

Abstract
Bacterial pathogens have evolved a sophisticated arsenal of virulence factors to modulate host cell biology. Enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC) use a type III protein secretion system (T3SS) to inject microbial proteins into host cells. The T3SS effector cycle inhibiting factor (Cif) produced by EPEC and EHEC is able to block host eukaryotic cell-cycle progression. We present here a crystal structure of Cif, revealing it to be a divergent member of the superfamily of enzymes including cysteine proteases and acetyltransferases that share a common catalytic triad. Mutation of these conserved active site residues abolishes the ability of Cif to block cell-cycle progression. Finally, we demonstrate that irreversible cysteine protease inhibitors do not abolish the Cif cytopathic effect, suggesting that another enzymatic activity may underlie the biological activity of this virulence factor.
Keywords: type III secretion, virulence, cell cycle, Cif, crystallography
Introduction
Many pathogenic bacteria of both plants and animals inject virulence factors into host cells through a type III secretion system (T3SS). These translocated proteins possess a diverse array of biochemical activities, reprogramming eukaryotic cell biochemistry to serve the requirements of the pathogen. Examples of translocated effector proteins hijacking essential host functions such as cytoskeleton assembly, vesicular transport, and apoptosis are numerous.1 However, until recently, less progress has been made with bacteria that also possess virulence mechanisms that target the host cell-cycle. Such cyclomodulins are a growing family of bacterial toxins and effectors that interfere with the eukaryotic cell-cycle.2,3
Enteropathogenic (EPEC) and most enterohemorrhagic Escherichia coli (EHEC) possess a T3SS encoded by the locus of enterocyte effacement pathogenicity island. EPEC and EHEC share similar virulence strategies with other mucosal pathogens: colonization of the mucosal site, multiplication, evasion of host defenses, and subversion of the host cell. EPEC is the leading cause of infantile diarrhea in developing countries. EHEC causes bloody diarrhea and, if serious, hemolytic uremic syndrome with possible kidney damage and failure.4 The locus of enterocyte effacement-encoded T3SS translocates effector proteins that are encoded at different locations in the genome and has an essential role in infection and disease.5
One substrate of the EPEC and EHEC T3SS has been shown to inhibit cell-cycle progression. The cycle inhibiting factor (Cif) is an effector protein encoded by a lambdoid prophage that is incorporated into the bacterial genome.6 Cif is a modular protein composed of an N-terminal secretion and translocation signal and a C-terminal domain with effector activity in the host cell.7 Upon translocation into the host cell, Cif induces a cytopathic effect characterized by cell-cycle arrest at the G2/M transition associated in HeLa cells with the formation of stress fibers and focal adhesion recruitment.8,9 The cell-cycle inhibition induced by Cif is not a consequence of cytoskeleton alterations, because inhibition of the formation of stress fibers by Rho inhibitors does not prevent accumulation of G2-arrested cells.8 Cif-induced cell-cycle arrest occurs with the sustained inhibitory phosphorylation of the mitosis inducer CDK1.6 In contrast to other cyclomodulins, such as the cytolethal distending toxin,10 or colibactin,11 Cif is not a genotoxic effector nor does it activate transducers of the DNA-damage checkpoint that leads to CDK1 phosphorylation and G2-arrest.9 Thus, Cif is a unique example of cyclomodulin acting as a G2-checkpoint inducer independently of the DNA-damage pathway. The Cif pathway leading to cell-cycle inhibition remains to be elucidated.
In order to better understand the function of Cif, we have determined its crystal structure by X-ray crystallography to a resolution of 1.7 Å. We show that Cif is a divergent member of the family of enzymes that includes cysteine proteases, transglutaminases, and acetyltransferases, identifying a putative catalytic triad consisting of cysteine, histidine, and glutamine residues. We show that these residues are essential for the ability of Cif to cause the cytopathic effect when introduced into cells and during infection with pathogenic strains of E. coli.
Results
Overall structure of Cif
Following the screening of numerous N-terminal deletions of Cif (while maintaining the wild type C-terminus), a construct spanning residues 100–282 (henceforth simply Cif) was found to crystallize in a form amenable to structural determination (see Materials and Methods; Table 1). The crystal structure of Cif was determined by X-ray crystallography to a resolution of 1.7 Å. Cif has a mixed α/β fold, with a central, four-stranded β-sheet (B1–B4) surrounded on both sides by six α-helices (H1–H6) and connecting loops (Fig. 1). The N-terminus of the crystallized construct begins as an α-helix that packs with its axis nearly parallel with the strands in the β-sheet. The C-terminal helix (H6) is a thumb-like extension that points away from the core.
Table 1.
Data collection and refinement statistics
| SeMet SAD | Native | |
|---|---|---|
| A. Data collection | ||
| Space group | P4222 | P4222 |
| Cell dimensions | ||
| a (Å) | 111.515 | 111.768 |
| c (Å) | 106.808 | 107.518 |
| Wavelength (Å) | 0.979 | 1.000 |
| Resolution (Å) | 50–2.0 (2.07–2.00) | 50–1.70 (1.76–1.70) |
| Rsym | 4.7 (17.3) | 4.5 (39) |
| I/σI | 35.9 (12.7) | 28.7 (3.76) |
| Completeness (%) | 99.3 (100) | 98.86 (99.7) |
| Redundancy | 8.5 | 4.7 |
| B. Refinement | ||
| Resolution (Å) | 50–1.70 | |
| No. reflections | 70,819 | |
| Rwork / | 0.172 | |
| Rfree | 0.209 | |
| Average B-factor (Å2) | 26.63 | |
| RMSD from ideal | ||
| Bond lengths (Å) | 0.019 | |
| Bond angles (°) | 1.710 |
Rsym=ΣhΣi | Ih,i − Ih Σ | / hΣi Ih,i, for the intensity of i observations of reflection h.
R=Σ | FP − Fcalc | / ΣFP; Fcalc is the model structure factor and 5% data were omitted for Rfree. Bond and angle deviations are from ideal values; B-factor deviations are between bonded atoms. The SeMet SAD data set was used for phasing, and the structure was subsequently refined to 1.7 Å resolution using the Native data set. Values in parentheses are for the highest resolution shell.
Fig. 1.
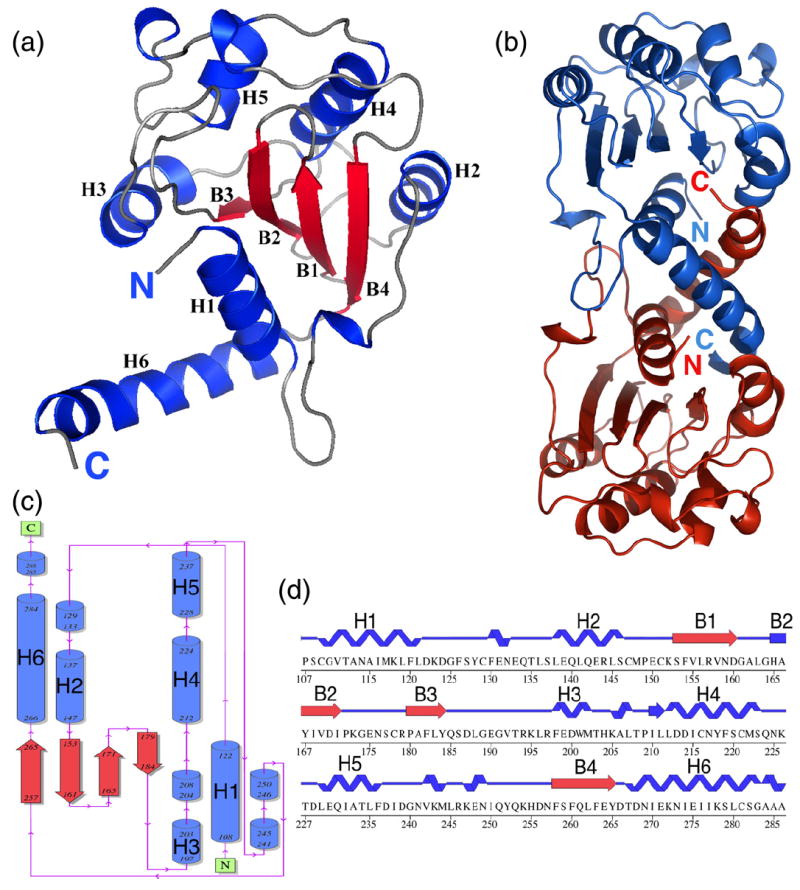
Overall structure of Cif. (a) A ribbon diagram of a Cif(100-282-His) monomer. (b) Two Cif molecules in the asymmetric unit shown as ribbon diagrams. (c) Topology diagram of Cif(100-282-His). (d) Sequence and secondary structure of Cif(100-282-His).
In the asymmetric unit of the crystal, Cif is present as a dimer that buries 1660 Å2 of surface area, similar to many physiological interfaces (Fig. 1b). Gel-filtration chromatography experiments indicate that both the full-length and the crystallized construct of Cif exhibit properties of a dimer, but only in low-salt conditions (Fig. 2a and b). In the crystal, both the N- and C-terminal helix of Cif contribute substantially to dimer contacts by interacting with the corresponding helix in the second Cif molecule. As the C-terminal helix has been shown to be essential for Cif activity,6,7 this raises the possibility that its functional role could be to form an active dimer. To address the role of the C-terminal helix of Cif in dimerization, we generated two truncated mutants: (i) Cif(1-265), which has the C-terminal helix deleted completely; and (ii) Cif(1-272), which has the last ten amino acids deleted. Both mutants were insoluble, and following refolding, ran as higher molecular mass aggregates on size-exclusion chromatography (Fig. 2c and d). These data confirm that truncations in the C-terminus greatly affect the solubility and folding of the protein,7 but do not clarify its role in dimer formation. Since dimer formation occurs only in crystallo or in low-salt conditions, it is possible that the biologically relevant state of Cif is monomeric, despite the large surface of interaction present in the structure.
Fig. 2.

The gel-filtration chromatography profiles of Cif C-terminal deletion mutants in: (a) 50 mM; and (b) 200 mM salt. Cif full-length and crystallized construct Cif(100-282)His gel-filtration chromatography profile in: (c) 50 mM and (d) 200 mM salt. Cif and various standard marker proteins were run on a Superdex 200 gel-filtration column using an AKTA FPLC. A standard calibration curve based on the elution volumes of standard markers was used to determine the experimental molecular mass of Cif. The calculated molecular mass of globular proteins based on the elution volume are indicated in parentheses.
Cif is a structural member of the cysteine protease superfamily
A search of known structures in the Protein Data Bank reveals that Cif shares structural homology with Pseudomonas syringae AvrPphB (PDB ID code 1ukf) with a Z-score of 4.0 and root-mean-square deviation (RMSD) of 3.8 over 88 residues.12 AvrPphB is a member of a superfamily of related enzymes containing cysteine proteases, acetyltransferases, and transglutaminases.13 While there is considerable divergence across this superfamily in the overall fold, a core anti-parallel β-sheet and an N-terminal helix, which packs against the β-strands, are always present (Fig. 3a).14 Several of the catalytic residues in these proteins are located on these elements as discussed below.
Fig. 3.

Structural comparison of Cif with cysteine protease superfamily members. (a) Ribbon diagrams of Cif, AvrPphB (PDB ID code 1ukf), and NAT (PDB ID code 1e2t). The catalytic triad in the active site is rendered as a stick model, the structurally conserved α-helix is colored green, and the β-sheets are colored orange. (b) Alignments of the catalytic cores of Cif (gray), AvrPphB (red), and NAT (blue) using the putative catalytic triad residues C109-H165-D187 as a reference. (c) Alignments of the same structures using C109-H165-Q185 as a reference.
As judged by Z-score and RMSD, the fold closest to Cif, AvrPphB belongs to the YopT family of cysteine proteases that are involved in bacterial pathogenesis. Like other members of the cysteine protease superfamily, AvrPphB and YopT have an invariant catalytic triad consisting of cysteine, histidine, and aspartate.15 Initially, a potential catalytic triad (Cys109-His165-Asp187) was identified in the Cif crystal structure based on the spatial proximity between these three residues and the assumption that the third residue was either aspartate or asparagine. Aligning this putative catalytic triad with that of other members of the superfamily resulted in a poor overall alignment in which the common core fold was misaligned (Fig. 3b). When the core fold of Cif and the members of superfamily are structurally aligned, the catalytic triad of Cif is shifted by several ångström units. A far better structural alignment was obtained by aligning only the side chains of Cys109 and His165 with the corresponding catalytic cysteine and histidine from the cysteine proteases (Fig. 3c). However, in this new alignment, Asp187 no longer superimposes with the Asp/Asn of the other catalytic triads. Instead, Gln185 occupies this position, making hydrogen bonds to the catalytic histidine that are very similar to those seen by the Asp/Asn residues in other members of this superfamily. In fact, the carboxamide group of Gln185 in this alignment superimposes well with the functional groups in the Asn/Asp side chains in other proteases. For example, the potential Cif catalytic triad (Cys109-His165-Gln185) can be superimposed well with the catalytic triad of AvrPphB (Fig. 4a),16 other cysteine proteases such as papain (PDB ID code 1ppn),17 and staphopain (PDB ID code 1cv8),18 and arylamine N-acetyltransferase (NAT) from Salmonella enterica serovar Typhimurium (PDB ID code 1e2t) (Fig. 4a).19
Fig. 4.
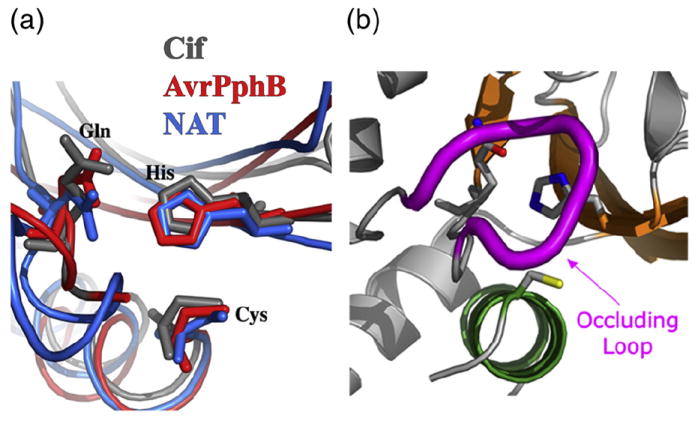
Putative catalytic triad of Cif superimposes well with triads of AvrPphB and NAT. (a) Superposition of the catalytic triads of Cif (gray), AvrPphB (red), and NAT (blue). (b) The catalytic triad of Cif, with the occluding loop shown in magenta.
In Cif, the nucleophile Cys109 is located on the N-terminus of α-helix H1 and His165 is located on the N-terminus end of the B2 β-strand. The third catalytic residue Gln185 lies on the C-terminal end of the B3 β-strand. The catalytic cysteine residue of AvrPphB lies on the N-terminus of a helix positioned similarly with respect to the β-sheet as helix H1 of Cif. As the His165 of Cif, the histidine residue of AvrPphB is located on a β-strand, and an aspartate residue is positioned on the β-strand.
A unique structural feature of Cif relative to other superfamily members is the presence of an occluding loop situated near the active site (Fig. 4b). The occluding loop (residues 189–195) obstructs part of the active site, generating a narrow protein-binding cleft (Fig. 5). As a consequence, the loop possibly restricts the active site accessibility to a polypeptide substrate. Such an occluding loop feature is not present in the structure of AvrPphB or NAT. These two proteins possess deep and extended substrate-binding clefts. To visualize Cif without the occluding loop, we generated an image of Cif with the loop residues 189–195 deleted. As shown in Fig. 5, the removal of the loop created a deeper catalytic cleft, which would allow the substrate to bind more efficiently.
Fig. 5.
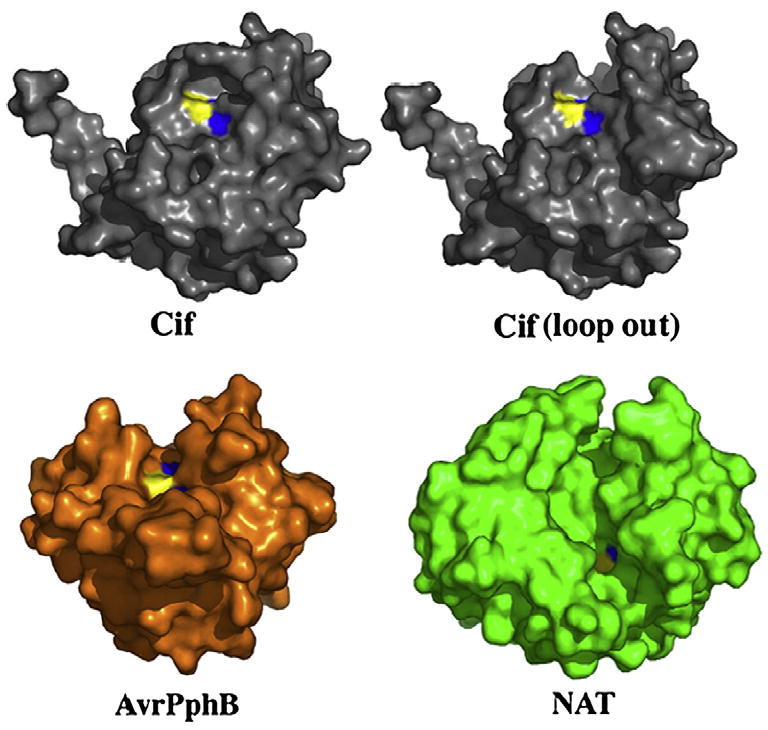
The Cif active site is occluded relative to other members of cysteine protease superfamily. Molecular surface representation of Cif, AvrPphB, and NAT. The catalytic triad residues are colored with the cysteine in yellow, the histidine in blue, and the glutamine in red (although it is not visible due to its buried nature). In silico removal of the occluding loop (residues 189–195) creates an extensive cleft, reminiscent of the substrate-binding grooves of AvrPphB and NAT.
The Cif catalytic triad is critical for actin stress fiber formation and cell-cycle arrest
To test the role of the putative catalytic triad in Cif function, single-point mutants, namely C109A and H165A were constructed by site-directed mutagenesis of native Cif. HeLa cells were infected with EPECΔcif mutant expressing plasmid-encoded wild-type Cif (Cif-WT) or Cif mutants. As expected, Cif-WT caused enlargement of cells, actin stress fibers formation and cell-cycle arrest at the G2/M phase transition (Fig. 6a). In contrast, cells infected with EPECΔcif expressing C109A or H165A mutants behaved like the control cells (Fig. 6a), indicating that these residues are critical for Cif activity and/or translocation. Despite many efforts in replacing Q185 with leucine, alanine, serine, asparagine, glutamate, and glycine, point mutations at this residue were insoluble in the bacterial recombinant protein expression systems and thus not tested in the infection assay (although mutants were tested in the BioPORTER assay, see below).
Fig. 6.
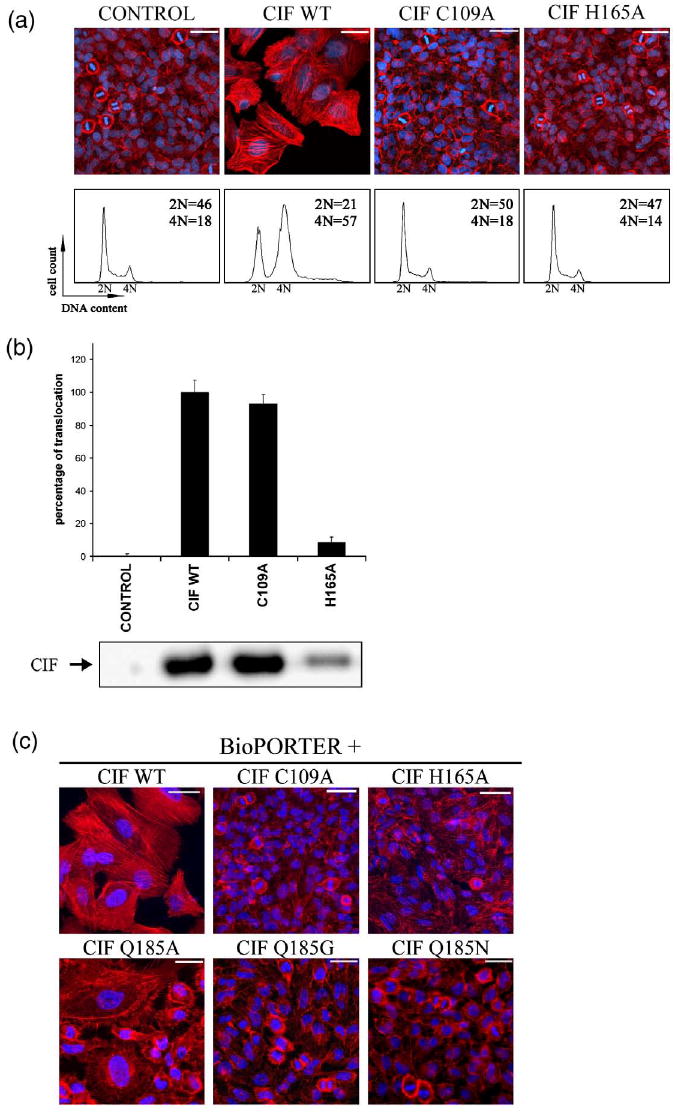
Cysteine and histidine from the Cif catalytic triad are critical for stress fiber formation and cell-cycle arrest. (a) G1/S synchronized HeLa cells were exposed for 90 min to EPEC E22Δcif hosting empty plasmid (control) or plasmid expressing wild-type Cif (WT), mutant C109A, or H165A. Upper panels: F-actin was labeled with phalloidin-rhodamine (red) and DNA was labeled with DAPI (blue) at 72 h post-infection. The scale bars represent 50 μm. Lower panels: cell-cycle distribution according to DNA content was analyzed by flow cytometry at 48 h post-infection. Percentages of 2 N and 4 N populations are indicated. (b) HeLa cells were exposed for 90 min to E22Δcif hosting plasmid expressing β-lactamase TEM-1 protein (control) or plasmid expressing the translational fusion between TEM-1 and Cif-WT, mutant C109A, or H165A. Upper panel: translocation levels were determined using CCF2-AM as a substrate for intracellular TEM-1 enzyme as described.7 Results are indicated as percentages compared to the translocation level of Cif-WT, which was set to 100%. The error bars represent the standard error of the mean from three independent experiments. Lower panel: the level of Cif proteins accumulated in bacteria just before the translocation assay was detected by Western blotting analysis using anti-Cif antibodies. (c) HeLa cells were exposed for 4 h to the indicated purified proteins in combination with a lipidic delivery agent (BioPORTER) and incubated for a further 72 h. F-actin was stained with phalloidin-rhodamine (red) and DNA was stained with DAPI (blue). The scale bars represent 50 μm.
To examine whether the loss of activity observed with the C109A and the H165A mutants was due to impaired translocation into HeLa cells, we monitored the injection of the mutated forms of Cif using the TEM1 β-lactamase fusion assay.7 The C109A mutant was translocated at a level similar to that of Cif-WT (Fig. 6b). In contrast, the H165A mutant was translocated at a tenfold lower level into infected HeLa cells. The poor translocation level of the H165A mutant could be explained by its lower level of expression and/or stability in bacteria as revealed by Western blotting analysis of bacterial lysates (Fig. 6b). These results confirmed that the cysteine residue of the putative catalytic triad is critical for Cif activity, since the C109A mutant is as well translocated as Cif-WT but fails to inhibit host cell-cycle progression. However, the lower level of translocation observed with the H165A mutant impaired analysis of the role of these residues in Cif function using the infection model.
To bypass the T3SS and the need to infect cells with bacteria, Cif-WT and mutant proteins were purified and introduced into HeLa cells using BioPORTER (Genlantis), a lipid-mediated protein delivery agent.9 Treatment of HeLa cells with a mixture of BioPORTER and purified Cif-WT protein resulted in cell enlargement and formation of actin stress fibers. In contrast, treatment with a mixture of BioPORTER and Cif C109A or H165A mutants did not affect the morphology of the cells (Fig. 6c). This alternative delivery method confirmed the inactivity of the C109A mutant. In addition, these results also demonstrated the essential role of the H165 residue, since its mutation abolished the phenotype upon lipid delivery. However, the mixture of BioPORTER and refolded Cif Q185A mutant induced a partial effect with both enlarged cells harboring actin stress fibers and unaltered cells, while Cif Q185N and Q185G did not affect the morphology of the cells (Fig. 6c). Q185 is buried in the interior of the protein, and thus mutation of this residue may compromise the protein’s integrity, resulting in improper folding as well as altered activity. Taken together, these results clearly indicate that the cysteine and histidine residues of the catalytic triad are critical for Cif cytotoxic activity. The poor solubility of Q185 mutants complicates the analysis of its role in the activity of Cif.
Cif remains active in the presence of protease inhibitors
We next attempted to demonstrate protease activity of purified recombinant Cif in vitro. Cif, catalytic triad mutants and various N-terminal deletion mutants were tested with a non-specific protease substrate (see Materials and Methods). In some trials, Hela cell extract was included in the assay buffer to examine whether eukaryotic host factor(s) were required for Cif’s activity. No promising result was obtained from either assay (Fig. 7). To test whether the occluding loop could restrict the accessibility to the active site, we generated a Cif mutant lacking residues 189–195, but this did not increase protease activity in the in vitro assay (Fig. 7b). Treatment of HeLa cells with a mixture of BioPORTER and the Cif Δ189-195 mutant protein did not induce stress fibers or cell cycle arrest (Fig. 8a), although the poor solubility and stability (data not shown) of this construct makes any interpretation of the results difficult, as the deletion likely compromised the protein at the structural level. In a further attempt to assess protease activity of Cif, we used E-64 (l-trans-epoxysuccinyl-leucylamido(4-guanidino) butane, C15H27N5O5), a non-competitive, irreversible inhibitor of cysteine proteases. However, addition of E-64 in the mixture of BioPORTER and Cif-WT did not impair Cif activity (Fig. 8b). This latter experiment suggests that cysteine protease activity is not required for the Cif-induced phenotype.
Fig. 7.
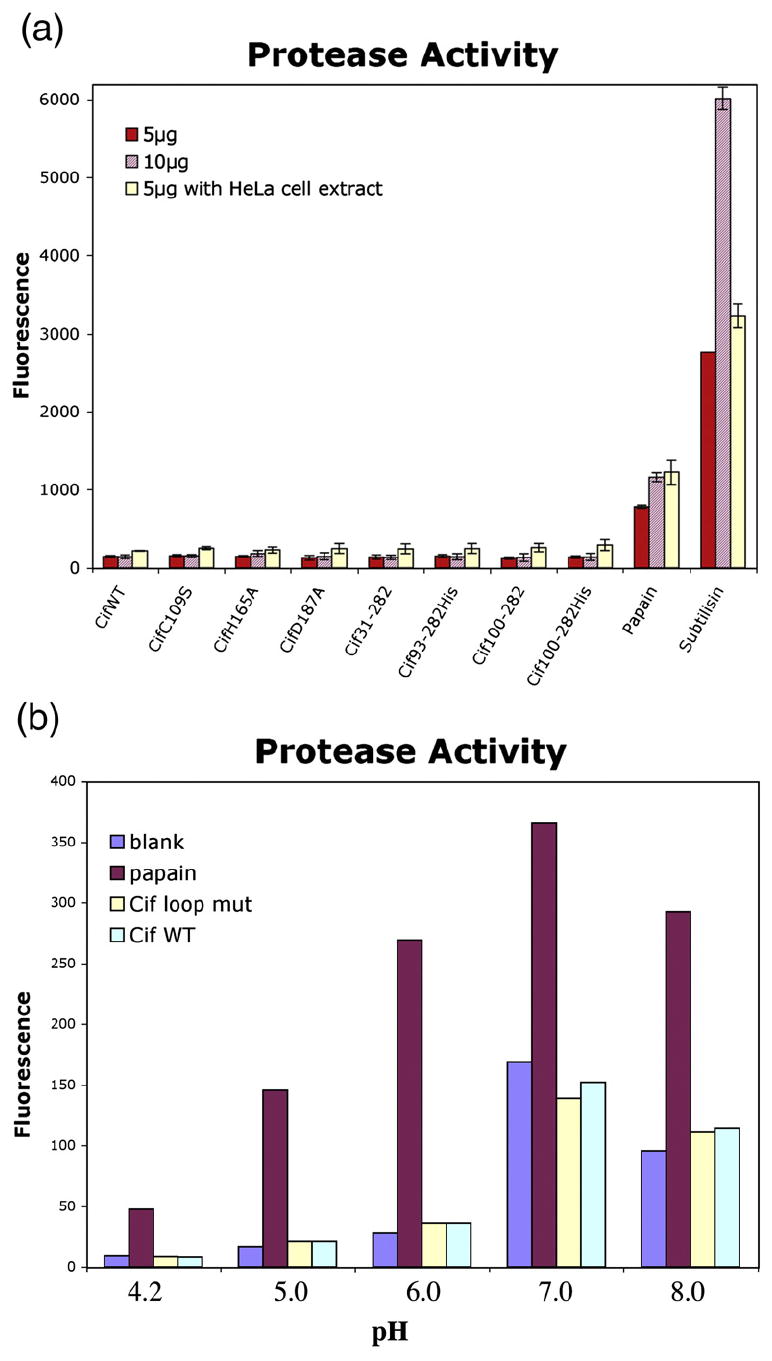
Cif does not cleave general protease substrate casein in vitro. (a) Cif does not possess general protease activity in vitro. Either 5 μg or 10 μg of purified Cif constructs were incubated with BODIPY FL conjugated casein for 1 h at room temperature with or without HeLa cell extract in the buffer. The fluorescence of BODIPY FL-labeled protease hydrolysis products was measured (excitation 485 nm, emission 530 nm). Either 5 μg or 10 μg of cysteine (papain) and serine (subtilisin) proteases were included as positive controls. The error bars represent the standard error of the mean from three independent experiments. (b) The Cif loop mutant Δ189-195 was incubated with BOD-IPY FL casein at various pH values.
Fig. 8.

Cysteine protease activity is not required for the Cif-induced phenotype. Cellular morphology of HeLa cells treated with BioPORTER reagent combined with either mock (DMSO), wild type, or mutant Cif. F-actin was stained with phalloidin-rhodamine (red) and DNA was stained with DAPI (blue). The scale bars represent 20 μm. (a) The effect of the Cif Δ189-195 loop mutant. (b) Cif was first incubated for 10 min with the irreversible cysteine protease inhibitor E-64 (Sigma) at a concentration of 70 μM.
Discussion
Bacterial pathogens utilize a wide array of virulence factors to modulate host cell biochemistry. In particular, the protein substrates of T3SS possess remarkable properties, altering the host cytoskeleton, signal transduction, cell-cycle progression, and programmed cell death.20 One of these translocated effectors, Cif, from pathogenic strains of E. coli is associated with a cytopathic effect involving cell-cycle arrest and cell distension.6 The molecular basis for this activity has remained elusive.
The crystal structure of Cif presented here shows that this T3SS substrate is a divergent member of the cysteine protease superfamily. Cif possesses a conserved core fold containing a putative catalytic triad composed of cysteine, histidine, and glutamine, which aligns well with triads in other proteases and several acetyltransferases. Mutation of any of these residues results in a Cif molecule that cannot fully induce cell-cycle arrest during infection or when introduced exogenously into cells.
While all known catalytic triads of this superfamily studied to date possess either aspartic acid or asparagine in the third position, Cif possesses glutamine. Although longer than asparagine, glutamine has as its functional group the same carboxamide, and this functional group in Cif Gln185 and those of asparagine-containing members of the superfamily align well. As in these other enzyme structures, Gln185 aids in positioning the catalytic residue His165 through a hydrogen bond between the side chains. The close chemical similarities as well as the convincing structural alignment with Asn/Asp residues in other catalytic triads, suggests that Cif has evolved to use glutamine in this position.
Based on these homologies, Cif may possess either protease activity or acetyltransferase activity. In fact, as has been shown to be the case with the Yersinia effector YopJ,21,22 it could possess both activities. To date, we have been unable to detect any protease activity of purified recombinant Cif in vitro. This may be due to the fact that Cif possesses high specificity for its intracellular substrate, such that it does not cleave a general protease substrate in an in vitro assay. On the other hand, as with clostridial neurotoxins and anthrax lethal factor,23,24 Cif may interact with a specific structural motif to fully engage its substrate. It is possible also that full-length Cif could be inactive (as a zymogen) and require a eukaryotic host factor for activation through processing or modification. Such activation could resemble that of the auto-proteolytic activity of the Pseudomonas syringae effector AvrRpt2, which requires the eukaryotic host factor cyclophilin to induce self-cleavage.25 A specific structural feature of Cif relative to other proteins of the cysteine protease superfamily is a loop that occludes part of the active site. It is conceivable that interaction with and/or modification by eukaryotic partner(s) could induce conformational changes allowing more efficient access of the substrate to the catalytic site. Arguing against a cysteine protease activity for Cif, we have shown that the cytopathic effect induced by this virulence factor is not affected by the cysteine protease inhibitor E-64, suggesting that the Cif-induced phenotype is due to another enzymatic activity. In addition to cysteine proteases, Cif shares structural homology with arylamine N-acetyltransferases. As NAT is not inhibited even by high concentrations of E-64 or other cysteine protease inhibitors,26 this leaves open the possibility that such an activity for Cif is responsible for the cytopathic phenotype. The identification of the host cellular targets for Cif should provide a direct means to examine the enzymatic activities suggested by the structure.
The eukaryotic cell-cycle is a tightly regulated process, including checkpoints that govern the potential of a cell to undergo growth or death. Uncontrolled cell proliferation is associated with tumorigenesis and other pathologies. However, cellular proliferation is critical to health and development, particularly in the expansion of immune cells during infection and the turnover of infected tissues to clear them from the body. Cif prevents proliferation in cells by arresting the cell-cycle. For cells that are constantly undergoing replication, such those of the intestinal epithelium, Cif may prevent shedding in order to prolong bacterial colonization. It is possible also that this activity allows bacteria to evade the immune system by inhibiting clonal expansion of lymphocytes. In these regards, Cif shows similarity to bacterial genotoxins such as CDT and Colibactin, which induces DNA damage in the host cell leading to cell-cycle arrest.11,27 Therefore, despite very different mechanisms of inducing cell-cycle arrest, these factors may share a common biological purpose.
Cif belongs to the superfamily of enzymes that includes cysteine proteases and acetyltransferases, both categories of enzymes that have been shown to function in bacterial pathogenesis. The mutagenesis of the predicted catalytic triad residues in Cif results in loss of host cell-cycle arrest normally induced by the pathogen, linking the proposed protease or acetyltransferase function from the crystal structure to the cytopathic phenotype. Future studies will focus on finding the host cell substrate(s) for Cif and elucidating its functional role in causing cytopathic effects. Understanding how Cif is able to modulate the host cell-cycle may illuminate mechanisms of the host cell-cycle regulation from a new perspective.
Materials and Methods
Protein preparation
The 846 bp cif gene was synthesized using assembly PCR and subsequently subcloned into a modified pET21a vector. The cif gene oligos were designed using a web-based algorithm (DNAWorks). A total of 42 oligos with lengths ranging from 32 to 51 bases were generated. BamHI, HindIII, NotI, and SalI restriction sites were specifically excluded from the DNA sequence for subcloning purposes. A SalI sequence was included in the 5′ flanking sequence, and a NotI sequence was included in the 3′ flanking sequence. The synthesized oligos were combined and assembled using a two-step PCR method. The full-length cif PCR product was purified and cloned into the pCR2.1-TOPO vector (Invitrogen). Cif residues 100–282 was subcloned into modified E. coli expression vectors pET21a using SalI and NotI sites and then transformed into BL21(DE3) cells. This construct possessed His6 tags at both the N- and C-termini. All clones were verified by DNA sequencing.
E. coli BL21(DE3) harboring the Cif100-282 construct was grown at 37 °C to an A600 nm of 0.8, then induced with 1 mM IPTG and grown for an additional 4 h. The bacteria were first harvested then lysed by sonication. Cif100-282 was denatured in 8 M urea and purified using nickel chelating resin (GE Healthcare). The denatured protein was refolded into the native state by gradual removal of the denaturing agent through dialysis. The N-terminal His6 tag was removed by site-specific proteolytic cleavage, leaving the C-terminal His6 tag in place (henceforth, the construct is called Cif100-282-His to distinguish it from Cif100-282 without any tags, which is important below). The protein was further purified by Source 15Q anion-exchange and Superdex 75 gel-filtration chromatography (GE Healthcare). Purified protein was concentrated to 25 mg/ml in a buffer containing 20 mM Tris, 200 mM NaCl, and 2 mM DTT, at pH 8.0.
The importance of Cif C-terminal helix was studied by gel-filtration chromatography using a 120 mL Superdex 200 HiLoad 16/60 column (GE Healthcare) with an ÅKTA FPLC. Following anionic-exchange chromatography, 2 ml of various Cif constructs were injected separately onto an equilibrated column in either low-salt buffer (20 mM Tris pH 8.0, 50 mM NaCl, and 2 mM DTT) or high-salt buffer (20 mM Tris pH 8.0, 200 mM NaCl, and 2 mM DTT).
Crystallization, data collection, and structural determination
Crystals of Cif100-282-His appeared after two days at 4 °C in 1.85–2.4 M sodium malonate and 0.1 M sodium acetate anhydrous pH 4.2–5.2 or trisodium citrate dehydrate, pH 3.8. Cif crystals were first observed in a 96-well sitting-drop plate using Qiagen’s (formally Nextel) Anionic Screen and later reproduced and optimized in 24-well plates using the hanging-drop, vapor-diffusion method, with the largest crystals grown in 1.85 M sodium malonate, 0.1 M trisodium citrate dihydrate, pH 3.8. Despite repeated efforts, Cif100-282 without the C-terminal His6 tag could not be crystallized, even with seeding with crystals of Cif100-282-His. Selenomethionine (SeMet) was incorporated into Cif by transforming the Cif100-282-His-containing plasmid into methionine autotroph strain 834(DE3). The strain was grown and induced in minimal medium containing selenomethionine in place of methionine. SeMet Cif100-282-His was purified and crystallized by the protocols used for native Cif100-282-His.
Diffraction data for the native and SeMet Cif100-282-His crystals were collected at X29 and X9A beamlines, respectively, at the National Synchrotron Light Source. The crystals were cryo-protected with 2.6 M sodium malonate, pH 7.0. SeMet Cif diffracted to 2.0 Å, and native Cif diffracted to 1.7 Å. Data were processed with HKL2000.28 Phases were determined and improved using the programs SOLVE/RESOLVE by using the anomalous signal from the scattering of selenium sites incorporated into the protein.29 There were two Cif molecules per asymmetric unit. The initial model was built using Arp/Warp 6.1,30 and further refined with REFMAC5 using 1.7 Å resolution data.31 The final model was completed through iterative cycles of manual building with O,32 and refinement with REFMAC5 (Table 1).33 Images were generated using PyMOL†.
Mutagenesis of the putative catalytic triad
Point mutations C109S, C109A, H165A, Q185A, Q185G, and Q185N were made to probe whether these residues were members of a catalytic triad. Amino acid substitutions were introduced into the cif gene by in vitro site-directed mutagenesis using oligos containing specific base changes. The mutant plasmids were amplified by PCR using Pfu Turbo DNA polymerase (Stratagene). The template plasmid was removed by digestion with DpnI before transformation. The Cif full-length point mutants were sequence-verified and transformed into EPEC strain E22 mutated for cif (E22Δcif).6
Production of anti-Cif antibodies
The His6-Cif protein was purified as described,9 and used to immunize three rabbits. To enhance specificity, the final sera were affinity-purified on a column of cross-linked Cif-GST-glutathione agarose. Bound anti-Cif antibodies were eluted at pH 2, dialyzed and stored at −20 °C.
Infection and BioPORTER assay
Bacterial strains were cultured overnight in Luria-Bertani (LB) broth then subcultured 1:100 in interaction medium (DMEM with 25 mM Hepes and 5% (v/v) fetal calf serum (FCS)) for 3 h at 37 °C. HeLa cells (ATCC CCL-2), synchronized at the G1/S phase transition by a double thymidine block, were washed and infected in interaction medium with a multiplicity of infection (MOI) of 100:1. Following infection for 90 min, the cells were washed and cultivated for the indicated times in DMEM supplemented with 10% FCS and 200 μg/ml gentamicin.
BioPORTER (Genlantis) assays were performed with purified Cif proteins (WT and mutants) as described.9 When indicated, E-64 (Sigma) was used at 70 μm and incubated for 10 min with purified Cif (WT or mutant) before mixing with BioPORTER.
Stress fiber formation and cell-cycle analysis
Study of cell morphology and visualization of cytoskeleton was done as described;9 briefly, HeLa cells in chamber slides were fixed for 15 min in 4% (v/v) formaldehyde, permeabilized in 0.1% (v/v) Triton X-100 and then incubated with rhodamine-phalloidin (Molecular Probes) to stain F-actin and with 4′,6-diamidino-2-phenylindole (DAPI, Sigma) to stain DNA. Images were acquired with a DMRB fluorescence microscope equipped with a DFC300FX digital camera (Leica).
Cell-cycle distribution analysis was done with a FACScalibur flow cytometer (Becton Dickinson) as described.34 Data from at least 10,000 cells were analyzed using FloJo software v8.5 (Tree Star).
Translocation assay
C-terminal translational fusions with TEM-1 β-lactamase were constructed to analyze the translocation efficiency of the triad mutants. All constructs were sequence-verified and transformed into E22Δcif strain. HeLa cells were infected, and translocation levels were determined using CCF2 as a substrate for intracellular TEM-1 enzyme as described.7 To determine the level of Cif protein in bacteria, bacterial culture with identical A600 nm were boiled for 5 min in SDS-PAGE sample buffer and subjected to Western blot analysis with anti-Cif antibodies.
In vitro protease assay
A non-specific protease substrate casein derivative labeled with green-fluorescent BODIPY FL (EnzChek Protease Assay Kit, Molecular Probes) was used to test Cif protease activity. The assay was performed as suggested by the manufacturer. Briefly, 5 or 10 μg of Cif, active site mutants, various N-terminal deletion mutants, and protease controls (papain and subtilisin) were added to BODIPY FL casein and incubated at room temperature, protected from light, for at least 1 h in 10 mM Mes, pH6.5. The detection of cleavage products was followed by fluorescence (excitation 485 nm, emission 530 nm). HeLa extract (200 μg) was included in the assay buffer in some trials as indicated.
Protein Data Bank accession number
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 3EFY.
Acknowledgments
Protein N-terminal sequencing and mass spectroscopic analysis for Cif domain delineation were done by the Protein Resource Center of the Rockefeller University under the direction of H. Deng. We thank H. Mueller at Rockefeller University, R. Udupi of Brookhaven beamline X9A, and W. Shi of Brookhaven Beamline X29 for access to and assistance with crystallographic equipment. We thank Claude Watrin and Peggy Binaut for help in producing anti-Cif antibodies. This work was funded by research funds to C.E.S. from the Rockefeller University, to E.O. from the French Agence Nationale de la Recherche (ANR-05-MIIM-009) and to F.T. from the French Ligue Nationale Contre le Cancer. The authors declare that they have no competing financial interests.
Abbreviations used
- T3SS
type III secretion system
- EPEC
enteropathogenic Escherichia coli
- EHEC
enterohemorrhagic Escherichia coli
Footnotes
References
- 1.Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- 2.Oswald E, Nougayrede JP, Taieb F, Sugai M. Bacterial toxins that modulate host cell-cycle progression. Curr Opin Microbiol. 2005;8:83–91. doi: 10.1016/j.mib.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Nougayrede JP, Taieb F, De Rycke J, Oswald E. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005;13:103–110. doi: 10.1016/j.tim.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Nataro JP, Steiner T, Guerrant RL. Enteroaggregative Escherichia coli. Emerg Infect Dis. 1998;4:251–261. doi: 10.3201/eid0402.980212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garmendia J, Frankel G, Crepin VF. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect Immun. 2005;73:2573–2585. doi: 10.1128/IAI.73.5.2573-2585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marches O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, et al. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol Microbiol. 2003;50:1553–1567. doi: 10.1046/j.1365-2958.2003.03821.x. [DOI] [PubMed] [Google Scholar]
- 7.Charpentier X, Oswald E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J Bacteriol. 2004;186:5486–5495. doi: 10.1128/JB.186.16.5486-5495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nougayrede JP, Boury M, Tasca C, Marches O, Milon A, Oswald E, De Rycke J. Type III secretion-dependent cell cycle block caused in HeLa cells by enteropathogenic Escherichia coli O103. Infect Immun. 2001;69:6785–6795. doi: 10.1128/IAI.69.11.6785-6795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taieb F, Nougayrede JP, Watrin C, Samba-Louaka A, Oswald E. Escherichia coli cyclomodulin Cif induces G2 arrest of the host cell-cycle without activation of the DNA-damage checkpoint-signalling pathway. Cell Microbiol. 2006;8:1910–1921. doi: 10.1111/j.1462-5822.2006.00757.x. [DOI] [PubMed] [Google Scholar]
- 10.Nesic D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- 11.Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 12.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 13.Andreeva A, Howorth D, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawlings ND, Barrett AJ. Families of cysteine peptidases. Methods Enzymol. 1994;244:461–486. doi: 10.1016/0076-6879(94)44034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao F, Merritt PM, Bao Z, Innes RW, Dixon JE. A Yersinia effector and a Pseudomonas avirulence protein define a family of cysteine proteases functioning in bacterial pathogenesis. Cell. 2002;109:575–588. doi: 10.1016/s0092-8674(02)00766-3. [DOI] [PubMed] [Google Scholar]
- 16.Zhu M, Shao F, Innes RW, Dixon JE, Xu Z. The crystal structure of Pseudomonas avirulence protein AvrPphB: a papain-like fold with a distinct substrate-binding site. Proc Natl Acad Sci USA. 2004;101:302–307. doi: 10.1073/pnas.2036536100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamphuis IG, Kalk KH, Swarte MB, Drenth J. Structure of papain refined at 1.65 Å resolution. J Mol Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann B, Schomburg D, Hecht H-J. Crystal structure of a thiol proteinase from Staphylococcus aureus V-8 in the E-64 inhibitor complex. Acta Crystallogr. 1993;49(Suppl):102. [Google Scholar]
- 19.Sinclair JC, Sandy J, Delgoda R, Sim E, Noble ME. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nature Struct Biol. 2000;7:560–56. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- 20.Galan JE, Cossart P. Host-pathogen interactions: a diversity of themes, a variety of molecular machines. Curr Opin Microbiol. 2005;8:1–3. doi: 10.1016/j.mib.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 22.Orth K, Xu Z, Mudgett MB, Bao ZQ, Palmer LE, Bliska JB, et al. Disruption of signaling by Yersinia effector YopJ, a ubiquitin-like protein protease. Science. 2000;290:1594–1597. doi: 10.1126/science.290.5496.1594. [DOI] [PubMed] [Google Scholar]
- 23.Pellizzari R, Rossetto O, Lozzi L, Giovedi S, Johnson E, Shone CC, Montecucco C. Structural determinants of the specificity for synaptic vesicle-associated membrane protein/synaptobrevin of tetanus and botulinum type B and G neurotoxins. J Biol Chem. 1996;271:20353–20358. doi: 10.1074/jbc.271.34.20353. [DOI] [PubMed] [Google Scholar]
- 24.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 25.Coaker G, Falick A, Staskawicz B. Activation of a phytopathogenic bacterial effector protein by a eukaryotic cyclophilin. Science. 2005;308:548–550. doi: 10.1126/science.1108633. [DOI] [PubMed] [Google Scholar]
- 26.Rodrigues-Lima F, Delomenie C, Goodfellow GH, Grant DM, Dupret JM. Homology modelling and structural analysis of human arylamine N-acetyltransferase NAT1: evidence for the conservation of a cysteine protease catalytic domain and an active-site loop. Biochem J. 2001;356:327–334. doi: 10.1042/0264-6021:3560327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr D. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 33.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 34.Sert V, Cans C, Tasca C, Bret-Bennis L, Oswald E, Ducommun B, De Rycke J. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene. 1999;18:6296–6304. doi: 10.1038/sj.onc.1203007. [DOI] [PubMed] [Google Scholar]