

Abstract
Recent research on the chemistry of natural products from the author’s group that led to the receipt of the ACS Ernest Guenther Award in the Chemistry of Natural Products is reviewed. REDOR NMR and synthetic studies established the T-taxol conformation as the bioactive tubulin-binding conformation, and these results were confirmed by the synthesis of compounds which clearly owed their activity or lack of activity to whether or not they could adopt the T-taxol conformation. Similar studies with the epothilones suggest that the current tubulin-binding model needs to be modified. Examples of natural products discovery and biodiversity conservation in Suriname and Madagascar are also presented, and it is concluded that natural products chemistry will continue to make significant contributions to drug discovery.
My first real exposure to natural products chemistry came in my third and final year as an undergraduate at Cambridge University, when I attended a course of lectures on the chemistry of natural products by the Nobel Prize-winning chemist Sir Alexander Todd (later to become Lord Todd). The lectures included many references to his own work in the field, with stories of his early work on the structure of cholesterol, the structure and function of various vitamins, and the structures of the nucleotides and nucleosides, and I was fascinated by the complex structures and biological importance of these substances. It was during this course that I decided to study the chemistry of natural products, and this study has been one of the loves of my life for the last 48 years.
Within the large field of natural products chemistry, I was particularly drawn to those compounds with biological activity, especially anticancer activity, and much of my research has been centered around the study of naturally occurring anticancer agents. I was fortunate to be funded by NIH for work in this area soon after my move to Virginia Polytechnic Institute and State University in 1971, and this funding has been crucially important to my success in the study of natural products. My initial studies involved the isolation and structure elucidation of potential anticancer agents from plants supplied by the National Cancer Institute, and this work has continued to the present, but with a new focus on the combination of natural products chemistry and biodiversity conservation. The other major thrust of my research has been on the chemistry and bioactivity of natural products with tubulin-assembly activity, and this will be discussed first.
The Chemistry and Tubulin-Binding Properties of Taxol
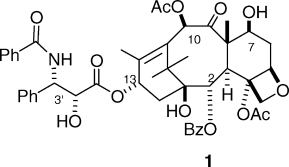
Although taxol1 (1) was first isolated by Wall and Wani in the late 1960s, and its structure published in 1971,(1) it was still very much a laboratory curiosity to most chemists in 1978. The oncologists at NCI were not initially enthusiastic about its prospects as a drug because it had two obvious drawbacks in spite of its clear activity; it was extremely insoluble in water, and it was difficult to obtain in quantity from the relatively scarce tree Taxus brevifolia. In addition to these problems, it had an unknown mechanism of action. Prospects for its development as an anticancer drug were thus very slim, but fortunately, some scientists within NCI, notably Matthew (Matt) Suffness, believed in its prospects and argued for its further development. These arguments were buttressed by some encouraging responses for taxol when treating solid tumor xenografts in nude mice, and in 1977, the NCI approved funds to develop a formulation of taxol for clinical use as well as funds to isolate enough taxol for this work. The following year, Fuchs and Johnson showed that it acted as a spindle poison,(2) and the year after this, in 1979, Susan Horwitz published her pivotal paper documenting that taxol caused the polymerization of tubulin to microtubules.(3) This discovery significantly increased the attractiveness of taxol as a potential drug and helped to maintain interest in its development when it encountered problems with toxicity in its initial clinical trials.
A conversation with my then colleague Bob Holton in 1978 started me on a new and particularly fruitful research area involving this novel compound. Bob had initiated an approach to the total synthesis of taxol, but he had no experience with the actual natural product, and so he suggested a research collaboration. We agreed that he would continue his total synthetic approach, while I would investigate the chemistry of taxol, about which very little was then known. We submitted a joint R01 grant proposal to NIH, but we were ahead of our time and the proposal was not funded. I thus began my studies on the chemistry of taxol on a shoestring budget, although a year or so later I was able to obtain some much needed support from the American Cancer Society, and later still (once taxol had become a hot property) I was able to obtain NIH funding for the work. From the earliest days, I did, however, receive strong support from Matt Suffness and the Natural Products Branch at NCI, who provided me with relatively large amounts of crude taxane mixtures, consisting of side-cuts from the purification of taxol for clinical trials by PolySciences, Inc. These supplies were crucial to my early work, which could not have been done without them.
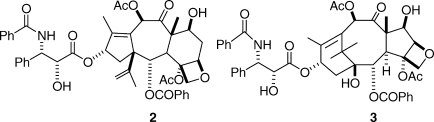
My group’s early studies of the chemistry of taxol have been reviewed on several occasions4–6 and will only be summarized briefly here. Their focus was on the systematic modification of the functional groups of the taxane ring system and on the effect of variations in the ring system itself on bioactivity. Among other discoveries, we found that removal of the C1 hydroxyl group,(7) the C2 benzoyl group,(8) and the C4 acetyl group(9) all produced analogues with significantly reduced bioactivities, but removal of the C7 hydroxyl group(10) or the C10 acetoxyl group(11) yielded products with much less activity loss. Contraction of the A-ring gave the A-nortaxol 2, which was several orders of magnitude less cytotoxic than taxol but which surprisingly retained much of taxol’s tubulin-polymerization activity.(12) Contraction of the C-ring by an interesting mechanism gave the C-nortaxol 3, which was significantly less active than taxol both in its cytotoxicity and in its tubulin-assembly activity.(13)
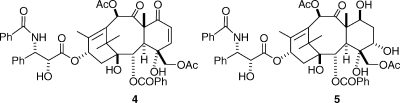
The oxetane ring was the focus of several studies. Oxidation at C7 allowed simple base-promoted opening of the oxetane ring to give the enone 4,(14) while treatment with Meerwein’s reagent gave the ring-opened product 5.(12) Both of these compounds were essentially completely inactive, and these findings led to the conclusion that the oxetane ring was essential for activity.
This conclusion was reinforced by the finding that the sulfetane analogue 6 was also much less active than taxol.(15) Later work from Dubois et al., however, suggested that the lack of activity of compounds 4 and 5 was due more to the lack of the C4-acetate group than of the oxetane ring per se, since the 5(20)-deoxydocetaxel analogue 7 was as active as taxol in promoting tubulin assembly.(16) The lack of activity of the sulfetane analogue 6 could then be explained by the fact that the large size of the sulfur atom prevented proper docking into the active site on tubulin.(17)
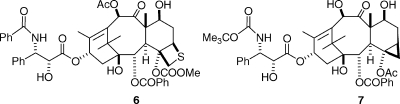
One of the most interesting observations to come from this early work was that changes to the C2-benzoate group had a profound effect on the activity of taxol. Para-substituents on the benzene ring uniformly made the resulting taxane much less active than taxol, but some ortho and meta substituents, especially the m-azido and m-methoxy substituents, significantly enhanced activity.(18) It was gratifying that this discovery has been incorporated into two taxanes in preclinical development, compounds SB-T-11033 (8) and SB-T-121304 (9).(19)
The work that my group did, combined with studies from several other research groups, especially those of Georg,(20) Ojima,(21) and Potier,(22) established the main outline of the structure−activity relationships of taxol. What remained to be determined was the nature of the crucial interaction between taxol and tubulin.
The pioneering work of Horwitz3,23 had shown that taxol bound stoichiometrically and noncovalently to tubulin, and the binding site was also shown to be on β-tubulin by labeling studies.(24) Photoaffinity labeling studies by Horwitz in collaboration with Swindell showed that 3′-(p-azidobenzamido)taxol photolabeled the N-terminal 31 amino acids of β-tubulin,(25) while studies by Horwitz in collaboration with my group showed that 2-(m-azidobenzoyl)taxol photolabeled amino acids 217−231 of β-tubulin.(26) This work did not, however, address the exact binding site or the conformation of taxol in the binding site. The complex of taxol with tubulin is polymeric and noncrystalline, and so the direct approach of examining the binding site by X-ray crystallography is not available. Fortunately, the structure of the tubulin dimer has been determined at 3.7 Å by electron crystallography of taxol-stabilized zinc-induced tubulin sheets,(27) and this result established the location of taxol on the protein. However, this structure lacked the resolution to define the detailed conformation of taxol on the tubulin polymer.
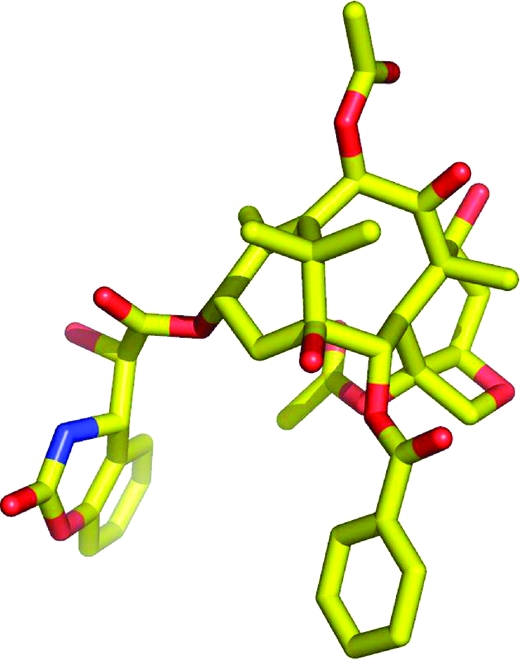
Taxol has several flexible side chains, and notably that at C13, so many possible binding conformations are possible. Several attempts to define these conformations have been made by studies of the solution NMR spectra of taxol. Thus, NMR studies in nonpolar solvents suggested a “nonpolar” conformation,28–30 while a “polar” conformation featuring hydrophobic interactions between the C2 benzoate, the C3′ phenyl group, and the C4 acetate was proposed on the basis of NMR studies in polar solvents.31–34 A combination of NMR studies using the NAMFIS deconvolution approach showed that taxol adopts 9−10 conformations in CDCl3,(35) and an analysis of the electron crystallographic data in combination with the NAMFIS results suggested that the actual binding conformation had a T-shaped structure, designated T-taxol (Figure 1).(36)
Figure 1.

T-taxol conformation. The C2-benzoate is in the lower middle and the C13-side chain is to the lower left in this perspective.
These studies, important as they were, did not provide direct experimental evidence for the actual conformation of taxol on the tubulin polymer. This requires a different technique, one that enables the determination of internuclear distances on the solid tubulin polymer sample. Fortunately, the relatively new technique of rotational-echo double-resonance (REDOR) NMR spectroscopy(37) was developed for precisely this situation, and so we entered into a fruitful collaboration with Professors Susan Bane (SUNY Binghamton), Jacob Schaefer (Washington University), and Jim Snyder (Emory University) to bring the combined forces of synthetic chemistry, biochemistry, REDOR NMR, and computational chemistry to bear on the problem of determining the binding conformation of taxol on β-tubulin. A knowledge of this binding conformation of taxol was an attractive goal because such a knowledge could guide the design of taxol analogues with improved activity by locking the molecule into the binding conformation. It had been suggested that “Taxol’s relatively weak association with tubulin may, in part, be due to the presence of an ensemble of nonproductive conformers”,(35) and these studies provided an opportunity to test this hypothesis. In addition, it was possible that simplified taxol analogues could be designed which might retain all or most of taxol’s anticancer activity.
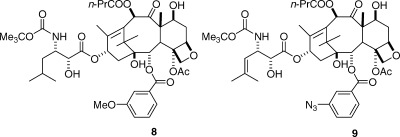
Our studies began with the synthesis of labeled taxols for REDOR NMR studies. The first compound investigated was the 13C- and fluorine-labeled analogue 10, and this yielded distances of 10.3 and 9.8 Å for the two distances a and b, respectively.(38) A later study with the deuterated and fluorinated analogues 11 and 12 gave distances of 6.3, 7.8, and >8 Å for the distances c, d, and e, respectively.(39) A careful analysis of these data and comparison with the other proposed conformations indicated that the T-taxol conformation provided the best fit to the REDOR data (Table 1).(39)
Table 1. Interatomic Distances for Various Taxol Conformations As Compared to Redor-Determined Separations for Taxol on Tubulin.
| distances, Å |
||||
|---|---|---|---|---|
| separation | Polarmodela | Nonpolarmodelb | T-Taxolmodeld,f | REDORdistance |
| a | 9.6 | 8.5 | 9.9 | 10.3i |
| b | 10.4 | 6.2 | 9.1 | 9.8i |
| c | 5.5 | 7.2 | 6.6 | 6.3h |
| d | 7.4 | 8.0 | 7.9 | 7.8h |
| e | 4.5 | 12.5 | 12.2 | >8h |
Simultaneously with these studies we also designed an approach to the synthesis of taxol analogues which would be locked into the T-taxol conformation. Several other investigators had prepared conformationally locked taxols, including those based on the nonpolar confomation40–43 and on the polar conformation,44,45 but with one exception(46)these bridged analogues were less bioactive than taxol. An important conclusion from analysis of the T-taxol structure was that the C4 acetate group and the C3′ phenyl ring were in close proximity; the centroid of the C4 acetate was only 2.5 Å from the ortho position of the C-3′ phenyl ring (Figure 2).(47) This conclusion informed our synthetic approach, which involved linking the C4 acetate to the C3′ phenyl ring using linkers of variable length.
Figure 2.
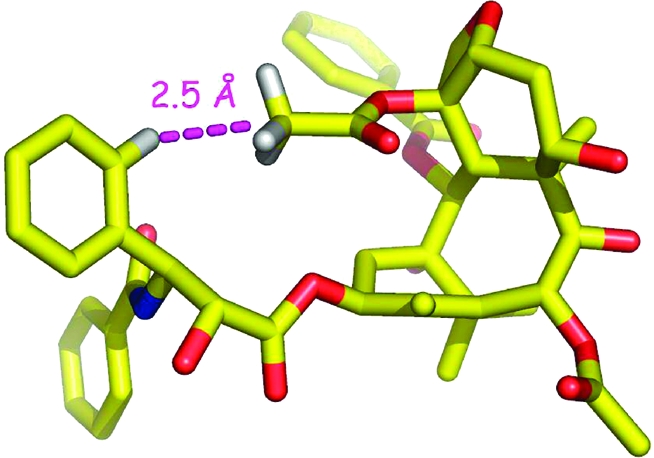
T-taxol conformation, illustrating the short H---H distance between the centroid of the C-4 acetate methyl group and the ortho position of the C-3′ phenyl ring.
Our retrosynthetic approach was based on using the flexible and versatile Grubbs’ metathesis reaction as the final ring-closing step; this reaction had previously been successfully used by Ojima in the synthesis of some bridged taxols.(45) The basic retrosynthetic approach is shown in Scheme 1, with the key diene 14 being prepared by coupling of the β-lactam 15 with the modified baccatin III 16. Olefin metathesis of 14 followed by deprotection would then give the bridged product 13.
Scheme 1.

Our first synthetic products were the compounds 17 and 18, in which the bridge was linked via the meta position of the C3′-phenyl ring. These compounds were both active, but disappointingly, they were significantly less active than taxol itself. The reason for this relative lack of activity became clear from a docking study of compound 17 into the taxol binding pocket of the electron crystallographic structure(48) of β-tubulin, which showed that the meta bridge was interacting with Phe272 of the protein, resulting in the displacement of 17 out of the binding pocket (Figure 3).(49) This finding also suggested an obvious solution, which was to remove the objectionable interaction by relocating the bridge to the ortho position of the C3′-phenyl ring.
Figure 3.

T-Taxol (brown) and 17 (blue) bound to β-tubulin. Compound 17 is seated higher in the same pocket as a result of close contact between the propene moiety of the tether and Phe272 of the protein (black) at the bottom of the illustration. Modified from ref (49). Copyright 2007 American Chemical Society.
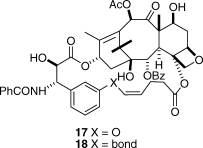
Gratifyingly, when this was done, and when the bridge was adjusted to the correct length, the activity improved dramatically. The two best derivatives were compounds 19 and 20, with just two carbons inserted between the C4 acetate methyl group and the ortho position of the C3′-phenyl ring.(50) Compound 19, for example, was 50-fold more potent than taxol to the A2780 ovarian cancer cell line and was twice as potent toward the PC3 prostate cancer cell line. It was also almost twice as effective as taxol at promoting the polymerization of tubulin.(47) Both 19 and 20 were also much more potent than taxol to taxol-resistant cell lines; compound 20, for example, was 150-fold more potent than taxol to the 1A9-PTX10 cell line with the Fβ270V mutation and almost 50-fold more potent than taxol to the 1A9-A8 cell line, with the Tβ274I mutation.(47) These results thus confirmed the T-taxol conformation as the bioactive tubulin-binding conformation of taxol. A combination of NAMFIS analysis of the conformation of 19 with docking into the β-tubulin binding site showed that this compound fit nicely into the taxol binding site and mapped well onto the T-taxol conformation (Figure 4). Not only was the objectionable interaction with Phe 272 removed, but a favorable interaction with a histidine residue was created.
Figure 4.

T-Conformations of taxol (blue) and 19 (red) in the β-tubulin binding site, the latter having been docked by the Glide software. Reproduced from ref (47). Copyright 2007 American Chemical Society.
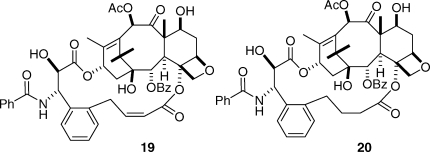
Although these results confirmed the T-taxol conformation as the tubulin-binding conformation, it was desirable to test the predictive power of the conformation by carrying out two further tests, one positive and one negative. As noted earlier, the A-nortaxol 2 was essentially noncytotoxic, although it retained moderate tubulin-assembly activity.(12) Could this compound be made cytotoxic by constraining it to the T-taxol conformation? The answer was a resounding yes! Compound 21 was prepared and was found to be approximately one-third as cytotoxic to PC3 cells as taxol, a far cry from the difference of at least 1000-fold in cytotoxicity between paclitaxel and compound 2. Interestingly, the bridged derivative 21 was approximately twice as effective at promoting the assembly of tubulin as taxol, a clear testimony to the importance of the T-taxol conformation.(51)
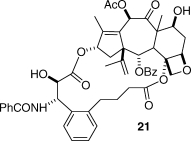
The second test was a negative one. Docetaxel (22) is a clinically used semisynthetic taxane discovered by Potier and differs from taxol only in the nature of the N-acyl and C10 substituents;(52) its 10-acetyl derivative 23 is equipotent.(53) If the carbamate oxygen of docetaxel were linked to the C3-phenyl group and the C10 hydroxyl group were acetylated, the resulting compound 24 would possess all the basic structural features necessary for bioactivity. It would not, however, be able to adopt the T-taxol conformation (Figure 5) and would thus be expected to be inactive in spite of its correct chemical connectivity. Compound 24 was prepared to test this hypothesis,(54) and satisfyingly, it was at least 2 orders of magnitude less active than taxol in the A2780 bioassay and was also significantly less active in a tubulin-assembly assay. This thus provided a conclusive “negative test” of the T-taxol conformation.
Figure 5.
Conformation of compound 24, viewed from the same perspective as T-taxol in Figure 1.

Given that the T-taxol conformation is the most probable tubulin-binding conformation for taxol, can this knowledge be used to design simplified compounds which mimic taxol’s tubulin-assembly activity? The answer at this point is a qualified yes. We synthesized the simplified analogue 25 and its diastereomer 26,(55) and both compounds, as well as some related ones, proved to have clear cytotoxic activity to the A2780 cell line and also to be effective in promoting the assembly of tubulin into microtubules. This activity in such drastically simplified compounds was encouraging, even though the cytotoxicities observed were over 2 orders of magnitude less than that of taxol. A part of the lack of activity may be due to solubility, since both compounds were very insoluble in water, but it is probably also due in part to the fact that the benzoyl group corresponding to the C2 benzoate of taxol is forced deeper into the hydrophobic binding pocket, leading to the conclusion that the compounds are not “sufficiently tailored to fully exploit the T-taxol concept.”(55) We are thus continuing our studies in this area in an attempt to discover simplified taxols that have superior biological profiles to compounds 25 and 26.
In addition to preparing simplified taxols, we have sought to develop improved ways of delivering taxol to the cancer. The most promising approach has been carried out in collaboration with colleagues at CytImmune, Inc., and involves the preparation of taxols with suitable linkers to bond to gold nanoparticles. The nanoparticles are also loaded with tumor necrosis factor alpha (TNFα), which targets the taxol-loaded nanoparticles to cancer cells, where the taxol is released.(56) Initial animal studies of this construct have been encouraging, and further development is in progress.
We have also investigated the epothilones. These exciting anticancer agents have very different structures and origin than taxol and yet have a very similar mechanism of action. Epothilones A (27) and B (28) were originally isolated as antifungal agents from the soil-derived mycobacterium Sorangium cellulosum in 1987,(57) but it was the discovery of their taxol-like tubulin-polymerization activity in 1995(58) and the elucidation of their absolute configuration in 1996(59) that led to a surge of interest in their development as potential anticancer agents.60–63 Epothilone B (Epo 906, patupilone) is in phase III clinical trials, and the lactam analogue of epothilone B (ixabepilone, 29) has been approved for clinical use for treatment of certain forms of breast cancer.(64)
Given the success of bridging in improving the activity of taxol, it was natural to ask whether this strategy could provide the same benefits to the epothilones. A model for the binding conformation of epothilone B on tubulin was proposed by Nettles and Snyder (Figure 6),(65) and this model juxtaposes the C4-methyl group with the C12-methyl group. We thus elected to prepare epothilones with bridges linking the C4 and C12 positions.
Figure 6.
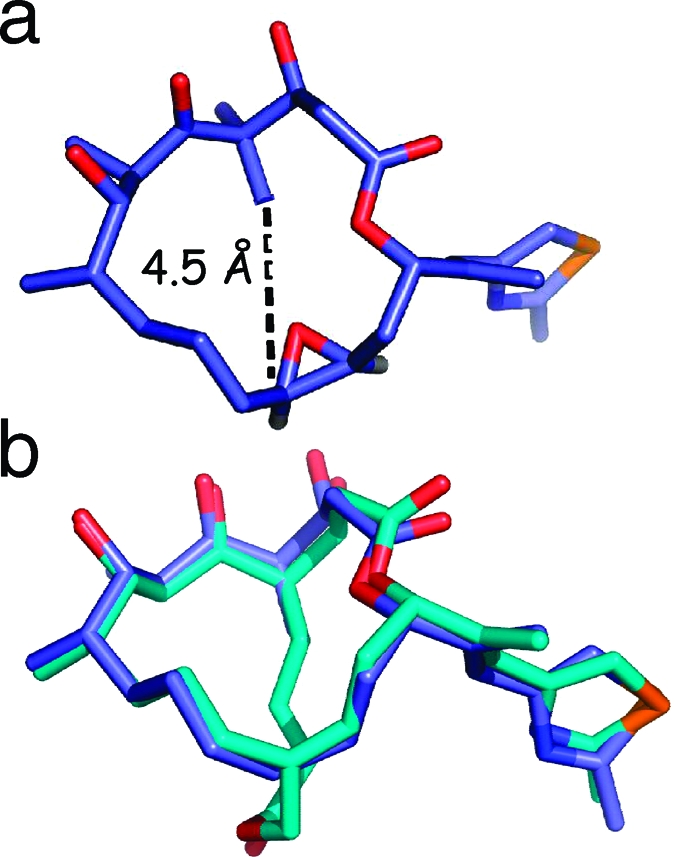
(a) Nettles−Snyder proposed epoA model based on EC density (left) with juxtaposition of the C4-methyl and C12-H (methyl for epoB); r(C---C) = 4.5 Å. (b) MMFF-energy minimized structures of the EC template and the designed bridged epoD analogue 31 (overlayed with three-point selection in PyMol).
Extensive synthetic studies, which will be reported elsewhere, led to the synthesis of the bridged epothilone 31 by olefin metathesis of the precursor 30. Compound 31 had a much lower antiproliferative activity toward the A2780 cell line than epothilone D, and it was also much less active than the deprotected precursor 32. These data suggest either that the original Nettles−Snyder model does not reveal the correct binding conformation of the epothilones or that some unusual structural features of compound 31 contribute to its lack of activity. We are thus continuing our studies in an effort to prepare conformationally constrained epothilones with improved bioactivity.
Biodiversity Conservation and Drug Discovery in Madagascar and Suriname
As indicated earlier, my initial studies on natural products were focused on the discovery of potential anticancer agents from plants, and this is the second important focus of my group’s work.
Drug discovery from natural sources requires continued access to plant, marine, and microbial biomass, and as far as plants are concerned this biomass is concentrated in the tropical rainforests of the world. These forests cover less than 7% of the earth’s surface, but hold 50% of its plant biomass. Sadly, tropical forests are disappearing fast; their coverage is down from 16% of the earth’s land surface in 1950 to less than 7% today. The preservation of tropical rainforests and tropical reef systems is or should be an important part of any comprehensive drug discovery program. One way to encourage preservation is to demonstrate the value of tropical forests and reef systems as potential sources of new pharmaceutical or agrochemical products. The Rio Convention on Biological Diversity (CBD) in 1992 established three main goals: the conservation of biological diversity, the sustainable use of its components, and the fair and equitable sharing of the benefits from the use of genetic resources. These objectives were subsequently incorporated into the innovative International Cooperative Biodiversity Group (ICBG) program based at the Fogarty International Institute at NIH, and we were fortunate to receive an award under this program to carry out drug discovery and biodiversity conservation work in Suriname and later in Madagascar. Some recent chemical and conservation results from this work will be described below. These results are all from my laboratory and do not include other discoveries made by our ICBG partners Eisai Research Institute and Dow AgroSciences.
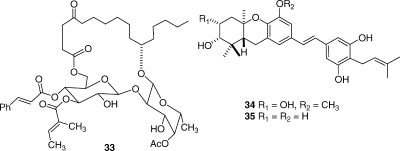
The ipomoeassins are a series of related resin glycosides isolated from the Suriname plant Ipomoea squamosa. Their isolation was challenging, but eventually 60 mg of the most abundant compound (ipomoeassin A, 33) was obtained, and its structure was elucidated by NMR and mass spectra combined with chemical conversions and Mosher ester formation for determination of stereochemistry. The compounds were of interest in part because of their unusual structures but primarily because they showed potent but selective antiproliferative activity to the A2780 cell line.(66) Ipomoeassin A showed selective antiproliferative activity in the NCI 60-cell line screen, and its pattern of activity did not match other known anticancer agents when subjected to a COMPARE analysis. These compounds have been synthesized(67) and are currently being considered for development by the National Cancer Institute.
The schweinfurthins are a class of highly potent cytotoxic agents that were originally discovered by Beutler.(68) We isolated four new compounds, of which schweinfurthins E (34) and G (35) had submicromolar antiproliferative activity against the A2780 ovarian cancer cell line.(69) These compounds are being evaluated by a pharmaceutical partner against additional cell lines, with a view to discerning their development potential. Schweinfurthin F has recently been synthesized by Wiemer,(70) and the relative simplicity of these compounds makes them attractive candidates for synthetic chemistry and SAR studies should their biology warrant it.
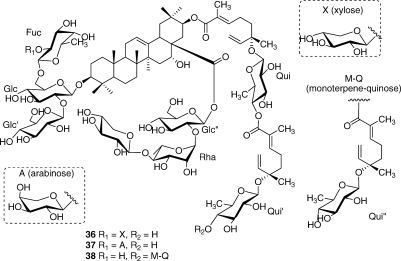
The three particularly complex cytotoxic triterpenoid saponins 36−38 were obtained from Albizia gummifera; their structure elucidation required a demanding interpretation of complex NMR spectra.(71) Although these compounds had micromolar antiproliferative activity against the A2780 ovarian cancer cell line, they are unlikely to be attractive development candidates because of their complex structures and presumed unfavorable pharmacokinetics.
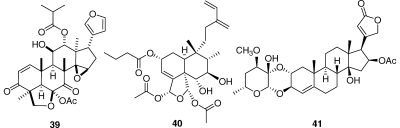
A Madagascar plant of the Malleastrum genus yielded several new bioactive diterpenoids, of which 39 is one example,(72) while Casearia nigrescens yielded several new diterpenoids with submicromolar activities, including compound 40.(73) Finally, the new and relatively unusual cardenolide 41 from an Elaeodendron sp. had potent antiproliferative activity to the A2780 cell line.(74)
In addition to the chemical work, the Suriname and Madagascar ICBG programs have made significant contributions to conservation and economic development in their respective countries. In Suriname, the ICBG Program contributed to the establishment of the Central Suriname Nature Reserve, a UNESCO World Heritage Site.(75) This important conservation success was brought about primarily through a cooperative agreement between Conservation International, a partner in the Suriname ICBG program, and the Government of Suriname, but the ICBG program provided important scientific justification for the value of the rain forest and thus of establishing this protected area.
In Madagascar, the Montagne des Français is an area of outstanding natural beauty and importance in the north of the country. The work to achieve protected area status for this area was complex and included making a botanical and economic evaluation, developing a plan for an ecotourism concession, establishment of steering committees to lead the process of setting up the new protected area, development of a community structure to assist with security and conflict management, consultation with local and regional authorities, development of the legal tools needed for protected status, and compilation of a complete initiative dossier documenting these and other steps for submission to the necessary government departments. As a result of all these steps, the Montagne des Français was granted temporary protected status in the Système d’Aires Protégées de Madagascar (SAPM) in 2006 and will achieve full protected status in mid-2008. These and other conservation achievements were accomplished by the ICBG Madagascar partners Centre National d’Application des Recherches Pharmaceutiques, Conservation International, and the Missouri Botanical Garden. These and other conservation and development successes have given significant added value to the Suriname and Madagascar ICBG programs, over and above the value of the novel bioactive natural products isolated.
Perspective
The foregoing account gives some taste of the joys and the challenges of natural products research in the 21st century. Natural products have proven to be the most reliable single source of new and effective drugs, and especially anticancer agents. Thus, Newman and Cragg have shown that 63% of anticancer drugs introduced over the last 25 years are natural products or can be traced back to a natural products source.(76) Natural products have not only yielded new and effective drugs, but they have also provided insight into new mechanisms of action, and cancer treatment would be immeasurably poorer without the insights and the compounds provided from Nature. It is thus instructive to ask why it is that natural products have proved to be such a prolific source of bioactive agents. There are several reasons, but certainly one of the most important is that plants and other organisms produce many biologically active substances for defense and other purposes, and so these substances are uniquely tailored to fit into a biological receptor of some kind.(77) Second, natural products are often large molecules with built-in chirality and are thus uniquely suited to bind to complex proteins and other biological receptors. As a result of these considerations, there is a high correlation between the properties of drugs and those of natural products.78,79 The misconception that natural products research has not produced many drugs recently is laid to rest by Butler, who writes “Another misconception has been that NP research has failed to deliver many new compounds that have undergone clinical evaluation over the last few years. However, in reality, 15 NP-derived drugs have been launched in the key markets of the United States, Europe, and Japan over the last three years, and an additional 15 NP-derived compounds were in Phase III clinical trials at the end of 2003.”(80)
Although it has been enormously successful, the pharmaceutical industry is under continuing pressure to speed up the process of drug discovery, and the natural products approach to drug discovery must compete in the scientific marketplace with other approaches which might appear to give more rapid short-term results. It is thus essential that natural products research become as efficient as possible. The three major components of any successful natural products drug discovery program are the availability of adequate source materials, the use of new and selective bioassays, and the use of rapid and efficient isolation and structure elucidation methods. The available source materials include not only the traditional microbial, plant, and marine organisms but also novel source materials such as extremophiles and “unculturable” microorganisms. The use of novel culturing environments or of heterologous DNA-based approaches, for example, has given encouraging results in the search for new antibiotics.(81) It goes without saying that any approach that involves collection of biomass must be done in a way that is fully consistent with the CBD and with full recognition of the rights of the host country. The second key component of a successful natural products based drug discovery program is the use of selective and predictive bioassays. In the anticancer area, these will include assays for individual enzymes such as the protein kinases and phosphatases82–84 rather than whole cell cytotoxicity assays, while in the antiinfective area a genetics-based fitness test holds promise for detecting novel agents with known and unusual mechanisms of action.(85) The third key requirement is that isolation and structure elucidation be carried out as efficiently as possible. This will usually require some level of dereplication to avoid the reisolation of known compounds, as well as the use of the best NMR and mass spectrometric instrumentation available today.
Two final considerations are the problem of compound supply, which can be severe in the case of plant and especially marine natural products, and of analogue synthesis. Although compound supply will always be a factor in evaluating potential drug candidates, it need not be a fatal one. The problem has been met in several ways in the past. In the case of taxol, a partial synthesis from 10-deacetylbaccatin III proved crucial in the early years,(86) while it is currently produced by plant tissue culture(87) as well as by direct isolation and semisynthesis. Another example is the marine natural product ecteinascidin (Yondelis), which is prepared by semisynthesis from the microbial natural product cyanosafracin.(88) In other cases, direct synthesis is possible; the most dramatic example of this is the approximately 70-step synthesis used for production of the phase III clinical candidate eribulin, a simplified derivative of the complex marine natural product halichondrin B.(89) It is thus safe to say that the combination of synthetic ingenuity with informed sourcing will succeed in providing adequate supplies of most if not all natural products of pharmaceutical interest.
The second consideration is that chemical synthesis can often produce modified natural products with improved properties. Natural products can thus also lead to new analogues with greater synthetic accessibility or improved activity, as exemplified by the many analogues of taxol that are in clinical trials(90) as well as numerous other examples such as the exciting activity of 26-trifluoro-(E)-9,10-dehydro-12,13-desoxyepothilone B as an improved epothilone analogue.(91)
In summary, the future of natural products research remains bright. If novel source organisms are combined with innovative bioassays and efficient structure elucidation, the natural products approach to drug discovery will continue to compete very effectively with other approaches, and will continue to contribute many novel bioactive agents for pharmaceutical use, as well as providing synthetic chemists with challenging targets for synthesis and for improvement.
Acknowledgments
The work described above has been the result of a happy combination of excellent graduate students and postdoctoral associates at VPISU with equally excellent external collaborators. The names of most of my group members who have made contributions to this work are included in the references cited, but I must also acknowledge the contributions made by my present group members Dr. Qiao-Hong Chen, who has made significant contributions to the synthesis of new epothilone analogues, only a few of which are recorded here, and Dr. Shugeng Cao, whose superb skills in isolation and structure elucidation have been a key to my work on natural products discovery over the past few years. Early collaborations with Professors Susan Horwitz and Bob Holton were stimulating and productive, and more recently, Professors Susan Bane at SUNY Binghamton, Jake Schaefer at Washington University, and Jim Snyder at Emory University have each made unique and crucial contributions to the work. The botanical work in Suriname and Madagascar was lead by Dr. James Miller of the Missouri Botanical Garden, with assistance from numerous colleagues in both countries. I am also most grateful to a succession of extremely supportive colleagues in the Natural Products Branch of the NCI, beginning with the late Jonathan Hartwell in the early 1970s and including John Douros and Matthew Suffness, both sadly no longer with us, and Gordon Cragg and David Newman. Yali Fu (formerly Yali Hallock) has been a most helpful Program Officer at the NCI, and Joshua Rosenthal has been a supportive Program Officer at the Fogarty Center. My gratitude to each of these friends and colleagues is sincere and unbounded. The work on taxol has been supported most recently by the NIH under Grant No. R01-CA-69571, and the work in Madagascar and Suriname has been supported by International Cooperative Biodiversity Group award U01-TW-00313; I am most grateful for the trust shown in me by the award of these funds. Cover art by Walter Hearn Associates LLC. The photographs of Sorangium cellulosum on the cover were kindly provided by Dr. Hans Reichenbach, and the artwork of Figures 1–6 was kindly provided by Dr. Jim Snyder, Ana Alcaraz, and Yi Jiang (Emory University).
Funding Statement
National Institutes of Health, United States
Footnotes
The name Taxol has been trademarked by Bristol-Myers Squibb for their formulation of the chemical compound formerly known as taxol. Because of the historical nature of this review, the name taxol is retained for compound 1. No infringement of the Bristol-Myers Squibb trademark is implied.
References
- Wani M. C.; Taylor H. L.; Wall M. E.; Coggon P.; McPhail A. T. J. Am. Chem. Soc. 1971, 93, 2325–2327. [DOI] [PubMed] [Google Scholar]
- Fuchs D. A.; Johnson R. K. Cancer Treat. Rep. 1978, 62, 1219–1222. [PubMed] [Google Scholar]
- Schiff P. B.; Fant J.; Horwitz S. B. Nature 1979, 277, 665–667. [DOI] [PubMed] [Google Scholar]
- Kingston D. G. I. J. Nat. Prod. 2000, 63, 726–734. [DOI] [PubMed] [Google Scholar]
- Kingston D. G. I. Pure Appl. Chem. 1998, 70, 331–334. [Google Scholar]
- Kingston D. G. I. Chem. Commun. 2001, 867–880. [Google Scholar]
- Kingston D. G. I.; Chordia M. D.; Jagtap P. G. J. Org. Chem. 1999, 64, 1814–1822. [DOI] [PubMed] [Google Scholar]
- Chaudhary A. G.; Chordia M. D.; Kingston D. G. I. J. Org. Chem. 1995, 60, 3260–3262. [Google Scholar]
- Neidigh K. A.; Gharpure M. M.; Rimoldi J. M.; Kingston D. G. I.; Jiang Y. Q.; Hamel E. Tetrahedron Lett. 1994, 35, 6839–6842. [Google Scholar]
- Chaudhary A. G.; Rimoldi J. M.; Kingston D. G. I. J. Org. Chem. 1993, 58, 3798–3799. [Google Scholar]
- Chaudhary A. G.; Kingston D. G. I. Tetrahedron Lett. 1993, 34, 4921–4924. [Google Scholar]
- Samaranayake G.; Magri N. F.; Jitrangsri C.; Kingston D. G. I. J. Org. Chem. 1991, 56, 5114–5119. [Google Scholar]
- Liang X.; Kingston D. G. I.; Long B. H.; Fairchild C. A.; Johnston K. A. Tetrahedron 1997, 53, 3441–3456. [Google Scholar]
- Magri N. F.; Kingston D. G. I. J. Org. Chem. 1986, 51, 797–802. [Google Scholar]
- Gunatilaka A. A. L.; Ramdayal F. D.; Sarragiotto M. H.; Kingston D. G. I.; Sackett D. L.; Hamel E. J. Org. Chem. 1999, 64, 2694–2703. [DOI] [PubMed] [Google Scholar]
- Dubois J.; Thoret S.; Gueritte F.; Guenard D. Tetrahedron Lett. 2000, 41, 3331–3334. [Google Scholar]
- Wang M.; Cornett B.; Nettles J.; Liotta D. C.; Snyder J. P. J. Org. Chem. 2000, 65, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Chaudhary A. G.; Gharpure M. M.; Rimoldi J. M.; Chordia M. D.; Gunatilaka A. A. L.; Kingston D. G. I.; Grover S.; Lin C. M.; Hamel E. J. Am. Chem. Soc. 1994, 116, 4097–4098. [Google Scholar]
- Ojima I.; Wang T.; Miller M. L.; Lin S.; Borella C. P.; Geng X.; Pera P.; Bernacki R. J. Bioorg. Med. Chem. Lett. 1999, 9, 3423–3428. [DOI] [PubMed] [Google Scholar]
- Georg G. I.; Harriman G. C. B.; Vander Velde D. G.; Boge T. C.; Cheruvallath Z. S.; Datta A.; Hepperle M.; Park H.; Himes R. H.; Jayasinghe L.. Medicinal Chemistry of Paclitaxel. In Taxane Anticancer Agents: Basic Science and Current Status; Georg G. I., Chen T. T., Ojima I., Vyas D. M., Eds.; American Chemical Society: Washington, DC, 1995; ACS Symposium Series Vol. 583, pp 217−232. [Google Scholar]
- Geney R.; Chen J.; Ojima I. Med. Chem. 2005, 1, 125–139. [DOI] [PubMed] [Google Scholar]
- Guenard D.; Gueritte-Voegelein F.; Potier P. Acc. Chem. Res. 1993, 26, 160–167. [Google Scholar]
- Manfredi J. J.; Horwitz S. B. Pharm. Ther. 1984, 25, 83–125. [DOI] [PubMed] [Google Scholar]
- Rao S.; Horwitz S. B.; Ringel I. J. Natl. Cancer Inst. 1992, 84, 785–788. [DOI] [PubMed] [Google Scholar]
- Rao S.; Krauss N. E.; Heerding J. M.; Swindell C. S.; Ringel I.; Orr G. A.; Horwitz S. B. J. Biol. Chem. 1994, 269, 3132–3134. [PubMed] [Google Scholar]
- Rao S.; Orr G. A.; Chaudhary A. G.; Kingston D. G. I.; Horwitz S. B. J. Biol. Chem. 1995, 270, 20235–20238. [DOI] [PubMed] [Google Scholar]
- Lowe J.; Li H.; Downing K. H.; Nogales E. J. Mol. Biol. 2001, 313, 1045–1057. [DOI] [PubMed] [Google Scholar]
- Williams H. J.; Scott A. I.; Dieden R. A.; Swindell C. S.; Chirlian L. E.; Francl M. M.; Heerding J. M.; Krauss N. E. Can. J. Chem. 1994, 72, 252–260. [Google Scholar]
- Dubois J.; Guenard D.; Gueritte-Voeglein F.; Guedira N.; Potier P.; Gillet B.; Betoeil J.-C. Tetrahedron 1993, 49, 6533–6544. [Google Scholar]
- Cachau R. E.; Gussio R.; Beutler J. A.; Chmurny G. N.; Hilton B. D.; Muschik G. M.; Erickson J. W. Supercomput. Appl. High Perform. Comput. 1994, 8, 24–34. [Google Scholar]
- Vander Velde D. G.; Georg G. I.; Grunewald G. L.; Gunn C. W.; Mitscher L. A. J. Am. Chem. Soc. 1993, 115, 11650–11651. [Google Scholar]
- Paloma L. G.; Guy R. K.; Wrasidlo W.; Nicolaou K. C. Chem. Biol. 1994, 1, 107–112. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Chakravarty S.; Inoue T.; Lin S.; He L.; Horwitz S. B.; Kuduk S. C.; Danishefsky S. J. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 4256–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima I.; Kuduk S. D.; Chakravarty S.; Ourevitch M.; Begue J.-P. J. Am. Chem. Soc. 1997, 119, 5519–5527. [Google Scholar]
- Snyder J. P.; Nevins N.; Cicero D. O.; Jansen J. J. Am. Chem. Soc. 2000, 122, 724–725. [Google Scholar]
- Alcaraz A. A.; Mehta A. K.; Johnson S. A.; Snyder J. P. J. Med. Chem. 2006, 49, 2478–2488. [DOI] [PubMed] [Google Scholar]
- Gullion T.; Schaefer J. Adv. Magn. Reson. 1989, 13, 58–82. [Google Scholar]
- Li Y.; Poliks B.; Cegelski L.; Poliks M.; Cryczynski Z.; Piszczek G.; Jagtap P. G.; Studelska D. R.; Kingston D. G. I.; Schaefer J.; Bane S. Biochemistry 2000, 39, 281–291. [DOI] [PubMed] [Google Scholar]
- Paik Y.; Yang C.; Metaferia B.; Tang S.; Bane S.; Ravindra R.; Shanker N.; Alcaraz A. A.; Johnson S. A.; Schaefer J.; O’Connor R. D.; Cegelski L.; Snyder J. P.; Kingston D. G. I. J. Am. Chem. Soc. 2007, 129, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima I.; Geng X.; Lin S.; Pera P.; Bernacki R. J. Bioorg. Med. Chem. Lett. 2002, 12, 349–52. [DOI] [PubMed] [Google Scholar]
- Geng X.; Miller M. L.; Lin S.; Ojima I. Org. Lett. 2003, 5, 3733–6. [DOI] [PubMed] [Google Scholar]
- Querolle O.; Dubois J.; Thoret S.; Dupont C.; Guéritte F.; Guénard D. Eur. J. Org. Chem. 2003, 542–50. [Google Scholar]
- Querolle O.; Dubois J.; Thoret S.; Roussi F.; Montiel-Smith S.; Guéritte F.; Guénard D. J. Med. Chem. 2003, 46, 3623–30. [DOI] [PubMed] [Google Scholar]
- Boge T. C.; Wu Z-J.; Himes R. H.; Vander Velde D. G.; Georg G. I. Bioorg. Med. Chem. Lett. 1999, 9, 3047–52. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Lin S.; Inoue T.; Miller M. L.; Borella C. P.; Geng X.; Walsh J. J. J. Am. Chem. Soc. 2000, 122, 5343–53. [Google Scholar]
- Barboni L.; Lambertucci C.; Appendino G.; Vander Velde D. G.; Himes R. H.; Bombardelli E.; Wang M.; Snyder J. P. J. Med. Chem. 2001, 44, 1576–1587. [DOI] [PubMed] [Google Scholar]
- Ganesh T.; Yang C.; Norris A.; Glass T.; Bane S.; Ravindra R.; Banerjee A.; Metaferia B.; Thomas S. L.; Giannakakou P.; Alcaraz A. A.; Lakdawala A. S.; Snyder J. P.; Kingston D. G. I. J. Med. Chem. 2007, 50, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E.; Wolf S. G.; Downing K. H. Nature 1998, 39, 199–203. [DOI] [PubMed] [Google Scholar]
- Metaferia B. B.; Hoch J.; Glass T. E.; Bane S. L.; Chatterjee S. K.; Snyder J. P.; Lakdawala A.; Cornett B.; Kingston D. G. I. Org. Lett. 2001, 3, 2461–2464. [DOI] [PubMed] [Google Scholar]
- Ganesh T.; Guza R. C.; Bane S.; Ravindra R.; Shanker N.; Lakdawala A. S.; Snyder J. P.; Kingston D. G. I. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10006–10011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S.; Yang C.; Brodie P.; Bane S.; Ravindra R.; Sharma S.; Jiang Y.; Snyder J. P.; Kingston D. G. I. Org. Lett. 2006, 8, 3983–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenard D.; Gueritte-Voegelein F.; Potier P. Acc. Chem. Res. 1993, 26, 160–167. [Google Scholar]
- Gueritte-Voegelein F.; Guenard D.; Lavelle F.; Le Goff M.-T.; Mangatal L.; Potier P. J. Med. Chem. 1991, 34, 992–998. [DOI] [PubMed] [Google Scholar]
- Hodge M. B.M. S. Thesis, Virginia Polytechnic Institute and State University, 2008.
- Ganesh T.; Norris A.; Sharma S.; Bane S.; Alcaraz A. A.; Snyder J. P.; Kingston D. G. I. Bioorg. Med. Chem. 2006, 14, 3447–3454. [DOI] [PubMed] [Google Scholar]
- Paciotti G. F.; Kingston D. G. I.; Tamarkin L. Drug Devel. Res. 2006, 67, 47–54. [Google Scholar]
- Höfle G.; Reichenbach H.. Epothilone, a Myxobacterial Metabolite with Promising Antitumor Activity. In Anticancer Agents from Natural Products; Cragg G. M., Kingston D. G. I., Newman D. J., Eds.; CRC Press: Boca Raton, FL, 2005; pp 413−450. [Google Scholar]
- Bollag D. M.; McQueney P. A.; Zhu J.; Hensens O.; Koupal L.; Liesch J.; Goetz M.; Lazarides E.; Woods C. M. Cancer Res. 1995, 55, 2325–2333. [PubMed] [Google Scholar]
- Höfle G.; Bedorf N.; Steinmetz H.; Schomburg D.; Gerth K.; Reichenbach H. Angew. Chem., Int. Ed. Engl. 1996, 35, 1567–1569. [Google Scholar]
- Nicolaou K. C.; Ritzen A.; Namoto K. Chem. Commun. 2001, 1523–1535. [DOI] [PubMed] [Google Scholar]
- Altmann K.-H. Org. Biomol. Chem. 2004, 2, 2137–2152. [DOI] [PubMed] [Google Scholar]
- Watkins E. B.; Chittiboyina A. G.; Avery M. A. Eur. J. Org. Chem. 2006, 4071–4084. [Google Scholar]
- Altmann K.-H.; Pfeiffer B.; Arseniyadis S.; Pratt B. A.; Nicolaou K. C. ChemMedChem. 2007, 2, 396–423. [DOI] [PubMed] [Google Scholar]
- Conlin A.; Fornier M.; Hudis C.; Kar S.; Kirkpatrick P. Nature Rev. Drug Disc. 2007, 6, 953–954. [Google Scholar]
- Nettles J. H.; Li H.; Cornett B.; Krahn J. M.; Snyder J. P.; Downing K. H. Science 2004, 305, 866–869. [DOI] [PubMed] [Google Scholar]
- Cao S.; Guza R. C.; Wisse J. H.; Evans R.; van der Werff H.; Miller J. S.; Kingston D. G. I. J. Nat. Prod. 2005, 68, 487–492. [DOI] [PubMed] [Google Scholar]
- Furstner A.; Nagano T. J. Am. Chem. Soc. 2007, 129, 1906–1907. [DOI] [PubMed] [Google Scholar]
- Beutler J. A.; Shoemaker R. H.; Johnson T.; Boyd M. R. J. Nat. Prod. 1998, 61, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Yoder B. J.; Cao S.; Norris A.; Miller J. S.; Ratovoson F.; Razafitsalama J.; Andriantsiferana R.; Rasamison V. E.; Kingston D. G. I. J. Nat. Prod. 2007, 70, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mente N. R.; Wiemer A. J.; Neighbors J. D.; Beutler J. A.; Hohl R. J.; Wiemer D. F. Bioorg. Med. Chem. 2007, 17, 911–915. [DOI] [PubMed] [Google Scholar]
- Cao S.; Norris A.; Miller J. S.; Ratovoson F.; Razafitsalama J.; Andriantsiferana R.; Rasamison V. E.; Kingston D. G. I. J. Nat. Prod. 2007, 70, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B. T.; Brodie P. J.; Slebodnick C.; Miller J. S.; Birkinshaw C.; Randrianjanaka L. M.; Andriantsiferana R.; Rasamison V. E.; TenDyke K.; Suh E. M.; Kingston D. G. I. J. Nat. Prod. 2008, 71, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. B.; Norris A.; Miller J. S.; Birkinshaw C.; Ratovoson F.; Andriantsiferana R.; Rasamison V. E.; Kingston D. G. I. J. Nat. Prod. 2007, 70, 206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S.; Brodie P. J.; Miller J. S.; Ratovoson F.; Callmander M.; Randrianasolo S.; Rakotobe E.; Rasamison V. E.; Kingston D. G. I. J. Nat. Prod. 2007, 70, 1064–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://whc.unesco.org/en/list/1017
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2007, 70, 461–477. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Stone M. J.; Hauck P. R.; Rahman S. K. J. Nat. Prod. 1989, 52, 1189–1208. [DOI] [PubMed] [Google Scholar]
- Feher M.; Schmidt J. M. J. Chem. Inf. Comput. Sci. 2003, 43, 218–227. [DOI] [PubMed] [Google Scholar]
- Ortholand J.-Y.; Ganesan A. Curr. Opin. Chem. Biol. 2004, 8, 271–280. [DOI] [PubMed] [Google Scholar]
- Butler M. S. J. Nat. Prod. 2004, 67, 2141–2153. [DOI] [PubMed] [Google Scholar]
- Clardy J.; Fischbach M. A.; Walsh C. T. Nat. Biotechnol. 2006, 24, 1541–1550. [DOI] [PubMed] [Google Scholar]
- Levitzki A. Acc. Chem. Res. 2003, 36, 462–469. [DOI] [PubMed] [Google Scholar]
- Hennessy B. T.; Smith D. L.; Ram P. T.; Lu Y.; Mills G. B. Nature Rev. Drug Disc. 2005, 4, 988–1004. [DOI] [PubMed] [Google Scholar]
- Lyon M. A.; Ducruet A. P.; Wipf P.; Lazo J. S. Nature 2002, 1, 961–976. [DOI] [PubMed] [Google Scholar]
- Xu D.; Jiang B.; Ketela T.; Lemieux S.; Veillette K.; Martel N.; Davison J.; Sillaots S.; Trosok S.; Bachewich C.; Bussey H.; Youngman P.; Roemer T. PLoS Pathogens 2007, 3, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton R. A.; Biediger R. J.; Boatman P. D., Semisynthesis of Taxol and Taxotere. In Taxol: Science and Applications; Suffness M., Eds.; CRC Press, Inc.: Boca Raton, FL, 1995; pp 97−121 [Google Scholar]
- Baumann K. Pharm. Unserer Zeit 2005, 34, 110–114. [DOI] [PubMed] [Google Scholar]
- Cuevas C.; Perez M.; Martin M.; Chicharro J. L.; Fernandez-Rivas C.; Flores M.; Francesch A.; Gallefo P.; Zarzuelo M.; de la Calle F.; Garcia J.; Polanco C.; Rodriguez I.; Manzanares I. Org. Lett. 2000, 2, 2545–2548. [DOI] [PubMed] [Google Scholar]
- Seletsky B. M.; Wang Y.; Hawkins L. D.; Palme M. H.; Habgood G. J.; DiPietro L. V.; Towle M. J.; Salvato K. A.; Wels B. F.; Aalfs K. K.; Kishi Y.; Littlefield B. A.; Yu M. J. Biorg. Med. Chem. Lett. 2004, 14, 5547–5550. [DOI] [PubMed] [Google Scholar]
- Kingston D. G. I.Taxol and Its Analogs. In Anticancer Agents from Natural Products; Cragg G. M., Kingston D. G. I., Newman D. J., Eds.; CRC Press: Boca Raton, 2005; pp 89−122 [Google Scholar]
- Rivkin A.; Yoshimura F.; Gabarda A. E.; Cho Y. S.; Chou T.-C.; Dong H.; Danishefsky S. J. J. Am. Chem. Soc. 2004, 126, 10913−–10922. [DOI] [PubMed] [Google Scholar]