves were floated, dermis side down, on a 0.5% ammonium thiocyanate solution (Sigma) for 30 minutes at 37°C. Dermis was separated from epidermis, gently rinsed in PBS then fixed in cold acetone for 30 minutes on a rotator at 4°C. Acetone was replaced every 10-minutes followed by 3 washes with cold PBS. Sheets were placed in a blocking antibody solution at a concentration of 4 μg/ml per sample (clone 2.4G2 hybridoma form ATCC) for 30 minute in 1% BSA at room temperature on a rotator. After the 30 minute incubation, FITC-conjugated anti I-A antibody or FITC-isotype control was added at 1 μg/sample (clone 2G9, BD Pharmingen) and incubated in the dark overnight on a rotator at 4°C. The next day sheets were washed 3 times in PBS then mounted in Flouromount-G (Southern Biotech, Birmingham, AL). Fluorescent images were captured at 20× or 60× magnification and analyzed with MetaMaorph Software (Universal Imaging Corp. Downington, PA) provided by the UAB Imaging Core Facility or captured with a DP70 digital camera (Olympus) and further analyzed using ImagePro Plus software v6.0 (Media Cybernetics, Inc., Silver Springs, MD). At least 3 sheets per treatment group were analyzed, and 10 - 20 fields per sheet imaged for enumeration. The number of I-A positive cells was enumerated after setting a standardized green intensity threshold to enhance contrast and reduce background staining.
Western blot analysis
Cytoplasmic fractions were obtained following manufacturers instructions of a Cytoplasmic Extraction Kit (#78833 from Pierce Biotechnology, Inc, Rockford IL). This kit stringently clears supernatants of mitochondria. Blots of 10% SDS-PAGE gels loaded with 20 ± 5μg protein were first blocked for 1 hour with 5% non-fat milk TBST, then incubated further with antibodies to β-actin (1:5,000; mouse mAb #A5316, Sigma) and cytochrome c (1:500; mouse mAb #13156, Santa Cruz) overnight at 4°C. HRP-conjugated goat-anti mouse antisera (1: 8,000; #31400 Pierce) was incubated on for 1 hour at RT. ECL treated membranes were wrapped in plastic and exposed to X-ray film for 45 seconds (actin) and 1.5 minutes (cytochrome c). The quantitative analysis was performed on a Macintosh computer using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Cutaneous migratory DCs from split ear cultures
Aged match female mice (WT and BID KO, n=5/group) were anesthetized and exposed to UVB at 120mJ/cm2. Control mice had their ears tape-stripped 10× on both dorsal and ventral sides to activate LC migration. After 4 hours, mice were sacrificed and ears recovered and split into dorsal (UV exposed) and ventral (non exposed) sides to float in culture as described previously (30). After 3 days of culture, cells were counted in trypan blue to determine cell recoveries and viability, then adjusted to 4×105 cells/ml PBS. Samples of 100 μl were subjected to cytospin centrifugation at 1500 rpm for 5 min. Cytopsin slides were fixed in cold acetone for 10 min and stored at -20°C until staining. Immunofluorescence assays for enumeration of CPD+ in migratory DCs.
To detect CPD positive cells in migratory cells from the UVB exposed ears, immunostaining was done as described previously (31), with a slight modification. Briefly, fixed cytospin slides were rehydrated in PBS for 10 min. DNA was denatured using 70 mM NaOH in 70% ethanol for 2 min, followed by neutralization with 100 mM Tris-HCl (pH 7.5) in 70% ethanol for 1 min. Slides were washed with PBS 2 times for 5 min each and then incubated for 30 min with 10% goat serum in PBS to block non-specific binding. Slides were then incubated with anti-CPD mAb (0.5 μg/slide, Kamiya) or an isotype control (murine IgG1) for 1 hr and washed with PBS 3 times for 5 min each. Then, slides were incubated with FITC-conjugated rat anti-mouse IgG for 30 min and washed with PBS 3 times for 5 min each, followed by incubation with fluorescence dye 4’, 6’-diamidino-2-phenylindole (DAPI) for nuclear staining. A parallel set of cytospin slides were stained with anti-MHC II (I-A) Ab. Slides were incubated with an anti-CD16/CD32 Ab (2.4G/2) and 3% BSA to block non-specific binding and then stained with FITC-labeled anti-I-A (clone 2G9, BD Biosciences. Slides were counterstained with DAPI, washed then cover slipped over Gel/Mount medium (Biomeda). Photomicrographs were taken with a 20× objective. CPD-positive or IA positive cells were counted in 12 or 5 fields, respectively, for each group. The mean and standard deviations were calculated and the difference between groups was analyzed statistically using the Student’s t-test.
Local immune suppression of contact hypersensitivity (CHS)
Mice (WT or Bid KO) were exposed to UVB at 100mJ/cm2 daily for 4 consecutive days on the shaved dorsal skin. Ears were protected during UVB exposure. On day 5, mice were sensitized on UV or mock treated dorsal skin with 25 μl of either vehicle (4:1 Acetone and olive oil) or 0.5% DNFB emulsified in vehicle. On day 5 post sensitization mice were challenged with DNFB antigen by painting ears with 10 μl of 0.3% DNFB on the dorsal and ventral side of each ear. CHS swelling responses were measured each day with an engineer’s micrometer (Mitutoyo Precision, USA Inc., Elk Grove Village, IL). The averages of 3 readings per ear over the base line measurement for each ear were recorded daily during the indicated period and expressed in μm. Groups of at least 5 mice were measured per treatment.
UV-induced tolerance induction to hapten
Mice (WT or Bid KO) were exposed to UVB at 100mJ/cm2 daily for 4 consecutive days on the shaved dorsal skin. Ears were protected during UVB exposure, unless otherwise noted. On day 5, mice were initially sensitized on UV or mock treated dorsal skin with 25 μl of either vehicle (4:1 Acetone and olive oil) or 0.5% DNFB emulsified in vehicle (S1). Two weeks post sensitization (day19), mice were sensitized again (S2) on unexposed shaved ventral skin (abdomen) with 25 μl of 0.5% DNFB. After 5 days, (d24) mice were challenged with 10 μl of 0.3% DNFB on the dorsal and ventral side of each ear. CHS swelling responses were measured each day with an engineer’s micrometer as above. Groups of at least 5 mice were measured per treatment and the specific ear swelling responses are reported as the difference in ear thickness from baseline measurements obtained for each ear.
Detection of CPD positive Langerhans cells in vivo
UVB-induced DNA damage in Langerhans cells was determined using procedure described previously (32). WT or BID KO mice were shaved dorsally with electric clipper on day -1. Mice had both backs and ears exposed to 100 mJ/cm2 UV for 4 consecutive days. On day 5, backs and ears were painting with vehicle only or 0.5% DNFB in vehicle as described. After 3 days mice were sacrificed and superficial parotid (ear draining) and subilliac (often referred to as inguinal) LNs (see ref(33) for nomenclature) were pooled per animal (n=3 per test group). LNs were digested by collagenase D (2 mg/ml in HBSS containing HEPES buffer, Roche) for 30 minutes at 37°C. RPMI-10%FBS medium was added to neutralize Collagenase D and filtered through 150μm nylon mesh. Cell suspensions from each mouse were washed, counted then stained for flow cytometric analysis. Cells (5 × 106 per sample) were blocked with hamster and goat gamma globulin (Jackson ImmunoResearch, West Grove PA) (5μg/sample) then stained with 0.5 μg PE-CD11c or PE-Hamster isotype. Washed cells were treated with Fix/Perm solution, then stained in Perm/Wash solution with 0.2μg Alexa 647-CD207 or 0.5 μg CPD mAb or isotype controls for 30 minutes on ice as per BD-Biosciences intracellular staining protocol. FITC-goat anti-mouse (1:100 dilution, Jackson ImmunoResearch) in Perm/Wash was added to washed samples and incubated another 30 minutes on ice. Cells were washed in PBS and fixed in 1% paraformaldehyde prior to flow cytometric analysis.
FITC painting to track skin-derived cells in LN. Mice (n=3/group) were painted on the dorsal side of ears, with 10μl of 0.5% FITC in vehicle (acetone:dibutylphthalate, 1:1), or vehicle alone. After 2 hours, mice were exposed to daily doses of 0, 120, or 400 mJ/cm2 UVB for 4 days. Parotid, ear-dLNs were harvested 3 days later, and pooled for each individual mouse. Single cell suspensions were counted and 5×106 cells per sample were stained with PE-anti-CD11c and CD8a. Multicolor flow cytometric analysis was performed on 106 cells per sample. Student’s t-test was applied to data for determining significant differences between groups.
Statistical Analysis
Paired student’s t-test (two-tailed, unless noted otherwise) was performed using Prism 4.0 for Mac (GraphPad Software, Inc. San Diego CA) or Excel (Microsoft) software to calculate significant differences. Significant values are reported in the text and figure legends.
Results
UV-induced apoptosis is reduced in Bid KO epidermis
UV radiation leads to morphological and functional alterations in epidermal cells, including apoptosis. To determine whether Bid KO mice sustained more or less UV damage than WT mice, we compared levels of apoptosis in the epidermis from both strains 2 days after UV exposure. Damaged cells were identified as sunburn cells by H&E or apoptotic cells by TUNEL staining. Figure 1a of H&E stained ear skin sections revealed normal structural integrity of Bid KO epidermis, indicating that Bid is not required for normal keratinocyte differentiation. More sunburn and TUNEL positive (+) cells (black arrows) were identified in UV-treated WT epidermis than Bid KO epidermis. The number of TUNEL+ cells that developed in the epidermis after exposure to different doses of UV were compared (Figure 1b). The mean numbers of TUNEL+ cells/mm2 ± SEM for WT versus Bid KO epidermis were as follows: 36 ± 2 versus 22 ± 2 (n=38) and 54 ± 5 versus 30 ± 2 (n=65) for 100 and 200 mJ/cm2 doses, respectively. Therefore, the level of UV-induced apoptosis in Bid KO epidermis was reduced by 38% and 44%, respectively. In other studies performed with cultures of primary skin keratinocytes, we observed that Bid KO cells maintained resistance to UV-induced apoptosis at in vitro doses up to 100 mJ/cm2, as compared to WT cells, over longer times (eg. 3 days in culture) (data not shown). These data suggest that Bid KO cells are not delayed in their apoptotic response. Bid KO cells also demonstrated reduced levels of cytochrome c release in the cytoplasmic fraction, as compared to WT derived samples, where 2-fold over control levels were measured 4 hours after UVB exposure. (Figure 1c and d). However, at higher doses of UVR (up to 200 mJ/cm2) necrotic cell death was observed at levels that were not significantly different between Bid-deficient and WT UV exposed cells (data not shown). Collectively, these data indicate that Bid expression is not required for UV-induced necrosis, but is critical for mediating UV-induced apoptosis in epidermal keritinocytes.
Figure 1.
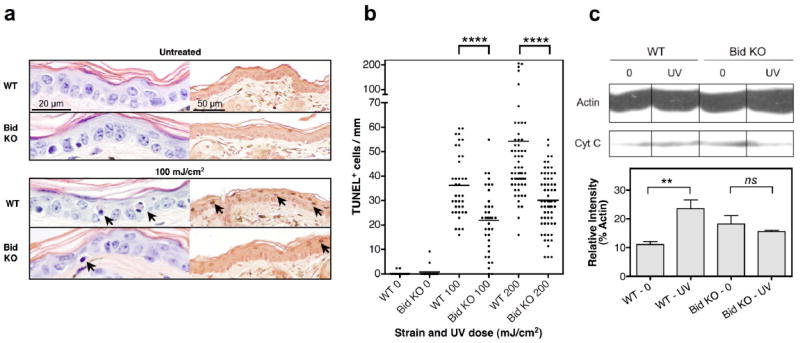
Resistance to UVB-induced apoptosis is exhibited in Bid KO skin. Ear skin samples were examined 2 days following a single UV dose, as indicated, (a) Histology of UV exposed skin reveals fewer sunburn and TUNEL+ cells in the epidermis of Bid KO mice. Mice were untreated (top panel) or treated with 100mJ/cm2 UV light (bottom panel). Representative H&E sections are shown in the left panel set, where sunburn cells (black arrow) were detected. The scale bar in the top left panel represents 20 μm on images originally captured at 60× magnification. Representative TUNEL stained sections are shown in the corresponding right panel, where TUNEL+ apoptotic cells (black arrow) were detected. The scale bar in the top right panel represents 50 μm on images originally captured at 20× magnification, (b) Scatter plot distribution of apoptotic TUNEL+ cells. TUNEL+ cells were enumerated in multiple views along the entire length of each sample at 20× magnification (437 μm). The same number of 20× views per section, per animal (≥ 3 mice/ treatment) was compared, and is shown cumulatively per group (n= 18, 36 and 60 for control, 100 and 200 mJ/cm2 respectively). Paired two-tailed student’s t-test p-values are shown. Cumulative results from 3 independent experiments are shown. (c) Cytoplasmic cytochrome c levels in WT and Bid KO mice. Sub-confluent cultures were exposed to 120 mJ/cm2 UVB, cultured further for 4 hours then prepared for analysis of cytoplasmic cell fractions by western blot analysis. Twenty μg of protein was loaded per sample, and blots labeled with anti-actin and cytochrome c antibodies. Film exposed for 1.5 minutes is shown. (d) Relative levels (integrated intensity) of cytochrome c and actin bands was determined using NIH Image 1.63 densitometry analysis software. Levels of cytochrome c released in the cytoplasm for each sample is expressed as % of the corresponding control actin band. Three measurements per sample are shown as the mean ± SD. Student’s t-test p value: **, 0.01; ns, no significant difference.
Epidermal LC densities are retained in Bid KO mice after UV-exposure
Since LC loss is an important marker of epidermal UV-damage responses, we compared the number of remaining LCs in WT and Bid KO mice ear skin 2 and 7 days after a single exposure to various doses of UVB radiation (0 - 400 mJ/cm2). LC depletion increased in a UVB dose-dependent manner. However, the extent of depletion was significantly reduced in UV exposed Bid KO epidermis as compared to WT epidermis at all UVB doses tested on day 2 (Figure 2b, left panel), and on all but the highest UVB dose (400 mJ/ cm2) on day 7 (Figure 2b, right panel). Notably, the morphology of Bid KO LCs was enlarged as compared to WT LCs after exposure to low dose UVB (50mJ/cm2) and was easily observed on day 7 suggesting that Bid KO LCs were activated as opposed to undergoing apoptosis. The morphology of WT LCs differed strikingly from Bid KO LCs on day 7. LCs developed enlarged rounded cell bodies with blebbing (see WT inset at 100mJ/cm2 in Figure 2a), as well as diffuse smeary (WT 50mJ/cm2) or blebby dendritic processes (WT 200mJ/cm2), providing morphological hallmarks of apoptosis in situ (indicated by the white arrow). We also noted that the tissue integrity of epidermis was maintained in Bid KO, but became quite fragile in WT mice after larger UVB exposure doses. Continued depletion of LCs was observed after 7 days for both strains at the high UV doses (200-400 mJ/cm2), but at low UV doses (50 mJ/cm2) Bid KO LC numbers remained stable while WT LC numbers declined. These findings suggest that most of the UV-induced LC depletion (migration out of the epidermis or apoptosis) occurs within 2 days, and continuous apoptosis may occur thereafter. However, the Bid KO LCs remaining in epidermal layers 7 days following UV treatment displayed cell morphologies consistent with cell activation, indicating they were resistant to UV-induced apoptosis.
Figure 2. Bid KO LCs resist UV-induced epidermal depletion and apoptosis.
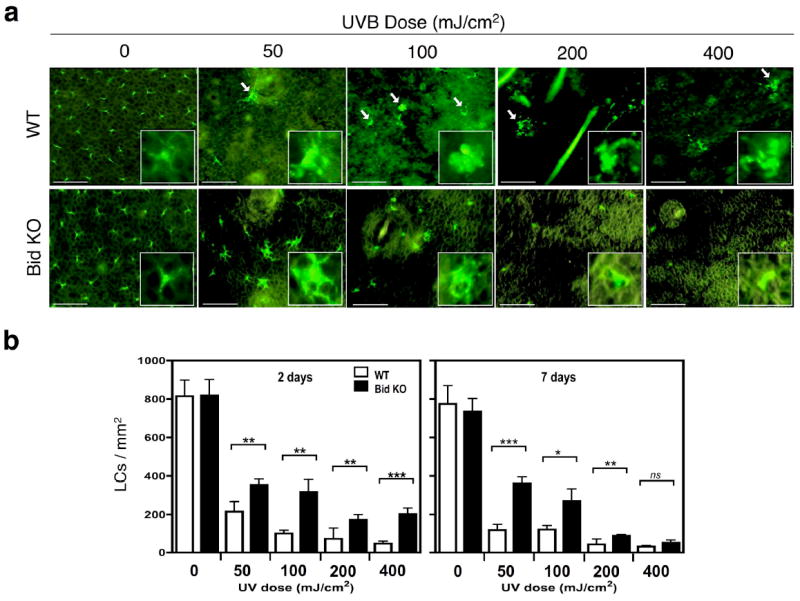
Whole mount epidermal ear skin samples were obtained 2 and 7 days after treatment and stained for I-A expression (FITC+). (a) Visualization of LCs within WT and Bid KO epidermal sheets after 7 days. Bar indicates 50μm of images captured at 20× magnification. Inset figures display 14.7μm × 14.7μm enlargements of a representative cell in the corresponding view. White arrows point out apoptotic cell bodies seen in WT samples, but rarely in Bid KO samples. (b) Enumeration of LC densities in response to UV dose on days 2 and 7 after exposure. The numbers of I-A positive cells were determined in 6 views (20×) per specimen (3 specimens per group) and the average used to calculate the density/mm2. Student’s t-test was performed on the cumulative views per group (n=18) with the following P values: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Results from one of 3 independent experiments are shown.
DNA damaged DCs from WT and Bid KO mice exhibit identical capacities for migration from UV-exposed skin explant cultures
The different levels of LC depletion seen in the epidermis of UV-exposed WT and Bid KO mice suggested that we would observe correlative differences in the number of photodamaged cells that migrate from the skin of these two strains. Therefore, we examined migratory cells derived from ear skin harvested 4 hours after single dose of 120 mJ/cm2 UVB radiation. Skin subjected to tape-stripping to activate LC migration served as a UV-negative control, and a positive control for LC migration. Ears were split and cultured in groups of dorsal skin (fully exposed to UVR and an indicator of direct UV damage) and ventral skin (a control for partial UV exposed skin) for 3 days, then samples were pooled for each treatment group and assessed for total cell numbers, viability, CPD positive cells and I-A-positive cells. We observed that dorsal skin cultures accumulated similar total numbers of cells, (Figure 3a and b) (7.9 × 103 cells and 9.5 × 103 cells per dorsal side respectively for WT and Bid KO samples), as well as numbers of CPD+ and I-A+ cells from both mouse strains. Interestingly, we observed a significant increase in the both the number of dead cells (Figure 3b, bottom left panel) and CPD+ cells recovered from WT cultures of ventral ear skin as compared to cells from parallel cultures of Bid KO ventral skin (Figure 3b). Whether the increase in both dead and CPD+ subsets is related remains to be determined. All CPD+ staining was located in the nuclei of cells, consistent with direct photodamage. We failed to detect CPD staining in the cytoplasm of migratory cells, which might identify cells that were not directly damaged but had secondarily incorporated photodamaged cell DNA. Thus, our staining technique is limited to detecting cells that have sustained direct DNA photodamage. Collectively, data from UV-treated dorsal skin cultures indicate that WT cutaneous DCs are as viable and as active as Bid KO DCs during this 3-day time span after a single dose UVB treatment. This observation is consistent with our data presented in Figure 2, showing that LC apoptosis is only detected in the epidermis late (5 – 7 days) after UVB treatment.
Figure 3.
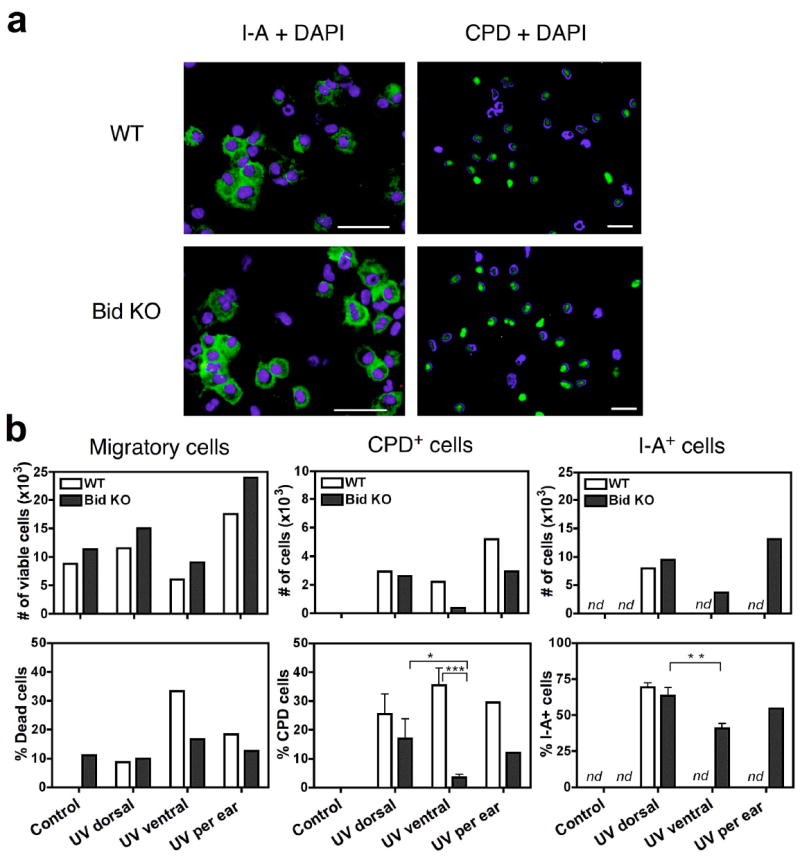
Equivalent numbers of CPD+ and I-A+ cells migrate from UV exposed dorsal ear skin in WT and Bid KO mice. Four hours after treatment with UVB 120mJ/cm2, mice were sacrificed and ear skin was harvested for ex vivo culture. Dorsal (UV-exposed) and ventral (variable UV exposure) split skin samples were cultured for 3 days. Pooled cells for each group were assessed for cell counts and viability prior to making cytospin slides. (a) Fluorescence microphotographs are shown for sets of slides stained for I-A or CPD (green) and nuclei with DAPI (blue). Merged images demonstrate that CPD staining is detected only in the nuclei of cells. I-A is detected only in the cytoplasm. Images were captured at 20× magnification, 50 μm bar is shown. (b) The number of I-A+ and CPD+ cells in migratory populations from dorsal skin cultures are equivalent for UV treated Bid KO and WT strains. Student’s t-test p value designations are *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. nd, not determined (due to low cell yields from WT).
Bid KO mice accumulate greater numbers of LCs in draining LNs after 4 days of UV exposure
Because Bid KO LCs exhibited resistance to UV-induced apoptosis in situ, we investigated whether the numbers of LCs found in draining LNs might also differ between UV-treated WT and Bid KO animals. Mice were exposed to 100mJ/cm2 UV on 4 consecutive days, then draining LNs harvested for the following consecutive 3 days to enumerate LCs by flow cytometric analysis (Figure 4). Consistent with our previous findings (30), steady-state LC numbers were the same in untreated mice from both strains (50,000 LCs/LN, Figure 3b bottom panel, Day 0). A modest difference in the percent of Langerin+ cells was observed (7.4% versus 5.3% in Bid KO versus WT, Figure 4a) on day 0, but no consistent differences in the percentage of CD11c levels were observed at any time (Figure 4b). However, opposite patterns of cellular composition were observed in LNs from these two mouse strains following UV exposure. One day after the final UV dose (Day 1), increases in both the total numbers of LN cells (from 7.4 × 106 to 11.3 × 106 cells per LN) and the percent of Langerin+ cells resulted in a 3-fold increase in LC numbers (in comparison to steady state levels) within Bid KO LNs. In contrast, within WT LNs, total numbers of LN cells declined modestly and the percent of Langerin+ cells dropped 2-fold resulting in a 1.7-fold decrease in LC numbers. On the following day (Day 2), the numbers of LCs from both strains dropped but at different rates, decreasing from Day 1 levels by 50% and 70% for Bid KO and WT, respectively. On Day 3, Bid KO LC numbers dropped 30% to control levels, while WT LC numbers rebounded 2-fold achieving 50% of control levels. Mechanisms that may contribute to the increased numbers of Bid deficient LCs in LNs include a greater capacity to (1) migrate to dLNs, (2) recruit Langerin+ cells from non-epidermal sources and/or (3) resist mechanisms of DC attrition observed in LNs of UV-exposed WT mice.
Figure 4.
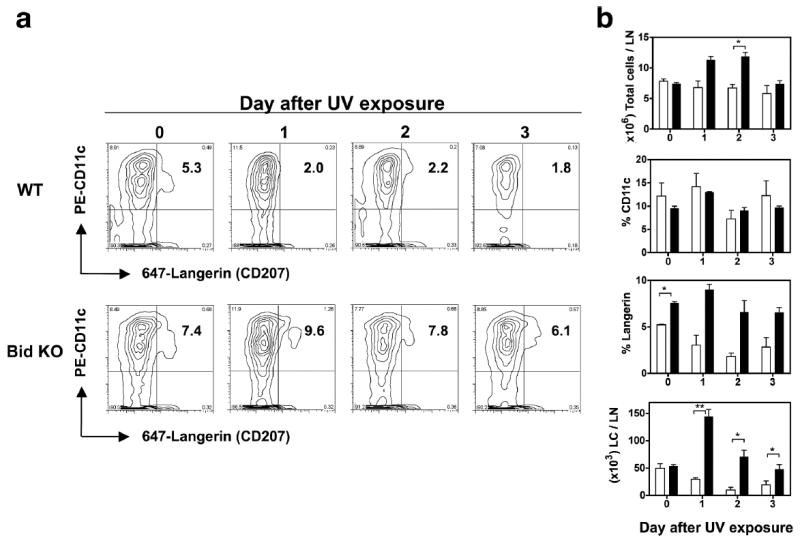
Enhanced accumulation of LCs detected in skin dLNs of UV exposed Bid KO mice compared to WT mice. Mice were irradiated with 100 mJ/cm2 UVB (on ears and shaved backs) daily for 4 days. Mice were sacrificed on the days indicated following the last UV dose, and draining LNs (6/mouse, 2 mice/group) were pooled and prepared for flow cytometric analysis. Data from untreated control mice is shown as day “0” samples. (a) Contour plots of LN cells stained with PE-CD11c and Alexa647-Langerin. Upper right quadrant number indicates % Langerin+ cells within the CD11c subset. (b) Kinetics of LC cell accumulation differs markedly in UV exposed Bid KO (black bars) versus WT mice (white bars). Total cell numbers, % CD11c, % Langerin and number of Langerin+ cells (LCs) per node were determined. Mean ± SD for 2 mice/group are shown. Values from duplicate animals showed significant differences between strains as shown (t-test. *, p< 0.05). One of 2 similar experiments is shown.
Bid KO mice are resistant to UV-induced local immune suppression
To test if resistance to UV-induced apoptosis would alter UV-induced local immune suppression, we subjected Bid KO and WT mice to 100 mJ/cm2 of UVB daily for 4 consecutive days on shaved backs, while ears were protected. The day following the last UV dose, dorsal skin was sensitized with hapten by painting with 0.5% DNFB or vehicle alone as a control. Five days post sensitization, ears were treated with hapten by applying 10μl 0.3% DNFB per side and the elicitation of a swelling response due to contact hypersensitivity (CHS) was measured each day following (Figure 5). An enhanced CHS response was observed in Bid KO mice as compared to WT mice under normal conditions, as described previously (30). However, while UV exposure completely abrogated the CHS response in WT mice, only a modest reduction in CHS was observed in Bid KO mice.
Figure 5. Impaired local UV-suppression of CHS responses in Bid KO mice.
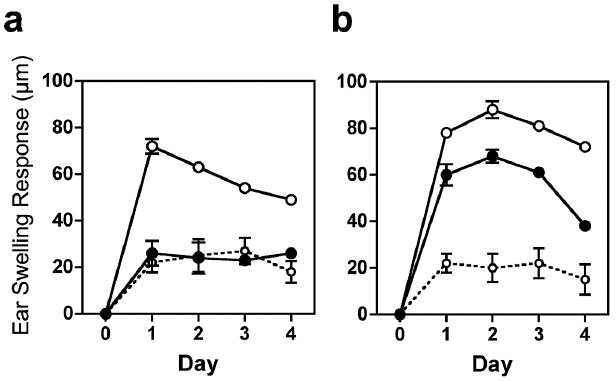
Mice were exposed to daily UVB for 4 days to induce local suppression to DNFB sensitization (as outlined in Materials & Methods ears were shielded). After 5 days, ears were painted with DNFB (0.3%) and specific ear swelling responses (CHS) were measured as differences from baseline ear thickness in μm. CHS responses are shown for WT mice (a), and Bid KO mice (b). DNFB (0.5%) was applied on normal shaved backs (open circles), UVB exposed shaved backs (closed circles), or vehicle was applied UVB treated backs (open circles with dotted lines) as a control. Four to 5 mice (8 – 10 ears) were measured per CHS group, while control groups had 2 mice (4 ears) per group. The mean and SEM values are shown. SEM values were usually smaller than the width of the circle). Results from one of 2 similar experiments are shown.
Bid KO mice do not develop UV-induced tolerance to hapten specific-responses
We next tested whether the Bid deficiency might alter susceptibility to UV-induced tolerance. Following well established protocols (32), the dorsal skin of UV-treated mice (UVB 100 mJ/cm2 for 4 consecutive days) was sensitized with 0.5% DNFB (S1) or vehicle alone as control. Two weeks later the same mice were secondarily sensitized (S2) on the ventral side (UV untreated) with 0.5% DNFB or vehicle alone, as indicated (Figure 6). UV-protected mouse ear skin was challenged 5 days later with 0.3% DNFB and CHS responses were measured by specific ear swelling responses on day 2. Striking differences in appearance of dorsal skin from UV tolerized WT and Bid KO mice were observed. Bid KO mice that underwent the UV-tolerizing regime (Group 3) develop inflamed lesions at sites of hapten sensitization especially on UV-treated dorsal skin (indicated by the white arrows in Figure 6a), while WT mice did not. This correlated with dramatic differences in ear swelling responses (Figure 6b,c). Increased ear swelling responses were observed by Bid KO mice as compared to WT mice for all treatment groups, outside of the control group. The ear thickness measured for WT versus Bid KO per treatment group are listed as follows (Mean specific ear swelling response in μm ±SEM, n=10): Control, 20 ± 2.4 versus 22 ± 3.4; Group1, 69 ± 3.4 versus 98 ± 2.0; Group 2, 49 ± 3.8 versus 60 ± 3.6; Group 3, 27 ± 4.2 versus 72 ± 5.0, respectively. In Group 3, the WT swelling response was minimal, attaining only a 7 μm increase over control (non-specific response) levels, while the Bid KO response swelled to 50 μm over control levels, a 7-fold greater specific CHS response than WT. These results demonstrated that Bid KO mice were unable to develop UV-induced tolerance to hapten.
Figure 6. Bid KO mice are resistant to UV-induced tolerance induction.
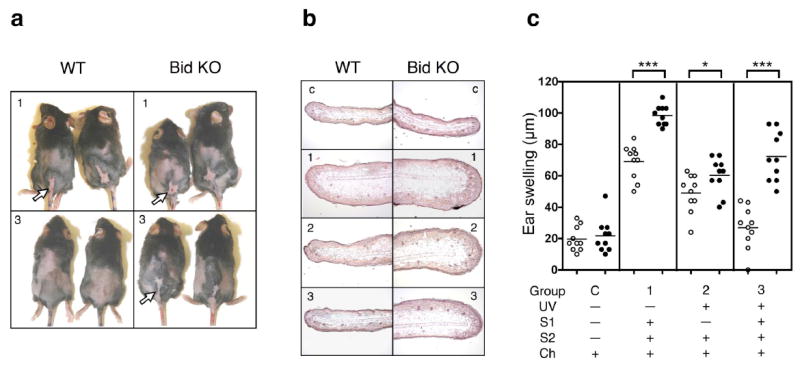
Mice were subjected to UVB for 4 consecutive days (with ears shielded, as outlined in Materials and Methods) on shaved backs (dorsal), or mocked treated (shaved, no UV). The first sensitization (S1) with 0.5% DNFB was applied to dorsal skin on day 5 and a second sensitization with 0.5% DNFB on non-UV-treated ventral skin (abdomen) was performed 2 weeks later. After 5 days, ears were painted with 0.3% DNFB and ears swelling responses measured. (a) Sensitized dorsal skin shows evidence of inflammation and ulceration (white arrows) in UVB-treated Bid KO but not UVB-treated WT mice. Numbers indicate the treatment group – outlined in the text and (c). (b) H & E ear skin sections provide corroborative evidence of ear swelling response measurements. Numbers indicate the treatment group. (c) Scatter plot of specific ear swelling responses from WT (open circles) and Bid KO (closed circles) mice (n=10 ears) are shown above descriptions of treatment group designation. Student’s two-tailed t-test p values designations are as follows: *, p < 0.05; ***, p < 0.0001. This experiment was repeated and provided identical results.
LCs that accumulate in LNs of UV treated Bid KO mice are devoid of CPD staining
Cyclobutane pyrimidine dimer (CPD) specific antibodies can detect UVB-induced thymine dimer formation within genomic DNA. LC staining with CPD specific antibody has been shown to correlate with UV-induced suppression and tolerance induction (32). Therefore, we examined the levels of CPD+ cells that accumulate in LNs of Bid KO and WT mice after 4 consecutive days of UV exposure (on ears and back) and subsequently sensitized with DNFB or vehicle as a control. Three days after the last UV dose, cells from parotid and subilliac LNs were pooled per individual mouse and triple stained for CD11c, Langerin and CPD DNA adducts and examined by flow cytometry (Figure 7a). Consistent with our previous experiments (shown in Figure 3) fewer LN cells were recovered from WT mice and more cells were recovered from Bid KO mice treated with UV, but not DNFB (Figure 7b, top panel). Similarly, LC numbers from Bid KO mice were at control levels in UV-treated groups but significantly higher levels after treatment with DNFB alone, while in WT mice diminished numbers of LCs were recovered for all treatment groups (Figure 7b, middle panels). Of the few remaining LCs recovered from WT mice, up to 26% were CPD positive in UV-treated groups, while only 3% were CPD positive in DNFB treated demonstrating a correlation with UV and CPD levels (Figure 6b lower panels). Surprisingly, only 1% of the Bid KO LCs were CPD positive in UV-treated groups, which dropped to approximately 0.2% in control groups. These results show that LCs from UV-treated WT mice have high levels of CPD DNA adducts and undergo rapid loss – presumably due to Bid-dependent apoptosis – and this phenomena correlates with suppressive function. In contrast, Bid KO LCs initially accumulate to high levels then drop down to control levels over 3 days, where only 1% of LCs are CPD positive. Our results demonstrate that it is the presence of high numbers of CPD negative LCs, not the absolute number of CPD+ LCs (which was equivalent to WT; Figure 7b, bottom panel) that correlates with resistance to mechanisms of UV-induced tolerance.
Figure 7. High numbers of LCs with undetectable CPD+ staining accumulate in UV-treated Bid KO LN.
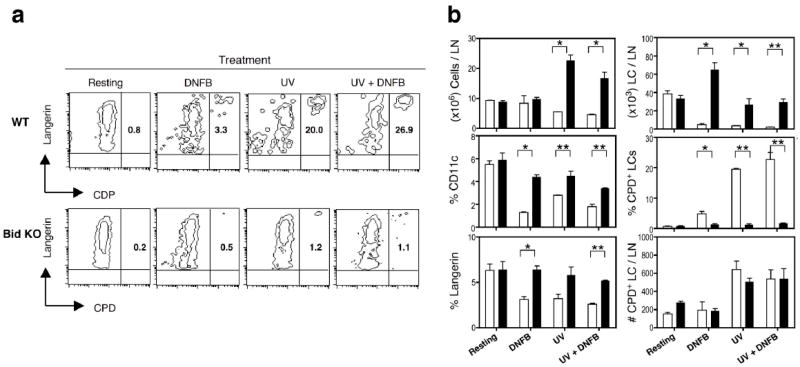
Superficial parotid LN cells were analyzed for expression of CD11c, Langerin and CPD from mice exposed once with UVB (100mJ/cm2) and/or 0.5% DNFB 3 days prior. (a) Contour plots of CD11c gated cells show percentage of CDP+ cells within the Langerin+ population in the upper right quadrant. (b) Summary and statistics of the phenotype and LN cell numbers (total, LCs and CPD+ cells per node) are shown. Mean and SEM (n=3) are displayed for each treatment of Bid KO (black bars) and WT (white bars) mice. Student’s one-tailed t-test p values indicate where significant differences between strains were evident. P value designations are *, p < 0.05; **, p < 0.001. A result from one of two similar experiments is shown.
Bid KO cutaneous DCs exhibit resistance to turnover in LNs of UV treated mice
To address whether the increased numbers of LCs found in dLNs of UV-treated Bid KO mice were due to increased turnover and recruitment of hematopoietic DCs or whether skin-derived DCs demonstrated longer life spans, UV-treated and untreated mice were painted with the hapten FITC on ear skin after the last UV exposure. LN cells were examined 3 days later, to provide time for FITC+ cells to migrate, and to mimic conditions used for tolerance induction. We observed that a daily UV dose of 400mJ/cm2 over 4 days induced substantial necrosis in mouse skin of both Bid KO and WT strains. Therefore, we used this dose to compare to a standard UV dose of 120 mJ/cm2 and untreated mice to test the effect of necrotic epidermal environment on the turnover rate of skin derived DCs. Our data presented in Figure 8 demonstrates that Bid DCs maintain a profound resistance to apoptosis, even when skin is exposed to high doses of UV. Thus, the necrotic epidermal environment, per se, does not significantly effect the turnover rate of Bid KO DCs, in contrast to the depletion seen by WT DCs.
Figure 8.
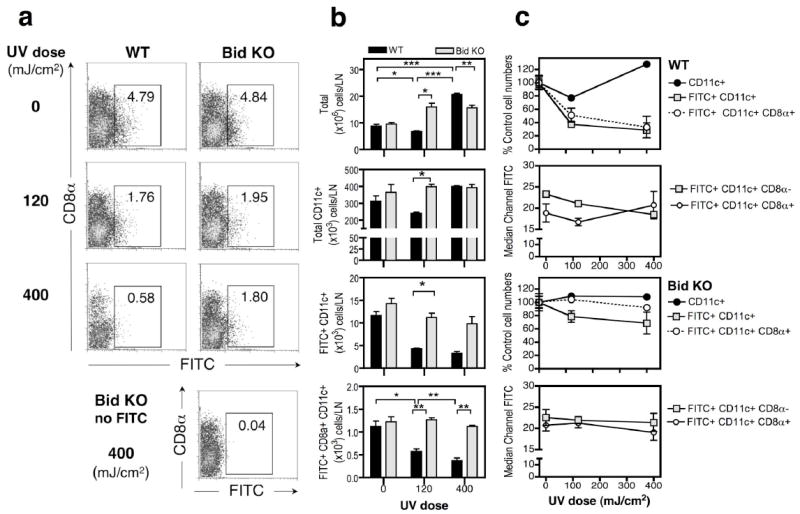
Resistance to UVB induced loss of skin-derived FITC+ cells in dLNs of Bid KO mice. Cohorts of non-shaved mice (3 per group) were exposed to daily UVB doses of 0, 120 or 400 mJ/cm2, as indicated, for 4 days. Mouse ears were painted with 0.5% FITC or vehicle (no FITC) after the last UV dose. Parotid LNs were harvested 3 days later. Single cell suspensions were counted and stained for CD11c and CD8α expression. (a) Bivariate plots of CD8α and FITC fluorescence are shown for CD11c+ gated cells, and the percentage of FITC+ cells is indicated within the designated gate for each sample. (b) Enumeration of cells recovered from the parotid, ear skin draining, LNs from WT (black bars) or Bid KO (light gray bars). Recoveries for total cells, CD11c+ cells and CD8α+ or CD8α− CD11c+ cells is shown. Mean ± SE for 3 mice/group are shown. Significant differences between groups are shown (one-tailed t-test. *, p < 0.05; ** p <0.01, *** p <0.001). One of 2 similar experiments is shown. (c) Susceptibility to UV-induced cell loss of DC subsets differs between WT and Bid KO mice. The % of control cell numbers obtained from dLNs of UV treated WT (top panel set) and Bid KO (bottom panel set) mice are shown for total CD11c+ DCs (filled circle), FITC+ DCs (square) and FITC+ CD8α+ DCs (open circle). Median channel fluorescence levels of FITC staining is shown for CD8α− and CD8α+ subsets of CD11c+ DCs for WT (top panel set) and Bid KO (bottom panel set) cells.
To begin to address if differences in DC subsets might account for differences in Bid KO and WT phenotypes, samples were co-stained for CD8a expression, and the frequency of DC subsets determined. In mice treated with 120 mJ/cm2 UV, WT CD11c+ DCs were depleted in LNs by 23%, while in Bid KO mice a 9% increase in CD11c+ DCs was observed. Moreover, the depletion rate of cells was greatly enhanced in FITC+ DC subsets from LNs of WT, but Bid KO mice. FITC+ DCs were depleted by 63% and FITC+ lymphoid DC subset (CD8α+CD11c+) were depleted by 49% in WT mice, whereas, Bid KO FITC+ DCs were depleted by only 22%, and FITC+ CD8α+ CD11c+ DCs remained at control levels. The enhanced depletion seen within the total FITC+ fraction suggests these cells were more susceptible to UV-induced apoptosis. Interestingly, in response to high dose irradiation treatment, elevated numbers of CD11c+ DCs were present in dLNs WT mice (WT, 127% increase versus Bid KO, 109% of control numbers, Figure 8c, top panel), consistent with the increase in total cells recovered (Figure 8b, top panel). Increased numbers of total cells was observed for both mouse strains in response to high dose UV radiation, and this was evident for Bid KO at the lower dose as well. However, for Bid KO, the increased cellularity did not contribute to an increase in CD11c cells (Figure 8b) and was consistent with our previous studies (Figure 4b).
We noted that the median fluorescence level of FITC staining of CD8α+ DCs tended to be lower than CD8α− DCs, consistent with the model that CD8 α+ DCs are resident LN cells that specialize in endocytosing apoptotic debris (e.g. dying FITC stained migratory cells) for cross presentation to T cells. The increased FITC levels within CD8α+ DC subset in LNs of WT mice observed in response to 400mJ/cm2 of UV radiation provides further evidence for this. (Figure 8c, upper panel set). The FITC fluorescence intensity measured for both DC subsets of Bid KO mice remained unchanged, regardless of UV radiation dose.
Discussion
Our study reveals that the Bid molecule plays a prominent role in UV-induced apoptosis in vivo, and that this Bid-dependent process is strongly linked to immune suppression and tolerance induction. We found that Bid deficiency resulted in marked resistance to apoptosis in both keratinocytes and LCs following UV exposure. Our data also indicate that Bid activation is required to initiate the mitochondrial signaling pathway. In addition, Bid deficiency abrogates UVB-induced local immune suppression of hapten sensitization and systemic tolerance induction. These studies provide the first evidence for the importance of Bid-dependent pathways in cutaneous immune regulation.
The link between apoptosis and UV-induced immune suppression have been examined in genetically engineered mice that either overexpress Bcl-2 (34), or are deficient for the CD95 (Fas) (35, 36). The immune suppression was shown to be significantly inhibited in these animals. The involvement of both intrinsic (Bcl-2) and extrinsic (CD95) apoptosis pathways supports our data demonstrating that Bid is an important mediator of this response. UV-induced death receptor (DR) signal initiation can occur through ligand-dependent and independent (receptor aggregation) mechanisms but to what extent the pathway depends on Bid was not known. Our findings provide evidence for a critical role of Bid in mediating epidermal cell apoptosis in response to UVR. Further investigations are required to understand the molecular mechanism underlying Bid’s role. Bid may act chiefly as a primary target of initiator caspase 8, unequivocally defining keratinocytes as ‘Type II cells’, or as a target of JNK-induced mechanisms of activation (37), or as a downstream target of effector caspases that cannot efficiently induce apoptosis without Bid activation to recruit mitochondrial mechanisms.
Our studies demonstrate that both local suppression and systemic tolerance induction are regulated by Bid-dependent apoptosis. These data contrast, in part, with studies in CD95 (Fas) deficient mice, in which inhibition of systemic tolerance but not local UV immune suppression was seen (38). Taken together, these data suggest that other death receptors may be critical in triggering local immune suppression mechanisms and is consistent with studies demonstrating that low doses of UVR sensitize human basal cell cultures to TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis but not to agonist anti-Fas antibody treatment (14).
A hallmark photodamage response is the depletion of LCs in the epidermis and a reduction of LC numbers recovered from draining LNs. The mechanism of this depletion is not fully understood. Migration out of the epidermis of these cells likely accounts for most of the epidermal depletion, but other mechanisms such as apoptosis, inhibition of migration from dermis to lymphatics (17), and/or increased susceptibility to apoptosis within the LN due to UV-induced cytokine production or T cell activation (39) have been proposed to contribute to low LC recovery. UV-induced apoptosis has been detected after in vitro irradiation of purified LCs after relatively long culture periods (48-72 hours) (19) but has been difficult to detect in vivo (40). However, most in vivo studies examine epidermal LCs within 24-48 hours after UV treatment, which may be too early to detect apoptosis. In accordance with previous findings in WT mice, we found that the majority of UV-induced LC depletion from the epidermal layer occurred within 48 hours and was UV dose dependent. However, significantly more epidermal LCs were retained in Bid KO mice. At this early time point, morphological evidence of LC apoptosis in situ was not observed (data not shown). However, when we examined UV-treated skin a week later, LC apoptotic morphology was prominently observed in all UV-treated samples from WT but not Bid KO mice. In striking contrast to the apoptotic blebs seen for WT LCs, the remaining epidermal Bid KO LCs exhibited enlarged cell bodies with thickened dendritic processes, consistent with an activated morphology. These observations support the notion that in WT mice, UV-induced LC apoptosis is delayed such that LCs can reach the LN before undergoing cell death.
The capacity of cells from WT or Bid KO mice to migrate from UV-exposed skin was not impeded and comparable to control samples (where tape stripping was used to activate migration). Further, the capacity of CPD+ cells to migrate from dorsal ear skin and remain viable after 3 days in culture was the same for both mouse strains. However, we did observe more dead cells in WT cultures of ventral ear skin as compared to Bid KO cultures, suggesting that a subset of cells was more susceptible to apoptosis. Taken together, the data support the notion that UV-induced LC apoptosis is delayed, and that resistance to apoptosis by Bid KO LCs can account for the increased numbers remaining in UV-treated epidermis. Furthermore, photodamaged LCs maintain their capacity to migrate from the epidermis to enter the lymphatics over the course of 3 days, since identical absolute numbers of CPD+ cells migrating from skin cultures and reaching lymph nodes were detected in samples from both strains.
The magnitude of CHS responses has been shown to correlate with epidermal LC density (18), so reduced LC numbers in the epidermis at the time of hapten application may play an important role in the mechanism of local immune suppression. However, while significant differences in LC density were observed between Bid KO and WT strains, we believe this does not completely account for the dramatic differences observed in their ability to sensitize UV-treated mice to hapten. Many reports indicate that UVR changes the quality of LC function such that antigen (hapten)-specific suppression is developed (22). In one study, purified epidermal LCs that were UV-treated and haptenated in vitro were shown to induce CHS suppression and long-lasting tolerance when injected into untreated mice (41). The nature of the relevant qualitative changes are under investigation by several laboratories, and a role for apoptosis has been implicated (38). Therefore, we tested whether Bid KO mice differed from WT mice in their susceptibility to UV-induced local immune suppression and systemic tolerance induction. We found that Bid KO mice were highly resistant to UVB-induced immune suppression and tolerance induction, demonstrating that the Bid-dependent apoptosis plays an essential role in these processes.
Several studies have shown that UV-induced DNA damage, in particular the generation of pyrimidine dimers present in migrating cutaneous DCs, correlates with immune suppression (29, 42). Therefore, we investigated the level of DNA damage found in LCs derived from UV-treated skin draining LNs from WT and Bid KO mice using flow cytometric methods (32, 43). Our analysis revealed three important differences between these strains: (1) In contrast to WT LCs, which contained greater than 25% CPD+ cells, the Bid KO LC population exhibited very low levels of DNA damage; (2) Greater numbers (2 to 3-fold) of Bid KO LCs were recovered from draining LNs of UV-treated Bid KO mice as compared to UV-treated WT mice; (3) and the lifespan of migratory cutaneous DCs was significantly extended in LNs of Bid KO mice. We addressed whether fewer CPD+ cells reached dLNs in Bid KO mice. Reduced CPD staining could be a secondary consequence of reduced epidermal cell apoptosis limiting a source of apoptotic CPD+ debris for LC uptake. However, we were unable to detect cytoplasmic CPD+ staining with our technique. Furthermore, we determined that the number of cells with CPD+ nuclei were not significantly different in populations of migratory cells from UV-treated skin explants derived from either strain. Thus, both strains are equally susceptible to DNA damage by UVR, and such damaged cells are fully capable of migration activity. Another consideration may be related to differences in DNA repair capacities between WT and Bid KO LCs. For example, IL-12 has been shown to be a key cytokine for the activation of DNA repair in response to UVR (44). It is possible that Bid KO mice produce enhanced levels of IL-12 in response to UVR, as compared to WT mice.
We favor the hypothesis that reduced levels of DNA damage observed by Bid KO LCs is likely due to an enhanced recruitment of non-damaged Langerin+ cells, as well as profound resistance to UV-induced apoptosis exhibited by Bid KO cutaneous DCs. The longer lifespan of such cells was shown convincingly by demonstrating that Bid KO LNs supported the maintenance of skin-derived FITC+ DCs at all UV doses tested. These findings are consistent with our previous report demonstrating that Bid KO LCs exhibited resistance to apoptosis induced by antigen-specific T cell activation (30). Additionally, increased numbers of LCs observed in LNs from UV-treated Bid KO mice likely did not originate from the epidermis and this may account for the low % of CPD+ cells observed. This interpretation is consistent with recent findings by three independent groups of investigators, demonstrating that a novel epidermal LC-distinct, blood-derived Langerin+ subset traffics through the dermis in the steady state and contributes to the majority of Langerin+ cells found in skin draining LNs (45-47). The contribution of dermal LCs also may explain why FITC labeled cells from skin exposed to toxic levels of UV (depleting epidermal LCs) could survive at all (Figure 8). Dermal cells are protected from direct photodamage due to limited skin penetration by the UVB bandwidth. Thus, the necrotic epidermal environment, per se, does not significantly affect the turnover rate of Bid KO DCs. The relative contributions made by dermal LCs and epidermal LCs in modulating cutaneous immune responses, including the induction of tolerance by UVR, will be an active area of future investigation.
Interestingly, equivalent numbers of CPD+ LCs per LN were recovered for UV-treated WT and Bid KO mice (Figure 7b, bottom panel), indicating that the absolute number of DNA damaged LCs does not necessarily correlate with tolerance induction, and that other factors contribute to the immune outcome. Our study establishes that Bid-dependent apoptosis plays an important role in the mechanism of UV-induced local immune suppression and systemic tolerance induction. The resistance to UV-induced suppression exhibited by Bid KO mice correlated with an increase in the number of healthy LCs present in both the epidermis and draining LNs after UVB exposure. The ratio of healthy LCs to DNA damaged LCs may be an important parameter in regulating the outcome of T cell development in UV-treated mice. It will be important to determine if Bid KO and WT mice differ in their ability to generate Treg cells, and if so, whether this qualitative difference is intrinsic to bona fide LCs (epidermis-derived Langerin+ cells) or to other DC subsets such as those derived from the dermis or those that are resident in draining LNs.
It is interesting to consider that cellular apoptosis by different pathways (e.g. extrinsic, intrinsic, Type I, Type II, JNK-dependent) may result in qualitatively different characteristics that affect reception by the immune system and subsequent immune function. This has important implications for understanding mechanisms of immune dysregulation (in autoimmunity and cancer) and for vaccine development. Our results in Bid KO mice provide strong evidence that Bid-dependent responses to UV damage are important in mediating suppression; but, whether this is a cell-type-dependent or apoptosis-pathway-dependent mechanism needs further investigation.
The mechanism of Bid-dependent resistance to UV-induced immune suppression and tolerance will be important to determine. Whether the altered response is due to indirect modulation of LCs by keratinocyte delivered signals or due direct modulation of LC responses is not known. An alternate set of compensatory responses may become activated in Bid KO mice that might repair cells that would normally undergo apoptosis and IL-12 may play an important role in this respect (32). Another keratinocyte-specific candidate to be investigated is TRANCE (or RANKL) which has been shown to increase in response to UVR, and is proposed to modulate local LCs to induce Treg cells (24). However, a direct consequence of LC resistance to apoptosis may alter suppression mechanisms. For example, the absence of LC-derived apoptotic vesicles that may be taken up by specialized LN resident DCs (eg. CD8α+ DCs) may alter the capacity to activate suppressor cells (e.g., Treg, Tr1, NK or NKT cells). Additionally, our data suggests that the ratio of normal antigen bearing LCs to DNA-damaged LCs may be important in driving suppressor cell development. Clarifying the mechanisms that alter photodamage responses in Bid KO mice will advance our understanding of tolerance induction and our ability to modulate cutaneous immune responses for therapeutic applications.
Acknowledgments
The authors wish to thank Josna Hartha, James Evans, and Taylor Preston for their dedicated efforts and assistance in these experiments.
Footnotes
This work was supported by NIH grants R01-AI50150, R01-CA86172, R01-ES015323, P30-AR050948, USAMRAA grant W81XWH-0510296 and a VA Merit Review Award.
Abbreviations used in this paper: Bid, BH3-interacting death domain protein; CHS, contact hypersensitivity; DC, dendritic cell; DNFB, 2-4-dinitrofluorobenzene, KO, knockout; LC, Langerhans cell; LN, lymph node; RANK, receptor activator of NFkB; TRANCE, TNF-related activation-induced cytokine; Treg, T regulatory cell; UVR, ultraviolet radiation; WT, wild type.
Conflict of Interest The authors state no conflict of interest.
Bibliography
- 1.Raj D, Brash DE, Grossman D. Keratinocyte apoptosis in epidermal development and disease. Journal of Investigative Dermatology. 2006;126:243–257. doi: 10.1038/sj.jid.5700008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nickoloff BJ, Qin JZ, Chaturvedi V, Bacon P, Panella J, Denning MF. Life and death signaling pathways contributing to skin cancer. Journal of Investigative Dermatology Symposium Proceedings. 2002;7:27–35. doi: 10.1046/j.1523-1747.2002.19633.x. [DOI] [PubMed] [Google Scholar]
- 3.Krajewska M, Zapata JM, Meinhold-Heerlein I, Hedayat H, Monks A, Bettendorf H, Shabaik A, Bubendorf L, Kallioniemi OP, Kim H, Reifenberger G, Reed JC, Krajewski S. Expression of Bcl-2 family member Bid in normal and malignant tissues. Neoplasia (New York) 2002;4:129–140. doi: 10.1038/sj.neo.7900222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, Kutok JL, Alt FW, Korsmeyer SJ. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes & Development. 2003;17:229–239. doi: 10.1101/gad.1045603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 7.Wang K, Yin XM, Copeland NG, Gilbert DJ, Jenkins NA, Keck CL, Zimonjic DB, Popescu NC, Korsmeyer SJ. BID, a proapoptotic BCL-2 family member, is localized to mouse chromosome 6 and human chromosome 22q11. Genomics. 1998;53:235–238. doi: 10.1006/geno.1998.5489. [DOI] [PubMed] [Google Scholar]
- 8.Barnhart BC, Alappat EC, Peter ME. The CD95 type I/type II model. Semin Immunol. 2003;15:185–193. doi: 10.1016/s1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 9.Hill LL, Ouhtit A, Loughlin SM, Kripke ML, Ananthaswamy HN, Owen-Schaub LB. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 1999;285:898–900. doi: 10.1126/science.285.5429.898. [DOI] [PubMed] [Google Scholar]
- 10.Aragane Y, Kulms D, Metze D, Wilkes G, Poppelmann B, Luger TA, Schwarz T. Ultraviolet Light Induces Apoptosis via Direct Activation of CD95 (Fas/APO-1) Independently of Its Ligand CD95L. J Cell Biol. 1998;140:171–182. doi: 10.1083/jcb.140.1.171. %R 10.1083/jcb 140.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang W, Ananthaswamy HN, Muller HK, Kripke ML. p53 protects against skin cancer induction by UV-B radiation. Oncogene. 1999;18:4247–4253. doi: 10.1038/sj.onc.1202789. [DOI] [PubMed] [Google Scholar]
- 12.Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 13.Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nature Cell Biology. 2002;4:842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 14.Qin JZ, Bacon P, Panella J, Sitailo LA, Denning MF, Nickoloff BJ. Low-dose UV-radiation sensitizes keratinocytes to TRAIL-induced apoptosis. Journal of Cellular Physiology. 2004;200:155–166. doi: 10.1002/jcp.20017. [DOI] [PubMed] [Google Scholar]
- 15.Kulms D, Zeise E, Poppelmann B, Schwarz T. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene. 2002;21:5844–5851. doi: 10.1038/sj.onc.1205743. [DOI] [PubMed] [Google Scholar]
- 16.Obata M, Tagami H. Alteration in murine epidermal Langerhans cell population by various UV irradiations: quantitative and morphologic studies on the effects of various wavelengths of monochromatic radiation on Ia-bearing cells. J Invest Dermatol. 1985;84:139–145. doi: 10.1111/1523-1747.ep12275379. [DOI] [PubMed] [Google Scholar]
- 17.Hamakawa M, Sugihara A, Okamoto H, Horio T. Ultraviolet B radiation suppresses Langerhans cell migration in the dermis by down-regulation of alpha4 integrin. Photodermatol Photoimmunol Photomed. 2006;22:116–123. doi: 10.1111/j.1600-0781.2006.00187.x. [DOI] [PubMed] [Google Scholar]
- 18.Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980;124:445–453. [PubMed] [Google Scholar]
- 19.Tang A, Udey MC. Effects of ultraviolet radiation on murine epidermal Langerhans cells: doses of ultraviolet radiation that modulate ICAM-1 (CD54) expression and inhibit Langerhans cell function cause delayed cytotoxicity in vitro. J Invest Dermatol. 1992;99:83–89. doi: 10.1111/1523-1747.ep12611871. [DOI] [PubMed] [Google Scholar]
- 20.Elmets CA, Bergstresser PR, Tigelaar RE, Wood PJ, Streilein JW. Analysis of the mechanism of unresponsiveness produced by haptens painted on skin exposed to low dose ultraviolet radiation. J Exp Med. 1983;158:781–794. doi: 10.1084/jem.158.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beissert S, Schwarz A, Schwarz T. Regulatory T cells. Journal of Investigative Dermatology. 2006;126:15–24. doi: 10.1038/sj.jid.5700004. [DOI] [PubMed] [Google Scholar]
- 22.Schade N, Esser C, Krutmann J. Ultraviolet B radiation-induced immunosuppression: molecular mechanisms and cellular alterations. Photochem Photobiol Sci. 2005;4:699–708. doi: 10.1039/b418378a. [DOI] [PubMed] [Google Scholar]
- 23.Rivas JM, Ullrich SE. Systemic suppression of delayed-type hypersensitivity by supernatants from UV-irradiated keratinocytes. An essential role for keratinocyte-derived IL-10. J Immunol. 1992;149:3865–3871. [PubMed] [Google Scholar]
- 24.Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, Schwarz T, Penninger JM, Beissert S. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med. 2006;12:1372–1379. doi: 10.1038/nm1518. [DOI] [PubMed] [Google Scholar]
- 25.Ullrich SE. Sunlight and skin cancer: Lessons from the immune system. Mol Carcinog. 2007;46:629–633. doi: 10.1002/mc.20328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beissert S, Hosoi J, Kuhn R, Rajewsky K, Muller W, Granstein RD. Impaired immunosuppressive response to ultraviolet radiation in interleukin-10-deficient mice. J Invest Dermatol. 1996;107:553–557. doi: 10.1111/1523-1747.ep12582809. [DOI] [PubMed] [Google Scholar]
- 27.Nishigori C, Yarosh DB, Ullrich SE, Vink AA, Bucana CD, Roza L, Kripke ML. Evidence that DNA damage triggers interleukin 10 cytokine production in UV-irradiated murine keratinocytes. Proc Natl Acad Sci U S A. 1996;93:10354–10359. doi: 10.1073/pnas.93.19.10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connor A, Nishigori C, Yarosh D, Alas L, Kibitel J, Burley L, Cox P, Bucana C, Ullrich S, Kripke M. DNA double strand breaks in epidermal cells cause immune suppression in vivo and cytokine production in vitro. J Immunol. 1996;157:271–278. [PubMed] [Google Scholar]
- 29.Vink AA, Moodycliffe AM, Shreedhar V, Ullrich SE, Roza L, Yarosh DB, Kripke ML. The inhibition of antigen-presenting activity of dendritic cells resulting from UV irradiation of murine skin is restored by in vitro photorepair of cyclobutane pyrimidine dimers. Proc Natl Acad Sci U S A. 1997;94:5255–5260. doi: 10.1073/pnas.94.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pradhan S, Genebriera J, Denning WL, Felix K, Elmets CA, Timares L. CD4 T cell-induced, bid-dependent apoptosis of cutaneous dendritic cells regulates T cell expansion and immune responses. J Immunol. 2006;177:5956–5967. doi: 10.4049/jimmunol.177.9.5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katiyar SK, Matsui MS, Mukhtar H. Kinetics of UV light-induced cyclobutane pyrimidine dimers in human skin in vivo: an immunohistochemical analysis of both epidermis and dermis. Photochemistry & Photobiology. 2000;72:788–793. doi: 10.1562/0031-8655(2000)072<0788:koulic>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Schwarz A, Maeda A, Kernebeck K, van Steeg H, Beissert S, Schwarz T. Prevention of UV radiation-induced immunosuppression by IL-12 is dependent on DNA repair. Journal of Experimental Medicine. 2005;201:173–179. doi: 10.1084/jem.20041212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van den Broeck W, Derore A, Simoens P. Anatomy and nomenclature of murine lymph nodes: Descriptive study and nomenclatory standardization in BALB/cAnNCrl mice. J Immunol Methods. 2006;312:12–19. doi: 10.1016/j.jim.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 34.Muller-Rover S, Rossiter H, Paus R, Handjiski B, Peters EM, Murphy JE, Mecklenburg L, Kupper TS. Overexpression of Bcl-2 protects from ultraviolet B-induced apoptosis but promotes hair follicle regression and chemotherapy-induced alopecia. American Journal of Pathology. 2000;156:1395–1405. doi: 10.1016/S0002-9440(10)65008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill LL, Shreedhar VK, Kripke ML, Owen-Schaub LB. A critical role for Fas ligand in the active suppression of systemic immune responses by ultraviolet radiation. Journal of Experimental Medicine. 1999;189:1285–1294. doi: 10.1084/jem.189.8.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aragane Y, Kulms D, Metze D, Wilkes G, Poppelmann B, Luger TA, Schwarz T. Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J Cell Biol. 1998;140:171–182. doi: 10.1083/jcb.140.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timares L, Katiyar SK, Elmets CA. DNA Damage, Apoptosis and Langerhans Cells-Activators of UV-induced Immune Tolerance. Photochemistry & Photobiology. 2008;84:422–436. doi: 10.1111/j.1751-1097.2007.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwarz A, Grabbe S, Grosse-Heitmeyer K, Roters B, Riemann H, Luger TA, Trinchieri G, Schwarz T. Ultraviolet light-induced immune tolerance is mediated via the Fas/Fas-ligand system. Journal of Immunology. 1998;160:4262–4270. [PubMed] [Google Scholar]
- 39.Kitajima T, Ariizumi K, Bergstresser PR, Takashima A. Ultraviolet B radiation sensitizes a murine epidermal dendritic cell line (XS52) to undergo apoptosis upon antigen presentation to T cells. Journal of Immunology. 1996;157:3312–3316. [PubMed] [Google Scholar]
- 40.Kolgen W, Both H, van Weelden H, Guikers KL, Bruijnzeel-Koomen CA, Knol EF, van Vloten WA, De Gruijl FR. Epidermal langerhans cell depletion after artificial ultraviolet B irradiation of human skin in vivo: apoptosis versus migration. J Invest Dermatol. 2002;118:812–817. doi: 10.1046/j.1523-1747.2002.01742.x. [DOI] [PubMed] [Google Scholar]
- 41.Cruz PD, Jr, Tigelaar RE, Bergstresser PR. Langerhans cells that migrate to skin after intravenous infusion regulate the induction of contact hypersensitivity. J Immunol. 1990;144:2486–2492. [PubMed] [Google Scholar]
- 42.Applegate LA, Ley RD, Alcalay J, Kripke ML. Identification of the molecular target for the suppression of contact hypersensitivity by ultraviolet radiation. J Exp Med. 1989;170:1117–1131. doi: 10.1084/jem.170.4.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vink AA, Strickland FM, Bucana C, Cox PA, Roza L, Yarosh DB, Kripke ML. Localization of DNA damage and its role in altered antigen-presenting cell function in ultraviolet-irradiated mice. J Exp Med. 1996;183:1491–1500. doi: 10.1084/jem.183.4.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwarz A, Stander S, Berneburg M, Bohm M, Kulms D, van Steeg H, Grosse-Heitmeyer K, Krutmann J, Schwarz T. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nature Cell Biology. 2002;4:26–31. doi: 10.1038/ncb717. [DOI] [PubMed] [Google Scholar]
- 45.Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, Hogquist KA. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204:3147–3156. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginhoux F, Collin MP, Bogunovic M, Abel M, Leboeuf M, Helft J, Ochando J, Kissenpfennig A, Malissen B, Grisotto M, Snoeck H, Randolph G, Merad M. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med. 2007;204:3133–3146. doi: 10.1084/jem.20071733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med. 2007;204:3119–3131. doi: 10.1084/jem.20071724. [DOI] [PMC free article] [PubMed] [Google Scholar]