

Abstract
Coibamide A (1) is a new, potent antiproliferative depsipeptide which was isolated from a marine Leptolyngbya cyanobacterium collected from the Coiba National Park, Panama. The planar structure of 1 was elucidated by a combination of NMR spectroscopy and mass spectrometry. Exhaustive 1D and 2D NMR spectroscopy included natural abundance 15N and variable temperature experiments; mass spectrometry included TOF-ESI-MSn and FT-MSn experiments. Chemical degradation followed by chiral HPLC- and GC-MS analyses was used to assign the absolute configuration of 1. This highly methylated cyclized depsipeptide exhibited an unprecedented selectivity profile in the NCI 60 cancer cell line panel and appears to act via a novel mechanism.
Marine organisms continue to yield a diverse array of biologically active molecules, a remarkable number of which are peptide-based cancer cell toxins of putative microbial symbiont biogenesis.1 Development of these as anticancer drugs has met with some success:2 ascidian-derived dihydrodidemnin B (aplidin®) has orphan drug status for the treatment of multiple myeloma and acute lymphoblastic leukemia; green algal isolate kahalalide F, and TZT-1027, a synthetic analog of the cyanobacterial metabolite dolastatin 10, reached phase II clinical trials. Other important cyanobacterial peptide leads include the cryptophycins and curacin A,3 and these organisms continue to produce a wealth of anticancer lead compounds.4 The high degree of N-methylation of many of these cyanobacterial peptides may improve their druggability since N-methylation has been shown to improve pharmacological parameters such as lipophilicity, proteolytic stability and duration of action, properties for which regular peptides are notoriously poor and which limits their bioavailability.5
In the context of our International Cooperative Biodiversity Groups program (ICBG) based in Panama, which focuses on drug discovery, biodiversity conservation and sustainable economic growth, we have isolated a potent cancer cell toxin with an unprecedented selectivity profile in the NCI 60 cell line panel. This cyanobacterial depsipeptide, named coibamide A in tribute to its discovery from the UNESCO World Heritage Site of Coiba National Park,6 highlights the importance of conserving pristine, unexplored repositories of diverse marine organisms.
The marine filamentous cyanobacterium Leptolyngbya sp. was collected by hand using SCUBA from the Coiba National Park, Panama. A crude organic extract of this material was subjected to bioassay-guided fractionation via normal phase vacuum liquid chromatography (NP-VLC) using a stepped gradient of hexanes to EtOAC to MeOH. In preliminary biological activity screening, the 100% EtOAc eluting fraction was cytotoxic (IC50 300 ng/mL) to NCI-H460 human lung tumor cells, and was also active against malaria, leishmaniasis and trypanosomal tropical disease parasites. This VLC fraction was separated by reversed-phase C18 solid phase extraction and isocratic HPLC to yield the optically active colorless oil ([α]D -54.1) coibamide A (1, 6.3 mg).
The molecular composition of 1 was established as C65H110O16N10 from FT-MS data ([M + H]+ m/z 1287.8156, Δ -0.2 mmu). The peptidic nature of 1 was evident from its complex 1H NMR spectra in all solvents (CDCl3, C6D6, DMSO, C5D5N). However, N-methyl conformations were minimized in CDCl3 which showed numerous α-proton multiplets (4.75-6.02), overlapped methyl doublets (δ 0.75-1.15), mutually coupled aromatic proton doublets (δ 7.14, 6.76), a broad 2H amide proton signal (6.65), and deshielded singlets integrating to 12 methyl groups attached to heteroatoms (δ 2.34-3.77). The 13C NMR spectra for 1 in CDCl3 and C6D6 featured an indeterminate number of resonances, with numerous ester/amide carbonyl 13C signals, due to localized symmetry (O-Me-Tyr, N,N-diMe-Val), steric constraints (N-Me-Thr), signal overlap and multiple conformations. These data suggested a high degree of N- and O-methylation, an observation supported by standard amino acid analysis which yielded only one alanine and one O-methyl tyrosine residue. These two amino acids, one hydroxy acid and eight N-methylated residues were assigned from 2D experiments (CDCl3) including COSY, TOCSY, multiplicity-edited HSQC, HSQC-TOCSY, HMBC, H2BC7 and 1H-15N gHMBC.
Elucidation of seven of the eight N-methylated residues began with HMBC correlations from each N-methyl singlet to the corresponding α-carbon, the side-chain spin systems of which were delineated by TOCSY experiments to give N-methylalanine, two N-methylleucines, N-methylisoleucine, two N,O-dimethylserines, and an N,N-dimethylvaline residue. The latter terminal residue was described by a 6H singlet (δH-64/65 2.34) that was HSQC-correlated to a prominent 13C resonance (δC-64/65 41.3) and 15N-gHMBC-correlated to a shielded 15N resonance (δN 24.6). Fortunately, nine of ten N atoms in 1 were observed in the latter 15N-gHMBC8 experiment which showed additional correlations from five N-methyls to δN 105.6, 108.5, 113.4, 117.5, 120.5, two α-methyls (Ala and N-Me-Ala) to δN 115.1, 122.4, and H2-7 of O-Me-Tyr to δN 118.2. Hydroxyisovaleric acid (HIV) was assigned on the basis of TOCSY correlations from deshielded CH-55 (δH 5.00, δC 74.7) to isopropyl methine (δH-56 2.21) and methyl (δH3-57/58 1.06) resonances. At this point, it remained to assign 114 mass units (interpreted as C5H8O2N = N-Me-Thr or N,O-diMe-Ser), to determine the carboxyl terminus and to establish the sequence of residues in the depsipeptide chain. COSY correlations were observed between an unassigned methyl doublet at δ 1.07 (H3-40) and an oxygenated methine multiplet at δ 5.50 (H-39). Strong ROESY correlations, but no COSY or TOCSY correlations, were observed between this methyl-oxymethine pair and a very broad, partially obscured signal (δ 2.89, CDCl3; 3.11 ppm, C6D6). Variable temperature experiments in CDCl3 (298-328K, 700 MHz, 1 mm cryoprobe) resolved this broad peak into a 3H singlet which was HSQC-correlated to an N-methyl resonance (δC-41 29.7). Furthermore, careful examination of C6D6 HSQC data revealed an additional heteroatom-substituted methine (δH-38 6.72, δC-38 56.6), which showed weak TOCSY correlations to the above described methyl-oxymethine pair. Hence, the remaining residue was assigned as N-Me-Thr.
Two partial structures (Figure 1, A and B) could be assembled based on a combination of mass spectrometric data and ROESY correlations between each N-methyl and the α-proton of the adjacent residue. Additionally, a ROESY correlation between N-CH3-36 and the N-Me-Thr β- and γ-protons (H-39, H3-40) positioned this residue at the N terminus of partial structure A.
Figure 1.
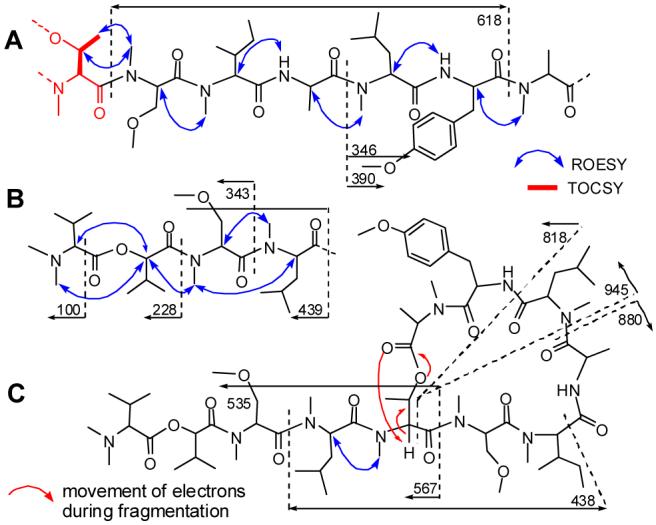
Partial structures A and B assembled from key ROESY correlations and mass fragments; C. Key mass spectrometric fragments supporting the position of N-Me-Thr and cyclization of 1.
A ROESY correlation between γ-H3-40 of the N-Me-Thr and α-H-43 (N-Me Leu) in combination with MS fragments of m/z 535 and 567 (Figure 1, C) oriented N-Me-Thr as the fifth residue in the depsipeptide backbone, thus linking partial structures A and B. This sequence of residues was also consistent with MS2 fragments observed by LC-MS of the base hydrolysate of 1, which comprised four major linear products (2-5, pS30). Finally, an HMBC correlation from H-39 to carbonyl C-1 (δC 170.4) indicated an ester linkage from N-Me-Thr to the C-terminal N-Me-Ala to complete the planar structure of coibamide A (1).
Acid hydrolysis of 1 followed by various HPLC-MS and GC-MS methodologies was used to determine the absolute configuration of coibamide A. While some standards were commercially available (N-Me-Leu, N-Me-Ile, N-Me-Ala, Ala, HIV and O-Me-Tyr), others required laboratory synthesis by standard methods (N-Me-Thr, N,N-diMe-Val, and N,O-diMe-Ser). Chiral HPLC (Phenomenex Chirex phase 3126 (D), 4.6 × 250 mm) established the presence of O-Me-L-Tyr, two N,O-diMe-L-Ser residues, N,N-diMe-L-Val, and L-Ala, while chiral GC-MS (CyclosilB, 30.0 m × 250 μm × 0.25 μm) of methylated standards and the natural product hydrolysate identified L-HIV. Treatment of the acid hydrolysate of 1 with Marfey’s reagent, followed by C18 HPLC established the presence of N-Me-L-Ile, N-Me-L-Leu, N-Me-L-Ala, and either N-Me-L-Thr or N-Me-L-allo-Thr. The presence of N-Me-L-Thr is proposed from computational models9 of the two possible coibamide structures, constrained by ROESY correlations between N-CH3-4 and CH3-40, and between α-H-2 and N-CH3-48.
Coibamide A displayed potent cytotoxicity to NCI-H460 lung cancer cells and mouse neuro-2a cells (LC50 < 23 nM), but did not interfere with tubulin or actin in cytoskeletal assays. Flow cytometric studies showed that 1 caused a significant dose dependent increase in the number of cells in the G1 phase of the cell cycle with little change in G2/M and a loss of cells in S phase (see supporting information). Coibamide A was evaluated against the NCI’s in vitro panel of 60 cancer cell lines, and produced mean cytostatic (GI50 and TGI with range) and cytotoxic (LC50 and range) parameters as follows: log GI50 -8.04 (2.96); log TGI - 5.85 (3.43); log LC50 -5.11 (2.66). These log mean values of < -4 with log range values of >2 indicate both potency and histological selectivity. Coibamide A showed highest potency (GI50) to MDA-MB-231 (2.8 nM), LOX IMVI (7.4 nM), HL-60(TB) (7.4 nM) and SNB-75 (7.6 nM), and good histological selectivity for breast, CNS, colon and ovarian cancer cells (see supporting information). Coibamide A was COMPARE negative,10 indicating that it likely inhibits cancer cell proliferation through a novel mechanism.
In summary, coibamide A (1) is a promising lead agent in cancer drug discovery, with a potentially new mechanism of action. Further investigation of the molecule is being pursued via chemical synthesis, since the producing organism has not been cultured successfully in the laboratory to date.
Supplementary Material
Acknowledgment
Financial support from the NIH Fogarty International Center (ICBG grant TW006634) and NCI (CA52955), and from the William Randolph Hearst Foundation (to SLM) is gratefully acknowledged. We thank the Smithsonian Tropical Research Institute, crew of R/V Urraca, T. Capson, C. Guevara and R. Thacker for collection of the cyanobacterium, and Autoridad Nacional del Ambiente (ANAM), Panama, for permission to make this collection, OSU Biochemistry NMR Facility for 600 MHz spectrometer time, J. Morre and the OSU EIHS Center for MS data acquisition (NIEHS P30 ES00210), Prof. F. D. Horgen for N,O-diMeSer standards, and C. Anklin (Bruker Biospin) for 700 MHz NMR data.
References
- (1).Simmons TL, Coates RC, Clark BR, Engene N, Gonzalez D, Esquenazi E, Dorrestein PC, Gerwick WH. Proc. Natl. Acad. Sci U.S.A. 2008;105:4587–4594. doi: 10.1073/pnas.0709851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rawat DS, Joshi MC, Joshi P, Atheaya H. Anti-Cancer Agents Med Chem. 2006;6:33–40. doi: 10.2174/187152006774755519. [DOI] [PubMed] [Google Scholar]
- (3).Gerwick WH, Tan LT, Sitachitta N. Alkaloids. 2001;57:75–184. doi: 10.1016/s0099-9598(01)57003-0. [DOI] [PubMed] [Google Scholar]
- (4).Tan LT. Phytochemistry. 2007;68:954–979. doi: 10.1016/j.phytochem.2007.01.012. [DOI] [PubMed] [Google Scholar]
- (5)(a).Loffert AJ. Pept. Sci. 2002;8:1–7. doi: 10.1002/psc.366. [DOI] [PubMed] [Google Scholar]; (b) Morishita M, Peppas NA. Drug Discov. Today. 2006;11:905–910. doi: 10.1016/j.drudis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- (6).Coiba National Park http://whc.unesco.org/en/list/1138.
- (7).Nyberg NT, Duus JØ, Sørensen OW. J. Am. Chem. Soc. 2005;127:6154–6155. doi: 10.1021/ja050878w. [DOI] [PubMed] [Google Scholar]
- (8).Martin GE, Hadden CE. J. Nat. Prod. 2000;63:543–585. doi: 10.1021/np9903191. [DOI] [PubMed] [Google Scholar]
- (9).Macromodel 9.1, see supporting information, pS6.
- (10).Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.