

Here, the crystal structure of TM0439, a GntR regulator with an FCD domain found in the Thermotoga maritima genome, is described.
Keywords: transcription regulation, GntR family, structural genomics, surface-entropy reduction
Abstract
The GntR superfamily of dimeric transcription factors, with more than 6200 members encoded in bacterial genomes, are characterized by N-terminal winged-helix DNA-binding domains and diverse C-terminal regulatory domains which provide a basis for the classification of the constituent families. The largest of these families, FadR, contains nearly 3000 proteins with all-α-helical regulatory domains classified into two related Pfam families: FadR_C and FCD. Only two crystal structures of FadR-family members, those of Escherichia coli FadR protein and LldR from Corynebacterium glutamicum, have been described to date in the literature. Here, the crystal structure of TM0439, a GntR regulator with an FCD domain found in the Thermotoga maritima genome, is described. The FCD domain is similar to that of the LldR regulator and contains a buried metal-binding site. Using atomic absorption spectroscopy and Trp fluorescence, it is shown that the recombinant protein contains bound Ni2+ ions but that it is able to bind Zn2+ with K d < 70 nM. It is concluded that Zn2+ is the likely physiological metal and that it may perform either structural or regulatory roles or both. Finally, the TM0439 structure is compared with two other FadR-family structures recently deposited by structural genomics consortia. The results call for a revision in the classification of the FadR family of transcription factors.
1. Introduction
Transcription regulators play a critical role in the biology of microorganisms (Huffman & Brennan, 2002 ▶). They repress, de-repress and activate gene transcription through tightly regulated direct interactions with cognate DNA sequences mediated by a variety of unique domains or motifs such as helix–turn–helix domains, zinc fingers, homeodomains, leucine zippers and β-sheet DNA-binding proteins. Within the helix–turn–helix (HTH) regulators, numerous superfamilies have been identified based on sequence similarities in the DNA-binding module. The GntR superfamily, Pfam PF00392 (Bateman et al., 2002 ▶), which was first described in 1991 and named after the gluconate operon repressor in Bacillus subtilis (Haydon & Guest, 1991 ▶), currently comprises over 6200 proteins found in diverse eubacterial genomes. The DNA-binding domains in this family share a significant level of similarity and all exhibit the winged helix–turn–helix (WH) topology with the canonical HTH motif followed by a β-hairpin. In contrast, the C-terminal regulatory ligand-binding domains vary significantly among individual proteins, providing a basis for the current classification of major families, i.e. HutC, MocR, YtrA, AraR, PlmA and, the largest family comprising ∼40% of all GntRs, FadR (Rigali et al., 2002 ▶; Lee et al., 2003 ▶; Franco et al., 2006 ▶). By far the best characterized GntR regulator is the fadR gene product, the founding member of the FadR family. It functions as a repressor of the fad regulon, which includes genes responsible for transport, activation and β-oxidation of long and medium-length fatty acids (DiRusso et al., 1992 ▶, 1993 ▶). The crystal structure of the apo repressor, as well as the structures of complexes with the dsDNA operon oligonucleotide and with an effector, myristoyl-CoA, have been determined (van Aalten et al., 2000 ▶, 2001 ▶; Xu et al., 2001 ▶). These studies revealed the mechanism by which the effector-induced conformation changes in the regulatory domain are transmitted to the WH domain and consequently disrupt the repressor–operon interaction, thereby relieving repression (van Aalten et al., 2001 ▶).
All known FadR-family transcription regulators are predicted to contain all-α-helical C-terminal domains with either seven or six α-helices. An accurate alignment has been elusive because of low levels of amino-acid similarity. However, the predicted number of helices serves as the basis for one classification scheme, sorting proteins into the FadR (seven helices) and VanR (six helices) groups (Rigali et al., 2002 ▶). Both groups appear to be involved at the crossroads of metabolic pathways, e.g. galactonate (DgoR), gluconate (GntR), vanillate (VanR), malonate (MalR) etc. An alternative classification of regulatory domains of FadR members is offered by the Pfam database (Bateman et al., 2002 ▶). The smaller FadR_C family (Pfam07840), represented by the C-terminal domain from FadR itself, comprises only ∼70 members exhibiting high amino-acid similarity. All proteins in this family have C-terminal domains of the FadR group, i.e. with seven helices. Interestingly, in the vast majority of cases there is one gene of this type per bacterial genome. The larger and more diverse FCD family (Pfam007729) has over 2800 known members in more than 400 species. It includes domains with both six and seven predicted α-helices, i.e. members of both the FadR and VanR groups.
Recently, atomic coordinates for three new structures of putative FadR-like transcription regulators were deposited in the PDB. Two of these were reported by structural genomics groups without accompanying publications: RO03477 from Rhodococcus sp. RHA1 (PDB code 2hs5; K. Tan, T. Skarina, A. Onopriyenko, A. Savchenko, A. Edwards & A. Joachimiak) and PS5454 protein from Pseudomonas syringae pv. tomato strain DC3000 (PDB code 3c7j; B. Nocek, A. Sather, M. Gu & A. Joachimiak). Both structures contain C-terminal domains with six α-helices, making them VanR-group members. The third structure, that of CGL2915 protein from Corynebacterium glutamicum (PDB code 2di3), is a FadR-group member as judged by the seven helices in its C-terminal domain (Gao et al., 2008 ▶). However, in spite of the size difference, all three proteins are annotated in the Pfam database as containing FCD domains.
In this paper, we describe the structure of TM0439, a putative transcriptional regulator from Thermotoga maritima. Based on amino-acid sequence, its regulatory domain was also annotated as an FCD-family member. We have compared the structure of TM0439 with those of FadR and the three newly deposited related transcriptional regulators and we show that, together with CGL2915 and PS5454, TM0439 is a member of a distinct yet previously unrecognized group of metal-binding transcription regulators in which a distinct variant of the FCD domain contains a metal-binding site. This domain is identified by a conserved fingerprint sequence motif: Arg-X 3-Glu-X 40-Asx-X 4-His-X ∼50-His-X ∼20-His. Although the metal in the TM0439 crystal structure is Ni2+, we determined experimentally that the protein can bind both Ni2+ and Zn2+, with K d values in the nanomolar (or lower) range, making Zn2+ the more probable biological ligand. Our study sets the stage for an improved annotation of the FadR family of transcription regulators and offers a structural rationale for the strict conservation of a unique sequence motif in a subset of these proteins.
2. Materials and methods
2.1. Protein expression and purification
The TM0439 gene was cloned as part of the structural genomics project on the T. maritima proteome (Lesley et al., 2002 ▶). As in other JCSG (Joint Center for Structural Genomics) expression vectors, there is a noncleavable N-terminal tag (MGSDKIHHHHHH) as well as both arabinose and T7 promoters. The wild-type protein, expressed and purified using routine methods, did not crystallize. To circumvent this problem, three mutants with reduced surface entropy, E118A,K119A,K122A (variant 1A), K2A,K3A (variant 2A) and E30A,K31A (variant 3A), were designed using the Surface Entropy Reduction Prediction (SERp) server (http://nihserver.mbi.ucla.edu/SER/) and created using the QuikChange protocol (Stratagene Inc.). Expression was carried out in Escherichia coli BL21 strain in M9 media with added SeMet for labeling. The protein was purified using nickel-affinity chromatography (Ni–NTA agarose column, Qiagen). Pure fractions were pooled together and dialyzed overnight against a buffer consisting of 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 2.5 mM β-mercaptoethanol (β-ME). Protein samples were concentrated to 15 mg ml−1 and stored at 193 K.
2.2. Crystallization and data collection
The mutant proteins were screened using the Wizard II crystallization matrix from Emerald Biosystems using reservoirs containing either the screen solution or 1.5 M NaCl (Newman, 2005 ▶). The triple mutant 1A yielded diffraction-quality crystals directly from the screen, i.e. 0.1 M acetate buffer pH 4.5, 35%(v/v) MPD. The crystals displayed C2 symmetry, with unit-cell parameters a = 85.19, b = 71.72, c = 43.32 Å, β = 104.6°. A MAD data set was collected on beamline 8.2.1 at ALS equipped with an ADSC Q315R detector. All data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶) with data statistics shown in Table 1 ▶.
Table 1. Crystallographic data.
(a).
Data-collection statistics.
| Peak | Edge | Remote | |
|---|---|---|---|
| Wavelength | 0.97960 | 0.97980 | 0.95370 |
| Resolution (Å) | 50–2.10 (2.18–2.10) | 50–2.10 (2.18–2.10) | 50–2.10 (2.18–2.10) |
| Total reflections | 77866 | 101252 | 94823 |
| Unique reflections | 12020 | 14439 | 14002 |
| Redundancy | 6.5 (3.6) | 7.0 (5.1) | 6.8 (4.2) |
| Completeness (%) | 81.7 (27.4) | 97.8 (84.2) | 94.5 (64.9) |
| Rmerge† (%) | 6.3 (35.8) | 5.4 (20.9) | 5.3 (28.6) |
| Average I/σ(I) | 31.2 (2.5) | 52.6 (5.5) | 42.4 (3.4) |
| Wilson B factor (Å2) | 29.7 | 34.0 | 33.5 |
(b).
Refinement statistics.
| Wavelength | 0.95370 |
| Resolution (Å) | 36.1–2.2 (2.42–2.20) |
| Completeness (%) | 97.6 (91.0) |
| Reflections (working) | 12586 |
| Reflections (test) | 620 |
| Rwork‡ (%) | 15.7 (16.7) |
| Rfree‡ (%) | 22.8 (27.7) |
| No. of waters | 81 |
| R.m.s. deviation from ideal geometry | |
| Bonds (Å) | 0.017 |
| Angles (°) | 1.31 |
| Average B factors§ (Å2) | |
| Main chain | 38.9 |
| Side chain | 38.1 |
| Waters | 50.2 |
| MOLPROBITY results | |
| Overall clashscore | 4.89 [98th percentile] |
| Ramachandran favored | 203 [98.1%] |
| Ramachandran outliers | 1 [0.5%] |
R
merge = 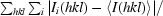
 , where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of the reflections. The values are for unmerged Friedel pairs.
, where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean intensity of the reflections. The values are for unmerged Friedel pairs.
R
work = 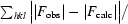
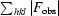 ; R
free is the same but for reflections belonging to a test set of randomly selected data.
; R
free is the same but for reflections belonging to a test set of randomly selected data.
B factors were refined using the TLS approximation (see §2).
2.3. Structure solution and refinement
The asymmetric unit contains one protein molecule, corresponding to a solvent content of 58.0%. Using MAD data, three selenium sites were located and phase calculations were carried out using SOLVE/RESOLVE (Terwilliger, 2003 ▶). Approximately two-thirds of the structure was built automatically. Model building and refinement of the SeMet structure were carried out using the data set collected at the remote high-energy wavelength, which was truncated at 2.2 Å to ensure completeness in the high-resolution shell (Table 1 ▶). Iterative refinement and model building were performed using RESOLVE and REFMAC5 (Murshudov et al., 1997 ▶). This process dramatically improved the maps and the missing fragments were identified in intermediate models. A combination of ‘cut-and-paste’ model building and manual refinement resulted in a complete structure. This iterative process allowed the refinement, which had previously stalled with an R free around 0.32, to converge with crystallographic R and R free values of 0.17 and 0.23, respectively. The final model was refined with PHENIX (Zwart et al., 2008 ▶) using the TLS (translation/libration/screw) approximation of thermal motion (Winn et al., 2001 ▶). Validation of the model was carried out using MOLPROBITY (Lovell et al., 2003 ▶). The corresponding refinement statistics are shown in Table 1 ▶. Figures were prepared with PyMOL (http://pymol.sourceforge.net/). The analysis of the dimer interface was performed using PISA v.1.15 (Krissinel & Henrick, 2007 ▶). Cavity volumes were calculated using VOIDOO (Kleywegt & Jones, 1994 ▶). For CGL2915, our cavity-volume calculation yielded results that differed from those reported in the literature (Gao et al., 2008 ▶).
2.4. Metal analysis
Stock metal concentrations and the metal content of TM0439 were determined using a PerkinElmer AAnalyst 400 atomic absorption spectrometer (AAS) with standard curves generated from NIST standards from Alfa Aesar (Ward Hill, Massachusetts, USA). Initial metal-content data were verified by ICP–OES (inductively coupled plasma–optical emission spectroscopy) at Dartmouth College Elemental Analysis Laboratory (Hanover, New Hampshire, USA). The complete removal of metal was accomplished by several rounds of extensive dialysis with 10 mM EDTA (ethylenediamine tetracetic acid) and 2 mM DTT (dithiotheitol) in 25 mM Tris and 100 mM NaCl at pH 8.0 and 277 K and was verified by AAS. Removal of DTT and EDTA was accomplished by four rounds of dialysis under an inert argon atmosphere with thoroughly degassed buffer (25 mM Tris and 100 mM NaCl at pH 8.0). Zn2+- and Ni2+-binding assays were performed by monitoring tryptophan fluorescence (λex = 287) on an ISS PC1 spectrofluorimeter under strictly anaerobic conditions. The concentration of TM0439 was 5.3 µM (25 mM Tris and 100 mM NaCl at pH 8.0 and 98 K). The data were fitted to appropriate chemical models (2:1 and 1:1, respectively) using DynaFit (Kuzmic, 1996 ▶) with metal–buffer interactions [logK ZnTris = 2.27; logK NiTris = 2.67; logβ2,Ni(Tris)2 = 4.6; NIST Standard Reference Database 46; http://www.nist.gov/srd/nist46.htm] included in the model.
3. Results and discussion
3.1. Design of crystallizable mutant
TM0439 was originally selected as one of the targets for a high-throughput pipeline at the Joint Center for Structural Genomics (Lesley et al., 2002 ▶). However, the wild-type protein did not yield X-ray-quality crystals. In order to overcome this problem, we used surface-entropy reduction (Derewenda, 2004 ▶) to generate variants of the protein with enhanced crystallizability. We used the SERp server (Goldschmidt et al., 2007 ▶) to predict suitable mutations to generate surface patches with reduced conformational entropy and enhanced ability to mediate crystal contacts and generate X-ray-quality crystals (Derewenda & Vekilov, 2006 ▶; Derewenda, 2004 ▶). Three mutants were suggested by the server: in order of ranking they were a triple mutant E118A,K119A,K122A, a double mutant K2A,K3A and another double mutant E30A,K31A. All three were expressed and screened for crystallization as described in §2. The triple mutant gave crystals with excellent morphology and diffraction properties directly from the crystallization screen and this crystal form was used in the subsequent analysis.
3.2. Overview of the structure and comparison with other FadR-family members
The crystal structure of TM0439 was determined by multiwavelength anomalous dispersion (MAD) using SeMet-labeled protein. The atomic model was refined to 2.2 Å resolution (Table 1 ▶; see §2). The protein has the canonical domain architecture of the GntR family, with an N-terminal WH domain and a C-terminal all-α-helical putative regulatory domain. The presence of only six α-helices within the C-terminal domain classifies TM0439 as a VanR member. Gel-filtration experiments (not shown) indicated that the protein was an obligate dimer in solution. The C2 space-group symmetry allows the formation of a head-to-head dimer via the crystallographic twofold axis, so that a large interface is buried between two C-terminal regulatory domains, with a resulting quaternary structure very close to that of FadR (van Aalten et al., 2000 ▶). In contrast, the two WH domains do not interact with one another, although they make limited crystal contacts with neighboring molecules in the unit cell. A comparison of TM0439 with FadR and with the recently deposited structures CGL2915, RO03477 and PS5454 shows dramatic differences in local tertiary and quaternary architectures, even though the individual domains are remarkably similar (Fig. 1 ▶).
Figure 1.

Overview of the structure and comparison with other FadR-superfamily transcription factors. VanR-group members are shown at the top of the figure and FadR-group members are shown at the bottom of the figure. The PDB codes for the proteins shown are TM0439, 3fms; Rhodococcus sp. protein RO03477, 2hs5; Pseudomonas protein PS5454, 3c7j; FadR, 1e2x; CGL2915, 2di3. The red and pink colors denote the DNA-binding domain, with the HTH motif highlighted in red. The FCD domain has been colored with a spectrum from blue to red, with the α0 helix of the FadR subfamily highlighted in magenta. The grey chain represents the second monomer in the dimer.
As pointed out above, TM0439, RO03477 and PS5454 can be classified in the VanR group based on secondary-structure prediction, which identifies only six α-helices in their C-terminal domains (Rigali et al., 2002 ▶). In all three structures, a short linker connects the second β-strand of the WH domain directly to the α1 helix of the regulatory domain, so that the α0 helix seen in FadR is absent. In the TM0439 and RO03477 structures the mutual disposition of the WH and regulatory domains is similar, with the two WH domains in close proximity; in contrast, the structure of PS5454 is distinctly different, with the two WH domains at opposite ends of the homodimer. The two FadR-group proteins (i.e. FadR and CGL2915) contain an extra α0 helix at the N-terminus of the regulatory domain. In FadR, this helix contains a sharp kink which reverses its course in the center, wedging it between the WH and regulatory domains. Consequently, the mutual disposition of the two domains of FadR is distinctly different from both TM0439 and RO03477 owing to a rotation of the regulatory domain relative to the WH domain. In CGL2915, the α0 helix is straight and as a consequence the two regulatory domains are swapped between the monomers (Gao et al., 2008 ▶).
The site of the three mutations made to enhance crystallizability is located in the loop between helices α2 and α3 of the C-terminal domain and is involved in a heterologous contact with a WH domain of a symmetry-related molecule. The site of the mutations is distant from functionally important structural elements.
3.3. The WH domain
The N-terminal portion of TM0439 (residues Val6–Val71) constitutes the winged-helix dsDNA-binding domain, with a canonical order of secondary-structure elements α1, α2, α3, β1, β2 (these are referred to henceforth as a1, a2, a3, b1, b2 in order to differentiate them from helices α0–α6 in the regulatory domain). The HTH (helix–turn–helix) motif is made up of helices a2 and a3 with the connecting loop; the antiparallel two-stranded β-sheet makes up the ‘wing’. Helix a1 provides a critical interface with the C-terminal regulatory domain in the same monomer. The WH domain is a hallmark of the GntR family. Not surprisingly, a structural comparison using DALI (Holm et al., 2006 ▶) identified a number of known WH domains with similar structure. The top hits, with Z > 8.0, include all of the known putative GntR structures, but also the Zα domain of the viral E3L protein (PDB code 1sfu), double-stranded RNA-specific adenosine deaminase (PDB code 1qbj), catabolite gene-activator protein (CAP; PDB code 1i6f) and LEXA repressor (PDB code 1jhf). The pairwise r.m.s.d. values for the Cα atoms are around 2.0 Å. The highest amino-acid sequence identity among proteins of known structure is observed for PDB entries 3c7j (PS5454) and 2di3 (CGL2915), at 35% and 32%, respectively.
Although all known structures of WH domains are very similar, their mode of interaction with dsDNA can vary considerably. While most of them use the second helix of the HTH motif to bind to the major groove of the cognate DNA sequence (Gajiwala & Burley, 2000 ▶), the FadR WH domain uses only the N-terminal fragment of this helix (Xu et al., 2001 ▶). Interestingly, residues Arg35, Arg45, Arg49 and Gly66, which are indispensable for DNA binding in FadR, are completely conserved in CGL2915. These observations suggest that CGL2915 may bind to DNA in a manner similar to FadR, which binds to TGGTN3ACCA (Xu et al., 2001 ▶). In fact, an identical sequence was identified in the C. glutamicum genome in the promoter of cgl2917 (Gao et al., 2008 ▶). However, in TM0439 the residue equivalent to Arg45 of FadR is Phe45, suggesting that the target DNA sequence for this protein is different. Both RO03477 and PS5454 also show differences from the putative dsDNA-binding consensus sequence (Fig. 2 ▶).
Figure 2.

The overall architecture of the HTH domain of TM0439, with putative DNA-binding residues shown. The DNA is modeled into this figure based on the superposition of the FadR–DNA complex (PDB code 1hw2) onto the HTH domain of TM0439.
3.4. The regulatory FCD domain
The FCD domain of TM0439, encompassing residues Glu76–Glu212, contains six α-helices, as predicted for the VanR group, arranged into an antiparallel bundle. The same tertiary fold is observed in the regulatory domains of RO03477 (PDB code 2hs5) and PS5454 (PDB code 3c7j), both of which are VanR-group members. The C-terminal domains of CGL2915 (PDB code 2di3) and FadR (PDB code 1hw1) also show a very similar fold, with the sole exception of the additional α0 helix characteristic of the FadR group (Fig. 3 ▶). Pairwise r.m.s. differences between Cα positions range from 2.2 to 2.9 Å. This structural similarity is particularly striking given the limited amino-acid sequence similarities of 18% between TM0439 and RO03477, 13% with PS5454, 17% with CGL2915 and only 11% with FadR. The FadR C-terminal domain is classified as a member of the FadR_C family (PF07840), while the remaining four domains belong to the FCD family (PFam 07729). Thus, the FadR and VanR groups are not equivalent to the FadR_C and FCD families, respectively, creating a confusing classification. We suggest that the FadR and VanR distinction should be discontinued.
Figure 3.

The regulatory domain of TM0439 and comparison with other FCD/FadR domains. The overall domain structure and a close-up of the kinked helix α4 is shown for each protein on the right and left, respectively. In each domain the kinked α4 helix is shown in red. The seventh helices of the FadR-group members, α0, are shown in yellow. The wire cages are the cavities calculated by VOIDOO (Kleywegt & Jones, 1994 ▶). The metals are displayed with a radius of 2.0 Å to highlight their position.
Although a fold comprising a six-helix antiparallel bundle is topologically simple, the FCD/FadR_C fold constitutes a unique family to the extent that DALI (Holm et al., 2006 ▶) shows no other structurally related domains with a Z score higher than 6. It seems that the distinction between the FadR_C and FCD families made in the Pfam database is insignificant and a single family, e.g. FCD, should comprise all these proteins; in the following discussion, the term FCD shall refer to all members of the FCD/FadR_C fold.
An interesting structural feature of the FCD fold is a conserved kink in the α4 helix. This helix is noteworthy because its N-terminal part is intimately involved in the dimerization of the domain (see below), while the C-terminal portion constitutes the main interface with the WH domain of the same monomer. In TM0439, the α4 helix has six full turns and the kink occurs approximately after the first three. The kink results in a strained secondary conformation of Ile153 (ϕ = −107°, ψ = 11°), which leaves the amides of Asp155 and Arg156, as well as the carbonyl of Lys164, free from intra-helical hydrogen bonds. Instead, the side-chain Glu58 from the WH domain positions itself so that O∊1 ‘caps’ the chain amides of both Asp155 and Arg156 (Fig. 3 ▶). An almost identical structural perturbation occurs in the corresponding α-helix in CGL2915, in which the kink at Leu167 (ϕ = −86°, ψ = −12°) leaves the amides of Leu169 and Ser170, as well as the carbonyl of Ala166, free; here, Ser81 from the WH domain performs the capping function (Fig. 3 ▶). A similar stereochemistry is reproduced in FadR, in which Met168 is at the center of the kink (ϕ = −78°, ψ = −23°), leaving the amides of Gly170 and Leu171 and the carbonyl of Gly167 uncapped but with no substitute hydrogen-bonding partners from the WH domain (Fig. 3 ▶). In RO03477 a similar kink occurs after the first two turns, not three as in the previous structures. Met168 is at its center (ϕ = −84° and ψ = −8°) and the free amides of Ser170 and Val171, as well as the carbonyl of Val167, are not involved in any hydrogen bonds (Fig. 3 ▶). The PS5454 structure is the only one in which the α4 helix is straight. It is also the only structure in which the WH domains are set apart. We will return to this point later.
3.5. The FCD domain as a dimerization module
The FCD domains are responsible for the dimeric architecture of the FadR transcription factors. The crystal structures of FadR and CGL2915 show an almost identical disposition of the FCD domains in the homodimers and suggest that the mode of dimerization is conserved (Gao et al., 2008 ▶). The TM0439 protein conforms to this paradigm. It forms a homodimer in which the interface is mediated exclusively by the α1 helix and the N-terminal portion of the α4 helix of the FCD domain. In each chain, 23 residues bury a surface of ∼950 Å2. The hydrophobic core of the interface is formed by Ile87, Met88, Met89, Phe92, Leu145, Leu146, Leu149 and Ile153. The residues that bury the largest solvent-exposed surface are Glu81, Glu84, Met88, Phe92, Asn143, Leu145, Leu149 and Lys152. A total of 14 hydrogen bonds and four salt bridges span the interface at its periphery (Fig. 4 ▶). Both the RO03477 and PS5454 structures have topologically very similar interfaces that are mediated by the α1 and α4 helices, although the buried solvent-accessible surfaces are smaller than in TM0439 (∼780 and ∼730 Å2, respectively). The same overall architecture is also seen in FadR and CGL2915, but their FCD domains contain the additional α0 helix, which contributes significantly to the dimer contact. In FadR, the surface buried on dimerization is ∼780 Å2 per monomer, of which 112 Å2 is contributed by Leu80, Ile82 and Leu83 from the α0 helix. In CGL2915, these buried surfaces are ∼950 and ∼145 Å2, respectively; the latter surface is contributed by Ala79, Leu80, Ser83, Val84 and Gln87.
Figure 4.

The dimerization interfaces of the FCD and FadR_C domains. For TM0439, two complete FCD domains are shown, with one monomer colored as in Fig. 3 ▶. Residues described in the text are represented as sticks. For the other structures only the helices that participate in dimerization are shown.
Thus, the mode of dimerization of all FCD domains is highly conserved, notably in the absence of any significant amino-acid sequence similarities between the individual proteins. The unique nature of each interface suggests that heterodimerization is not possible within this family.
3.6. A novel metal-binding subfamily of FCD
Based on the FadR paradigm, it is thought that the regulatory domains of the FadR family bind small organic ligands and as a consequence undergo conformational changes that reorient the WH domains and affect their binding to cognate DNA. We were therefore interested whether the structure of TM0439 might reveal a putative binding site for such a ligand. Indeed, we find an internal polar cavity in the FCD domain, at the bottom of which are three histidines (His134, His174 and His196) with imidazole groups arranged in a three-blade propeller with the N∊2 atoms pointing towards a strong peak of positive electron density. When a dummy atom was placed in this density and refined, it was found to be 2.0–2.2 Å from the three N∊2 atoms, which is consistent with the coordination stereochemistry of a metal ion.
Histidines primarily coordinate metal ions via the N∊2 atoms (Chakrabarti, 1990b ▶), even though they are preferentially protonated on these atoms in solution (Reynolds et al., 1973 ▶). Thus, histidines within metal-binding sites typically donate hydrogen bonds through their Nδ1 atoms to carboxyl side chains or other hydrogen-bond acceptors (e.g. main-chain carbonyls) to stabilize the less favorable tautomeric form that is unprotonated on N∊2 (Argos et al., 1978 ▶; Christianson & Alexander, 1989 ▶). In concert with this paradigm, two of the metal-binding histidines, i.e. His134 and His196, are stabilized in this form by hydrogen bonds to neighboring carboxylic acids (Glu173 O∊1 acts as an acceptor for His196 Nδ1 and Glu90 O∊1 for His134 Nδ1). In addition, His134 donates a Cδ2(H)⋯O bond to the main-chain carbonyl of Asp130 (3.1 Å; Fig. 5 ▶). Similar CH⋯O bonds involving the C∊1(H) group, which is modestly acidic, are commonly observed for histidines in proteins (Derewenda et al., 1994 ▶), but those involving Cδ2(H) are rare.
Figure 5.

Metal-binding sites of TM0439 (PDB code 3fms), CGL2915 (2di3) and PS5454 (3c7j). An OMIT map contoured at 5σ is shown for TM0439. This was generated by deleting the metal and acetate and truncating the histidines back to the Cβ atoms, shaking the coordinates to yield an r.m.s.d. of 0.3 Å and performing a round of refinement in phexix.refine.
The three imidazoles form a triangular propeller, with the angles at each N∊2 close to 60°. Further, the putative metal ion is elevated ∼1.25 Å above the plane defined by the N∊2 atoms, as expected for tetrahedral coordination. The putative fourth position in the coordination sphere is unoccupied, and above it we find electron density consistent with a carbonate or an acetate ion, which may have originated from the crystallization mixture. The refined B value for the metal (36 Å2) was consistent with a divalent ion such as Zn2+ or Ni2+. In order to identify the metal, we employed atomic absorption spectroscopy on the SeMet samples used for crystallization and found stoichiometric amounts of Ni2+. Metal removal was found to be kinetically impaired; greater than 48 h of dialysis against 10 mM EDTA and 2 mM DTT was required for its complete removal at 277 K. This slow removal may be a consequence of the inherently slow Ni2+ ligand-exchange kinetics as well as the relatively buried nature of the metal-binding site. We suspect that Ni2+ may have been inadvertently introduced during the purification protocol, i.e. Ni2+-affinity chromatography, and that Zn2+ is the physiological ligand; this is consistent with the tetrahedral coordination geometry, as well as the presence of histidines as coordinating residues, both of which favor Zn2+ (Dokmanić et al., 2008 ▶).
Using tryptophan fluorescence, we measured the metal affinity of TM0439 for both Zn2+ and Ni2+. Fig. 6 ▶(a) shows the fluorescence emission spectrum upon excitation at 287 nm, with a characteristic tryptophan peak at λem = 340 nM. We find that Ni2+ binding is stoichiometric, 1:1, with K = 1.47 ± 0.01 × 107 M −1 (K d = 68 ± 5 nM). Unexpectedly, Zn2+ binds with a stoichiometry of 2:1, with sequential binding constants of K 1 ≥ 1.4 ± 0.1 × 107 M −1 (K d ≤ 71 ± 5 nM) and K 2 ≥ 4.5 ± 0.4 × 105 M −1 (K d ≤ 2.0 ± 0.2 µM), respectively, with an approximately twofold increase in the Trp fluorescence (Fig. 6 ▶ b). The origin of the second binding site is unknown and it is not clear whether the lower affinity site is of functional significance. We note that the protein contains a His6 tag which in principle could influence the apparent metal-binding affinities and stoichiometries. However, the N-terminal localization of the polyhistidine sequence virtually rules out any potential influence on the quantum yield of Trp154, which is located at the kink in the α4 helix of the C-terminal regulatory domain. Both Zn2+ and Ni2+ bind to synthetic histidine-rich sequences with affinities of ∼104 (Whitehead et al., 1997 ▶). Since the measured Zn2+-binding constants are lower limits (see legend to Fig. 6 ▶ b), it is unlikely that there is significant competition from the polyhistidine tail. Since we did not observe a secondary low-affinity Ni2+-binding site, it may be possible that it is masked by competition from the His6 tail. Taken together and considering the relative abundance of Zn2+ compared with Ni2+ for most organisms (Outten & O’Halloran, 2001 ▶), it is reasonable to hypothesize that TM0439 is a Zn2+-binding protein, although our analysis did not include other transition metals, e.g. Co or Mn, which in principle might also be involved.
Figure 6.

Metal binding by TM0439 monitored by Trp fluorescence: (a) 200 µM Ni2+ and (b) 200 µM Zn2+ titrated into 5.3 µM TM0439. The inset plots the emission (λ = 340 nm) versus the metal:protein molar ratio and the red line indicates the best fit according to a one-site (Ni2+; K = 1.47 ± 0.01 × 107 M −1) or two-site (Zn2+; K 1 = 1.4 ± 0.3 × 107 M −1 and K 2 = 4.5 ± 0.4 × 105 M −1) sequential binding model in DynaFit (Kuzmic, 1996 ▶) accounting for appropriate metal–buffer interactions. Note that that the best fit shown in the inset of (b) represents a lower limit of K i values, because as long as K 1/K 2 remains constant larger K 1 and K 2 fit the data equally well (simulations not shown).
Interestingly, the structures of both CGL2915 (PDB code 2di3) and PS5454 (PDB code 3c7j) also contain metals bound in stereochemically analogous sites. In CGL2915 the coordinating histidines are His148, His196 and His218 and their imidazoles are stabilized in the Nδ1-protonated tautomers by Glu106, Gln193 and Glu195, respectively. His148 is additionally stabilized by a CH⋯O bond via its Cδ1, as is the case for His134 of TM0439. However, another protein atom, Oδ1 of Asp144 (analogous to Asp130 in TM0439), serves as an axial ligand (distal to His218), resulting in slightly distorted trigonal bipyramid coordination, with a water molecule completing the equatorial plane (Fig. 5 ▶). The same stereochemistry is preserved in the second, crystallographically independent, subunit. It is also interesting to note that Asp144 Oδ1 approaches the putative metal with the syn sp 2 orbital, as is usual in metal-binding sites (Chakrabarti, 1994 ▶, 1990a ▶). The ligand in CGL2915 is annotated as Zn2+ based on XAFS data (Gao et al., 2008 ▶).
In the P. syringae regulator (PDB code 3c7j), the coordinating histidines are His148, His192 and His214, while the fourth ligand, equivalent to Asp144 in CGL2915, is Asn144. The His214 and Asn144 side chains serve as axial ligands and the latter is oriented with its side-chain O atom towards the metal. His192 and His214 are stabilized in the required tautomeric forms by hydrogen bonds from Nδ1 to Asp191 and Gln189, respectively. The His148 residue has the same interesting CH⋯O bond to the carbonyl of Asn144 as its counterparts in CGL2915 and TM0439. In one subunit, a single water molecule is found in an equatorial plane, while in the second independent monomer two water molecules complete an octahedral coordination sphere (Fig. 5 ▶). The metal in this structure is annotated as Ni2+, consistent with the coordination preference and with reasonable B values.
Neither the FadR nor the RO03477 structures have metal-binding sites. In FadR, the three metal-coordinating histidines are replaced by Phe149, Tyr193 and Tyr215. In RO03477, one of the three histidines, His152, is present, but the other two are replaced by Asn196 and Tyr218, respectively, leaving no room for the metal.
An analysis of the genomic data for the FCD-domain family (PF07729) reveals that more than 2800 members have been identified to date in 402 species of eubacteria and four species of archaea. The amino-acid sequences show low average identity on full alignment (∼21%). A majority (>70%) contain a complete set of motifs with all four putative metal-binding residues that together make up a consensus fingerprint, R-X 3-ΦE-X 19-Φ-X 19-D/N-X 2-ΦH-X 3-Φ-X 2-S/T-X 2-N-X 2-Φ-X 6-Φ-X 20-H-X 6-Φ-X 3-D-X 3-A-X 6-H, where Φ denotes a hydrophobic residue, typically Leu, Met or Ile, and residues involved in metal coordination are shown in bold. Because of poor amino-acid sequence conservation in this family, this fingerprint is not readily identifiable by automated sequence alignment.
Numerous examples of bacterial species contain a number of FCD-family proteins: Mycobacterium smegmatis contains 46 of these regulators, Rhodococcus sp. RHA1 contains 49, Arthrobacter sp. (FB24) contains 28 and Agrobacterium tumefaciens contains 51. Interestingly, the sequences are very diverse within each species but in each case about two-thirds show conservation of all metal-binding amino acids. This situation is in stark contrast to the FadR_C family, for which there are only 71 annotated sequences in 70 species (with only one gene per organism) and an average amino-acid identity of 48%.
3.7. Functional implications
The structural evidence presented here strongly suggests that the majority of FCD domains and therefore the majority of FadR transcription regulators are metal (most likely Zn2+) dependent. What is not clear is whether these transcription factors are metal-sensing or whether the metal plays a structural role or perhaps is required for binding of other effector molecules through direct coordination bonds. Metal-sensing transcription factors are ubiquitous in prokaryotes, with seven major families characterized to date (Giedroc & Arunkumar, 2007 ▶). Five of these families, i.e. ArsR, MerR, CopY, Fur and DtxR, utilize WH domains, also found in the GntR regulators, for binding to dsDNA. Almost all of these proteins are dimeric and metals bind typically at or near dimer interfaces, enabling the metal-bound form of the regulators to repress, de-repress or activate the transcription of operons coding for metal-efflux pumps, transporters, redox machinery etc. (Giedroc & Arunkumar, 2007 ▶; Pennella & Giedroc, 2005 ▶; Silver & Phung, 2005 ▶). In the FCD domains, the metal-binding site is distinctly buried within an individual monomer and removal by dialysis takes a relatively long time, which would seems to argue against a role in sensing changes in metal concentration. It is therefore more plausible that the FCD domains bind carboxylic acids or small organic compounds containing carboxylic groups, so that the latter are buried and interact directly with the metal at the bottom of the ligand-binding cavity. The presence of acetate (or less likely carbonate) in the TM0439 structure is consistent with this hypothesis. However, the polar cavities observed inside the metal-binding FCD domains of TM0439 and CGL2915 are relatively small and do not appear to be able to bind larger organic compounds: calculations with a 1.4 Å probe resulted in only ∼130 Å3 for TM0439 and ∼72 Å3 for CGL2915. Interestingly, in PS5454 the volume of the cavity is difficult to estimate because one of the flanking loops is disordered in the crystal structure and the cavity appears to be open to bulk solvent. The loop that is disordered links the α4 helix with the α5 helix. We note that PS5454 is unique in that the α4 helix is straight, lacking the characteristic kink, and it is possible that the structure represents an ‘active’ conformer in which the cavities are open and able to bind a ligand, while the WH domains are ∼68 Å apart, i.e. ideally positioned to bind to major grooves separated by two complete turns of the dsDNA.
Further studies will be needed to fully characterize the new metal-binding subfamily of the FadR transcription regulators.
Supplementary Material
PDB reference: TM0439, 3fms, r3fmssf
Acknowledgments
This study was supported by NIH NIGMS grants U54 GM074946-01US (ISFI), U54 GM074898 (JCSG) and R01 GM042569 (to DPG). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. The authors would like to thank the staff at BL 5.0.2 managed by the Berkeley Center for Structural Biology (BCSB) at the Advanced Light Source (ALS) for technical support. The BCSB is supported in part by the National Institutes of Health, National Institute of General Medical Sciences. The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Aalten, D. M. van, DiRusso, C. C. & Knudsen, J. (2001). EMBO J.20, 2041–2050. [DOI] [PMC free article] [PubMed]
- Aalten, D. M. van, DiRusso, C. C., Knudsen, J. & Wierenga, R. K. (2000). EMBO J.19, 5167–5177. [DOI] [PMC free article] [PubMed]
- Argos, P., Garavito, R. M., Eventoff, W., Rossmann, M. G. & Brändén, C. I. (1978). J. Mol. Biol.126, 141–158. [DOI] [PubMed]
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002). Nucleic Acids Res.30, 276–280. [DOI] [PMC free article] [PubMed]
- Chakrabarti, P. (1990a). Protein Eng.4, 49–56. [DOI] [PubMed]
- Chakrabarti, P. (1990b). Protein Eng.4, 57–63. [DOI] [PubMed]
- Chakrabarti, P. (1994). J. Mol. Biol.239, 306–314. [DOI] [PubMed]
- Christianson, D. W. & Alexander, R. S. (1989). J. Am. Chem. Soc.111, 6412–6419.
- Derewenda, Z. S. (2004). Structure, 12, 529–535. [DOI] [PubMed]
- Derewenda, Z. S., Derewenda, U. & Kobos, P. M. (1994). J. Mol. Biol.241, 83–93. [DOI] [PubMed]
- Derewenda, Z. S. & Vekilov, P. G. (2006). Acta Cryst. D62, 116–124. [DOI] [PubMed]
- DiRusso, C. C., Heimert, T. L. & Metzger, A. K. (1992). J. Biol. Chem.267, 8685–8691. [PubMed]
- DiRusso, C. C., Metzger, A. K. & Heimert, T. L. (1993). Mol. Microbiol.7, 311–322. [DOI] [PubMed]
- Dokmanić, I., Šikić, M. & Tomić, S. (2008). Acta Cryst. D64, 257–263. [DOI] [PubMed]
- Franco, I. S., Mota, L. J., Soares, C. M. & de Sa-Nogueira, I. (2006). J. Bacteriol.188, 3024–3036. [DOI] [PMC free article] [PubMed]
- Gajiwala, K. S. & Burley, S. K. (2000). Curr. Opin. Struct. Biol.10, 110–116. [DOI] [PubMed]
- Gao, Y. G., Suzuki, H., Itou, H., Zhou, Y., Tanaka, Y., Wachi, M., Watanabe, N., Tanaka, I. & Yao, M. (2008). Nucleic Acids Res.36, 7110–7123. [DOI] [PMC free article] [PubMed]
- Giedroc, D. P. & Arunkumar, A. I. (2007). Dalton Trans., pp. 3107–3120. [DOI] [PubMed]
- Goldschmidt, L., Cooper, D. R., Derewenda, Z. S. & Eisenberg, D. (2007). Protein Sci.16, 1569–1576. [DOI] [PMC free article] [PubMed]
- Haydon, D. J. & Guest, J. R. (1991). FEMS Microbiol. Lett.63, 291–295. [DOI] [PubMed]
- Holm, L., Kaariainen, S., Wilton, C. & Plewczynski, D. (2006). Curr. Protoc. Bioinformatics, Unit 5.5. [DOI] [PubMed]
- Huffman, J. L. & Brennan, R. G. (2002). Curr. Opin. Struct. Biol.12, 98–106. [DOI] [PubMed]
- Kleywegt, G. J. & Jones, T. A. (1994). Acta Cryst. D50, 178–185. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Kuzmic, P. (1996). Anal. Biochem.237, 260–273. [DOI] [PubMed]
- Lee, M. H., Scherer, M., Rigali, S. & Golden, J. W. (2003). J. Bacteriol.185, 4315–4325. [DOI] [PMC free article] [PubMed]
- Lesley, S. A. et al. (2002). Proc. Natl Acad. Sci. USA, 99, 11664–11669.
- Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2003). Proteins, 50, 437–450. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Newman, J. (2005). Acta Cryst. D61, 490–493. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Outten, C. E. & O’Halloran, T. V. (2001). Science, 292, 2488–2492. [DOI] [PubMed]
- Pennella, M. A. & Giedroc, D. P. (2005). Biometals, 18, 413–428. [DOI] [PubMed]
- Reynolds, W. F., Peat, I. R., Freedman, M. H. & Lyerla, J. R. Jr (1973). J. Am. Chem. Soc.95, 328–331. [DOI] [PubMed]
- Rigali, S., Derouaux, A., Giannotta, F. & Dusart, J. (2002). J. Biol. Chem.277, 12507–12515. [DOI] [PubMed]
- Silver, S. & Phung, L. T. (2005). J. Ind. Microbiol. Biotechnol.32, 587–605. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Acta Cryst. D59, 1174–1182. [DOI] [PMC free article] [PubMed]
- Whitehead, I. P., Campbell, S., Rossman, K. L. & Der, C. J. (1997). Biochim. Biophys. Acta, 1332, F1–F23. [DOI] [PubMed]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed]
- Xu, Y., Heath, R. J., Li, Z., Rock, C. O. & White, S. W. (2001). J. Biol. Chem.276, 17373–17379. [DOI] [PubMed]
- Zwart, P. H., Afonine, P. V., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., McKee, E., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., Storoni, L. C., Terwilliger, T. C. & Adams, P. D. (2008). Methods Mol. Biol.426, 419–435. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: TM0439, 3fms, r3fmssf
PDB reference: TM0439, 3fms, r3fmssf