

Abstract
20-Hydroxyeicosatetraenoic acid (20-HETE) has been implicated as a potential mediator in epithelial cell proliferation and cyst formation in polycystic kidney disease (PKD). In the present study, we studied the effects of chronic blockade of 20-HETE synthesis in an orthologous rodent model of autosomal recessive polycystic kidney disease (ARPKD), the PCK rat. RT-PCR analysis indicated that the expression of CYP4A1, CYP4A2, CYP4A3, and CYP4A8 mRNA was increased two- to fourfold in cystic PCK compared with noncystic Sprague-Dawley rat kidneys. Daily administration of a 20-HETE synthesis inhibitor, HET-0016 (10 mg·kg−1·day−1 ip) for 4–7 wk significantly reduced kidney size by 24% from 4.95 ± 0.19 g in vehicle-treated PCK rats to 3.76 ± 0.15 g (n = 4). Collecting tubule morphometric cystic indices were reduced in HET-0016-treated PCK rats (2.1 ± 0.2; n = 4) compared with vehicle-treated PCK rats (4.4 ± 0.1; n = 4). The cellular mechanism by which 20-HETE may play a role in cyst formation has not been well characterized, but there was a significantly lower (P < 0.05) level of intracellular cAMP and decreased phosphorylation (activation) of ERK1/2 protein in PCK rat kidneys (n = 3) treated with HET-0016 . These studies indicate a potential role of 20-HETE in cyst formation in the orthologous rodent PCK model of ARPKD.
Keywords: cytochrome P-450, HET-0016, epithelial cell proliferation, polycystic kidney rat, pck, autosomal recessive polycystic kidney mouse
autosomal recessive polycystic kidney disease (ARPKD) is a genetic disorder associated with mutations in the polycystic kidney and hepatic disease 1 (PKHD1) gene that affects ∼1:20,000 newborn children each year. Approximately 25% of the newborns die early after birth, and those children that survive generally progress to end-stage renal disease during childhood or early adolescence. Most children with ARPKD also develop significant portal hypertension, with its attendant morbidity and mortality, secondary to a biliary plate developmental abnormality (1).
The main renal phenotype in ARPKD is the gross morphological enlargement of the kidney. This is due to the rapid expansion of renal epithelial cells originating from the collecting ducts leading to the formation of large, ectatic, fluid-filled collecting ducts which extend from the medulla to the cortex (3, 14, 16, 20). At present, there is no disease-specific therapy for this devastating disease. However, studies in preclinical animal models have demonstrated that slowing the progression of cyst formation enhances kidney and animal survival (1).
Numerous systems have been studied as potential therapeutic targets in mitigating the proliferative phenotype of PKD. Furthermore, blockade of several systems have been shown to reduce cyst formation and enhance renal function and animal survival (34, 37). At present, very little is known about the role of eicosanoids in PKD. Our laboratory has recently reported that 20-HETE promotes hyperplasia of renal epithelial cells leading to the formation of cysts in a murine model of ARPKD both in vitro and in vivo (26). These findings are consistent with previous work in which several investigators have shown the mitogenic role of 20-HETE in various normal (15, 18, 19) and oncogenic (4, 8) cell lines. We also found that chronic inhibition of 20-HETE synthesis using a phenotypic murine model of ARPKD, the Balb/C polycystic kidney (BPK) mouse, minimized cyst formation and reduced kidney size. These changes were associated with improved renal function and prolonged lifespan of the animal (26).
In the present study, we examined whether 20-HETE plays a similar role in promoting cystogenesis in another rodent model of autosomal recessive polycystic kidney disease (ARPKD), the PCK rat. The PCK rat is an orthologous model of human ARPKD, since it carries a deletion in the PKHD1 gene (40) and exhibits both renal and biliary abnormalities observed in humans affected by ARPKD. The experimental approach was to chronically inhibit 20-HETE synthesis using HET-0016, which is a specific pharmacological blocker of the CYP4A and CYP4F isozymes, and determine whether the reduction in 20-HETE formation attenuates the severity of polycystic kidney disease.
MATERIALS AND METHODS
Drugs.
HET-0016 was generously provided by Dr. N. Miyata (Taisho Pharmaceuticals, Tokyo, Japan). Two selective epoxygenase inhibitors, N-methylsulfonyl-6-(2-propargyloxyphenyl) hexanamide (MS-PPOH) and N-trifluorosulfonyl-6-(2-propargyloxyphenyl) hexanamide (FS-PPOH) were generously provided by Dr. J. R. Falck (Department of Biochemistry, University of Texas-Southwestern, Dallas, TX).
Animal experiments.
Male polycystic kidney (PCK) and noncystic Sprague-Dawley (SD) rats were obtained from breeding colonies housed at both Charles River Laboratories (Wilmington, MA) and the Medical College of Wisconsin (Ellis D. Avner/William E. Sweeney laboratory) between postnatal (PN) weeks 4 and 5. One PCK rat was treated at PN day 7 through PN day 56, and no difference was noted in the cystic indices compared with the PCK rats treated at PN 4–5 wk, so we grouped the entire HET-0016-treated animal together in our analysis. The animals were allowed free access to water and food during this study. All animal handling and experiments outlined in this study were approved by the Institutional Animal Care Committee following the guidelines set forth by the National Institutes of Health. HET-0016 was dissolved in an isotonic mannitol solution containing 11% captisol (wt/vol; CyDex Pharmaceuticals, Lenexa, KS) and intraperitoneally (ip) injected daily at a dose of 10 mg/kg for a period of 4–7 wk. In a separate group of animals, the effects of selective blockade EET synthesis with MS-PPOH (15 mg/kg ip) and FS-PPOH (5 mg/kg ip) were studied. MS-PPOH or FS-PPOH was administered to PCK rats at 4 wk of age for a period of 4 wk. The drugs were initially dissolved in 100% ethanol and then diluted 10-fold with 11% captisol. All rats were killed at 8 wk postnatal. At the end of the experiment, blood urea nitrogen (BUN) and serum creatinine was determined using an autoanalyzer (ACE; Alfa Wasserman, Fairfield, NJ).
Protein isolation and Western blot analysis.
Renal kidney pieces containing segments of the cortex, outer medulla, and inner medulla were homogenized in 1× RIPA buffer in the presence of protease inhibitors (Roche Applied Science, Indianapolis, IN) and phosphatase inhibitors (Pierce, Rockford, IL). Homogenates were centrifuged at 3,000 g for 5 min and subsequently at 10,000 g for 10 min at 4°C. The supernatant was collected and the protein measured by the Bradford method (Bio-Rad, Hercules, CA). Microsomal protein (50–100 μg) was loaded onto a 10% NuPAGE Bis-Tris Gel (Invitrogen, Carlsbad, CA) and size-separated for transfer to polyvinylidene difluoride membranes. Membranes were blocked in TBS-T containing 5% nonfat dry milk and subsequently incubated overnight with primary antibodies: rabbit polyclonal CYP4A1 (1:1,500 dilution; Daiichi Pure Chemicals, Tokyo, Japan), rabbit anti-phospho-p44/42 MAP kinase (9101, 1:1,500 dilution; Cell Signal), rabbit anti-p44/42 MAP kinase (4695, 1:1,500; Cell Signal), rabbit anti-Stat3 (9132, 1:1,000; Cell Signal), mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:2,000; Sigma, St. Louis, MO). Following the primary antibody, the membranes were incubated with secondary antibodies: goat anti-rabbit IgG conjugated with horseradish peroxidase (HRP; 7074, 1:1,500; Cell Signal) and goat anti-mouse IgG conjugated with HRP (1:1,500; Bio-Rad). The membranes were thoroughly washed in TBS-T and then placed into a chemiluminescent solution (GE Healthcare) for 120 s. The membranes were covered in Saran Wrap and placed on film for development of the protein bands using a Kodak X-OMAT 2000A processor.
Total RNA extraction and reverse transcription assay.
Total RNA was extracted from a section of the kidneys using TRIzol reagent (Invitrogen). The extracted total RNA was resuspended in DEPC-treated water and incubated in the presence of RNAse-free DNAse I (20 U; New England BioLabs, Boston, MA) for 30 min at 37°C. The total RNA was reextracted using TRIzol and re-suspended in DEPC-treated water and frozen at −80°C until use. The RNA concentration was determined by UV spectroscopy. For the reverse transcription assay, DNAse-treated RNA (1 μg) was reverse transcribed using random hexamers and SuperScript III RT (Invitrogen) for 50 min at 50°C and then terminated by heating at 85°C for 5 min. The synthesized RNA-DNA hybrid was eliminated by incubation with RNase H at 37°C for 20 min. Synthesized cDNA was placed on ice and used in the subsequent PCR assay.
Cytochrome P-450 primer sequences.
The following gene-specific primers for CYP4A and 4F isoforms were used (IDT Technologies, Coralville, IA): CYP4A1 (accession no. NM_175837) sense 5′-CTC TTA CTT GCC AGA ATG GAG AA-3′, antisense 5′-GAC TTG GAT ACC CTT GGG TAA AG-3′; CYP4A2 (accession no. NM_001044770) sense 5′-AGA TCC AAA GCC TTA TCA AT-3′, antisense: 5′-CAG CCT TGG TGT AGG ACC T-3′; CYP4A3 (accession no. NM_175760) sense 5′-CAA AGG CTT CTG GAA TTT ATC-3′, antisense 5′-CAG CCT TGG TGT AGG ACC T-3′; CYP4A8 (accession no. NM_031605) sense 5′-ATC CAG AGG TGT TTG ACC CTT AT-3′, antisense: 5′-AAT GAG ATG TGA GCA GAT GGA GT-3′; CYP4F1 (accession no. NM_019623) sense 5′-TCG TCC GAT CTA TCC TCA ATG-3′, antisense 5′-TTG TCA CCA GCA CTC ACC AG-3′; CYP4F5 (accession no. BC129085) sense 5′-GAG AAC TGC TCC CGC CTC AGA T-3′, antisense 5′-CAG AGA TGG ATA TCT CGG AAA GTC T-3′; and CYP4F6 (accession no. BC078713) sense 5′-GTG ATA TCC ACC TCT CTT GGG-3′, antisense 5′-GAG GGT CAT TTC CTT GGG TG-3′.
PCR of cytochrome P-450 mRNA isoforms.
One hundred nanograms of cDNA was amplified in the presence of 400 nM of the forward and reverse primers using SYBR GreenER qPCR SuperMix Universal (Invitrogen). For 18S reactions, only 1 ng of cDNA was amplified in the final concentration of 50 nM forward and reverse primers. PCR conditions for CYP4A1, CYP4A8, CYP4F1, CYP4F5, CYP4F6, and 18S was as follows: 50°C for 2 min (UDG incubation) and 10 min at 95°C for initial melting. This was followed by 40 cycles of 95°C for 30 s, 60°C for 1 min, and 72°C for 30 s. For the CYP4A2 and CYP4A3 isoforms, which have extremely high homology between one another (∼97%), a higher annealing temperature of 70°C was used for the first 10 cycles to increase the specificity. After the initial 10 cycles, the annealing temperature was reduced to 60°C for the remaining 30 cycles. Quantitative copy number values were obtained by comparing to a standard curve using serially diluted plasmids containing full-length cDNAs of the CYP4A and CYP4F isoforms, respectively, and the data were obtained from the Stratagene Mx3000P application (Stratagene, La Jolla, CA). Copy numbers were subsequently corrected according to the values obtained from the 18S amplification.
Liquid chromatographic/mass spectrometric eicosanoid profiling.
SD and PCK rats (6 wk of age) were injected with vehicle (11% captisol) or HET-0016 (10 mg/kg ip) 90–120 min before death and removal of the kidneys. In a separate assay, we measured the 20-HETE production in the PCK kidneys chronically treated with vehicle or HET-0016 (10 mg/kg ip). The harvested kidneys were homogenized, and renal microsomes were isolated as previously described (11). Renal microsomes (250 μg) were incubated in 0.5-ml reactions containing 0.1 M potassium phosphate buffer (pH 7.4) consisting of 1 mM EDTA, 10 mM MgCl2, 2 mM NADPH, 10 mM sodium isocitrate, 0.8 U isocitrate dehydrogenase, and 40 μM arachidonic acid. The reactions were incubated at 37°C for 30 min under an atmosphere of 100% O2. The reaction was stopped by acidification using formic acid (pH 3.5). The lipids were extracted using ethyl acetate, and 20-HETE-d6 (10 ng) was added as an internal control to calculate the extraction efficiency. The organic phase was dried down under N2 gas. Samples were reconstituted in 1:1 methanol-water mixture, and then the 20-HETE and other arachidonic acid metabolites were identified by API 3000 Liquid Chromatograph-triple quadrupole Mass Spectrometer (LC-MS; Applied Biosystems, Foster City, CA) as previously described by Williams et al. (41).
cAMP assay.
Kidney tissue slices were pulverized in liquid nitrogen using a mortar and pestle. A volume of 1 ml of HCl (0.1 N) was added to the kidney powder and homogenized. The kidney lysates were centrifuged at >600 g for 10 min, and the protein concentration was determined. cAMP levels were determined using a commercially available acid-tolerant EIA kit (Assay Designs, Ann Arbor, MI), and the values were reported as picomoles cAMP per milligram protein.
Renal histology and cystic indices.
Kidneys were cut coronally and embedded in plastic as previously described by Sweeney et al. (22, 30, 33, 35). Five-micrometer sections were made and stained with hematoxylin. The cystic indices for PCK rat kidneys were calculated by modifying the basic morphometric method utilized in BPK mice as developed by Sweeney et al. (22, 30, 33, 35) and more recently in our laboratory (26). In brief, the collecting tubule cystic indices (CT CI) was scaled from 0 to 10 based on the number and size of cysts normally found in untreated PCK rat kidneys beginning at PN days 0–150.
Statistical analyses.
Mean values ± SE are presented. The significance of differences in mean value between the treatment groups was evaluated using an ANOVA followed by the Tukey post hoc test (Prism 2.0 software). A P < 0.05 was considered to be statistically significant.
RESULTS
20-HETE production in noncystic SD and cystic PCK rat kidney microsomes.
To demonstrate that the dose of HET-0016 (10 mg/kg) used in our chronic studies was selective and effective in reducing 20-HETE production in vivo without altering the production of other metabolites of arachidonic acid, we administered HET-0016 (10 mg/kg ip) 90 min before euthanizing the rats and harvested their kidneys. The results of these experiments are presented in Fig. 1. We found that the acute administration of HET-0016 significantly reduced (P < 0.001) the renal production of 20-HETE by 69 ± 5 and 83 ± 6% in SD and PCK rats, respectively, vs. the levels seen in rats treated with vehicle. HET-0016 had no effect on the production of other HETEs or DiHETEs and tended to increase the formation of EETs although these values were nonsignificant from their respective vehicle control groups. These experiments demonstrated that the dose of the HET-0016 used in our chronic studies is effective in vivo and selectively reduces the renal formation of 20-HETE without altering the production of other metabolites of arachidonic acid.
Fig. 1.
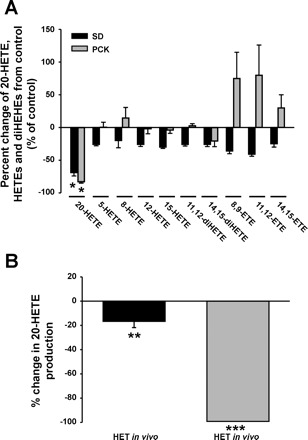
Inhibition of 20-HETE production in cystic PCK vs. noncystic Sprague-Dawley (SD) rat kidneys. A: HET-0016 (10 mg/kg) was administered acutely into the intraperitoneal space 90–120 min before death. Renal microsomes were isolated as described in materials and methods, and a cytochrome P-450 assay was performed and analyzed by liquid chromatography coupled to triple quadrupole mass spectrometry (LC/MS/MS). The data was graphed as a ratio of HET-treated vs. vehicle-treated rats (i.e., percent change from vehicle-treated control). *P < 0.001 significance between the PCK and SD rats treated with vehicle vs. HET-0016; n = 3–4 rats/eicosanoid group. B: LC/MS was performed on rat renal microsomes harvested from chronically treated PCK rats with vehicle or HET-0016 (HET) and graphed as percent change from the vehicle group (black bar). Renal microsomes were incubated with HET-0016 (1 μM) during the incubation phase and analyzed for 20-HETE synthesis inhibition in vitro (grey bar); n = 3–4 samples/group. **P < 0.05 significance between chronic vehicle- vs. HET-0016-treated kidneys. ***P < 0.001 significance between kidney samples treated with vehicle or HET-0016 during in vitro P450 assay.
Following the 4-wk period of chronic HET-0016 or vehicle administration, the kidneys were removed to document the effects on 20-HETE production. The kidneys in our study were harvested 24 h following the final administration of vehicle or HET-0016, and the LC/MS eicosanoid profiling was performed on the isolated renal microsomes. As shown in Fig. 1B, we detected that there was a mild, but significant (P < 0.05) 17 ± 5% reduction in 20-HETE production in the PCK rats treated with HET-0016 compared with the vehicle-treated rats, which correlated with the reduction in cystic kidney size (Table 1). To provide a positive control for the effect of HET-0016 on 20-HETE production, we performed the in vitro P-450 assay in the presence of HET-0016 at a dose of 1 μM, which significantly (P < 0.001) abolished the production of 20-HETE by the isolated renal microsomes (grey bar, Fig. 1B).
Table 1.
Biological parameters of PCK rats following 20-HETE synthesis inhibition
| PCK (VEH) | PCK (HET), 10 mg·kg−1·day−1 | |
|---|---|---|
| Total body weight, g | 384±12 (n = 4) | 362±7 (n = 4) |
| Kidney weight, g | 4.95±0.19 (n = 4) | 3.76±0.15* (n = 4) |
| KW/BW, % | 1.29±0.02 (n = 4) | 1.04±0.03* (n = 4) |
| CT CI | 4.4±0.1 (n = 4) | 2.1±0.2* (n = 4) |
| LW/BW, % | 5.76±0.2 (n = 4) | 5.02±0.27 (n = 4) |
| BUN, mg/dl | 12.4±1.8 (n = 3) | 10.2±0.2 (n = 3) |
| Serum creatinine, mg/dl | 0.49±0.19 (n = 3) | 0.37±0.03 (n = 3) |
Values are means ± SE; n = no. of rats. Body, kidney, and liver weights were measured in polycystic kidney (PCK) rats treated with vehicle (VEH; 11% captisol) or HET-0016 (HET; 10 mg·kg−1·day−1) to calculate the kidney weight/body weight ratio (KW/BW) and liver weight/body weight ratio (LW/BW). Kidneys were subsequently sectioned to evaluate the collecting tubule cystic indices (CT CI). Blood was collected at the end of the experiment to determine the blood urea nitrogen (BUN) and creatinine; n = 4 rats for each measurement.
P < 0.001 denotes significant difference between PCK rats treated with VEH vs. HET-0016.
Effects of chronic blockade in 20-HETE and EET formation on kidney mass.
Chronic administration of HET-0016 (10 mg·kg−1·day−1 ip) significantly decreased (P < 0.001) kidney weight by 24% (Table 1). Kidney weight-to-body weight (KW/BW) ratios significantly decreased (P < 0.001) from 1.29 ± 0.02% in vehicle-treated PCK rats to 1.04 ± 0.03% in rats treated with HET-0016. We also found that HET-0016 treatment had a mild, but nonsignificant tendency to reduce the liver weight-to-total body weight (LW/BW) ratio. The LW/BW ratio was reduced by 13% to 5.02 ± 1.95 g in the HET-0016-treated rat kidneys compared with the vehicle-treated rat kidneys (5.76 ± 0.02 g).
In a separate experiment, we demonstrated that chronic blockade of EET formation with MS-PPOH (15 mg·kg−1·day−1) and FS-PPOH (5 mg·kg−1·day−1) had no significant effect on kidney and liver weight compared with the vehicle-treated rats (Table 2).
Table 2.
Biological parameters of PCK rats following EET synthesis inhibition
| PCK (VEH) | PCK (MS-PPOH), 15 mg·kg−1·day−1 | PCK (FS-PPOH), 5 mg·kg−1·day−1 | |
|---|---|---|---|
| Total body weight, g | 302±6 | 292±11 | 281±19 |
| Kidney weight, g | 4.51±0.18 | 3.88±0.12 | 4.31±0.08 |
| KW/BW, % | 1.50±0.08 | 1.33±0.02 | 1.53±0.06 |
| CT CI | 3.75±0.08 | 3.13±0.07* | 3.61±0.16 |
| LW/BW, % | 6.29±0.14 | 6.29±0.32 | 6.44±0.64 |
| BUN, mg/dl | 17.5±2.5 | 15.9±1.9 | 15.5±1.4 |
| Serum creatinine, mg/dl | 0.36±0.12 | 0.41±0.07 | 0.31±0.02 |
Values are means ± SE; n = no. of rats. Body, kidney, and liver weights were measured in PCK rats treated with vehicle (35% ethanol:65% 11% captisol), N-methylsulfonyl-6-(2-propargyloxyphenyl) hexanamide (MS-PPOH; 15 mg·kg−1·day−1) and N-trifluorosulfonyl-6-(2-propargyloxyphenyl) hexanamide (FS-PPOH; 5 mg·kg−1·day−1) to calculate KW/BW and LW/BW. Kidneys were subsequently sectioned to evaluate CT CI. Blood was collected at the end of the experiment to determine BUN and creatinine; n = 4 rats for each measurement.
P < 0.01 significant difference between MS-PPOH- vs. VEH-treated PCK rats.
The representative appearance of the kidney from PCK rats treated with vehicle and HET-0016 is presented in Fig. 2. The cysts found in the PCK kidneys were exclusively found in the collecting ducts, and the size and number varied between rats. Chronic treatment with HET-0016 produced a marked reduction in the size and number of cysts. The cystic index was significantly reduced (P < 0.001) by ∼50% in PCK rats treated with HET-0016 compared with the values seen in PCK rats treated with vehicle. In contrast, chronic treatment of the rats with the epoxygenase inhibitors MS-PPOH and FS-PPOH did not reduce the CT CI as markedly as in the HET-0016-treated rat kidneys (Table 2).
Fig. 2.
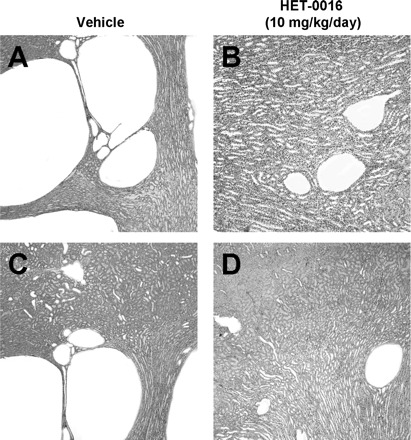
Effect of 20-HETE synthesis inhibition on kidney mass in a rat model of autosomal recessive polycystic kidney disease (ARPKD). HET-0016 (10 mg·kg−1·day−1), a specific inhibitor of 20-HETE synthesis, was administered daily to polycystic kidney (PCK) rats into the intraperitoneal space, and the animals were killed between postnatal days 56 and 63. The kidneys were harvested, fixed in plastic, and sectioned. Representative kidney sections from four different rats treated with either vehicle (A and C) or HET-0016 (B and D) are shown. Sections were stained with hematoxylin; n = 4 kidneys/group. Magnification ×20.
BUN and plasma creatinine concentration were in the normal range in PCK rats (Tables 1 and 2). Chronic treatment with either 20-HETE (HET-0016) or EET (MS-PPOH and FS-PPOH) synthesis inhibitors had no significant effect on BUN or plasma creatinine levels.
Cytochrome P-450 4A and 4F mRNA isoforms and P-450 4A protein expression in PCK rats.
The expression of the CYP4A mRNA was significantly higher (P < 0.001 to P < 0.01) by 2.6-to 4.6-fold in the kidneys harvested from vehicle-treated PCK rats compared with the levels seen in control SD rats. For the CYP4F mRNA isoforms, only the CYP4F1 mRNA was significantly increased (P < 0.05) in the PCK rat vs. the noncystic SD rat kidneys (Table 3).
Table 3.
Cytochrome P-450 4A and 4F mRNA isoforms in noncystic SD and PCK rat kidneys
| CYP Isoform | SD (VEH) (n = 4) | PCK (VEH) (n = 7) | PCK (HET) (n = 3) |
|---|---|---|---|
| CYP4A1 | 14,143±1,852† | 36,411±3,811 | 13,854±1,627† |
| CYP4A2 | 311,250±48,445† | 1,344,960±309,760 | 404,167±63,899* |
| CYP4A3 | 403,625±100,636† | 1,841,714±282,586 | 743,389±151,999* |
| CYP4A8 | 661,750±87,103‡ | 2,122,500±213,877 | 984,111±123,512† |
| CYP4F1 | 23,012±123* | 68,961±16,260 | 42,944±22,959 |
| CYP4F5 | 23.4±0.2 | 88.1±14.3 | 53.9±26.7 |
| CYP4F6 | 958±6 | 807±152 | 2,541±1,815 |
Values are means ± SE; n = no. of rats. Total RNA was extracted from noncystic Sprague-Dawley (SD) and PCK rat kidneys, and quantitative RT-PCR was performed.
P < 0.05 significant difference from vehicle-treated PCK rats.
P < 0.01 significant difference from vehicle-treated PCK rats.
P < 0.001 significant difference from vehicle-treated PCK rats.
We also examined the expression of CYP4A and 4F isoforms in PCK rats chronically treated with HET-0016. The expression of each of the CYP4A mRNA isoforms was significantly decreased (P < 0.01 to P < 0.05) in the kidneys of PCK rats treated with HET-0016 compared with the vehicle-treated counterparts.
Subsequently, we evaluated the CYP4A protein expression in the noncystic SD and cystic PCK rat kidneys treated with vehicle (VEH) or HET-0016 (HET) as shown in Fig. 3. The CYP4A antibody was generated using purified rat 4A1 protein, but it has cross-reactivity to CYP4A2 and CYP4A3. In our hands, it had minimal cross-reactivity to murine Cyp4a12, which is the rat homolog to CYP4A8, but it was capable of strongly detecting murine Cyp4a10 (data not shown). There was a tendency for the CYP4A protein to be higher in the PCK rats by densitometry, but no change was noted in the PCK rat kidney lysates whether vehicle or HET-0016 was administered chronically to the animals.
Fig. 3.
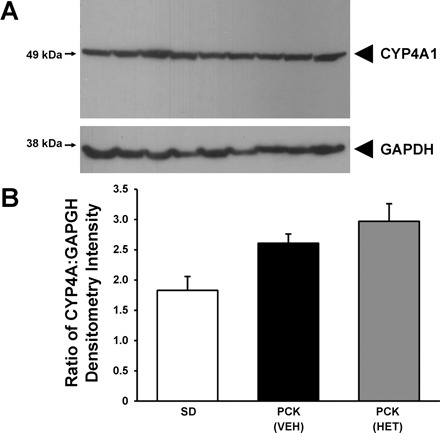
Immunoblot analysis of CYP4A protein in the kidneys. A: protein lysates from noncystic SD and PCK rats treated with vehicle (VEH) or HET-0016 (HET) were electrophoresed and immunoblotted with primary polyclonal antibody targeted to cytochrome P-450 4A (CYP4A). GAPDH was used to normalize equal loading of the lanes; n = 3 kidney samples/group. Small arrow, protein standard. B: graph demonstrating densitometry values for CYP4A:GAPDH. Arrows on the left indicate protein standard to document protein size of CYP4A and GAPDH.
Intracellular cAMP levels following 20-HETE synthesis inhibition.
The results of these experiments are presented in Fig. 4. Intracellular cAMP levels were significantly higher (P < 0.05) by 27% in PCK rats treated with vehicle compared with levels detected in PCK rats treated with HET-0016. The reduction in the cAMP levels correlated with the decreased KW/BW ratio and the cystic indices following HET-0016 treatment as shown in Table 1.
Fig. 4.
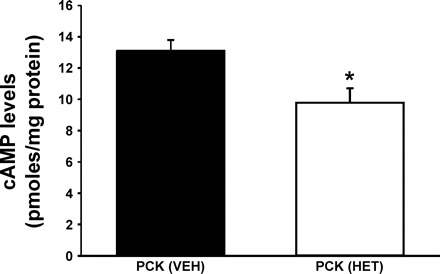
Renal cAMP levels in PCK rats following treatment with 20-HETE synthesis inhibitor. Protein lysates were extracted by 0.1 N HCl from PCK rat kidneys treated with vehicle (n = 7) or HET-0016 (n = 3), and the intracellular levels of the second messenger cAMP was measured by ELISA. *P < 0.05 significant difference between groups.
Effects of HET-0016 on activation of the ERK1/2 pathway.
As shown in Fig. 5, Western blot analyses of cystic PCK rat kidneys chronically treated with HET-0016 resulted in a significant 55% decrease (P < 0.02) in the phosphorylation (activation) of p44/42 MAPK (ERK1/2) protein (9,472 ± 682 densitometry units) compared with the values from the vehicle-treated PCK rat kidneys (4,254 ± 1,059 densitometry units). In contrast, total ERK1/2 protein was not significantly altered between the two groups.
Fig. 5.
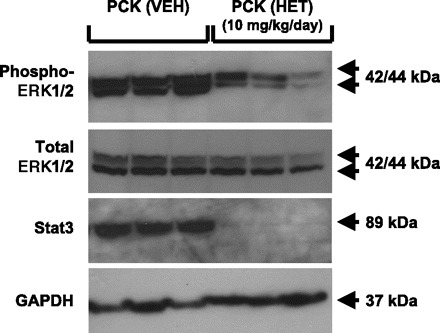
Immunoblot analysis of ERK1/2 and Stat3 in PCK rats. Protein lysates from the PCK rats treated with vehicle (VEH) or HET-0016 (HET) were electrophoresed and immunoblotted with primary antibodies targeted to phospho-ERK1/2 (p44/42 MAPK; A), total ERK1/2 (p44/42 MAPK; B), Stat3 (C), and GAPDH (D); n = 3 kidney samples/group.
The transcription factor Stat3, which has been found to be activated downstream of the ERK1/2 pathway, was significantly reduced (P < 0.01) in the HET-0016-treated rat kidneys (undetectable) compared with the vehicle-treated rats (4,626 ± 382). As shown in Fig. 5, no significant change (P > 0.05) was detected in the housekeeping protein GADPH between the vehicle-treated (9,309 ± 1,339 densitometry units) vs. HET-0016-treated PCK rats (10,800 ± 975 densitometry units), demonstrating equal loading of the protein samples.
DISCUSSION
Previous work has shown that 20-HETE is produced in both tubular and vascular cells in the kidney (31) by ω-hydroxylation of arachidonic acid. This enzymatic reaction to produce 20-HETE is catalyzed by a family of cytochrome P-450 enzymes, specifically CYP4A and CYP4F. 20-HETE is known to play an important role in the regulation of tubular sodium reabsorption, vasoactivity of renal blood vessels, angiogenesis, and more recently, renal epithelial cell mitogenesis (26, 31).
However, the latter role of 20-HETE to mediate a mitogenic response has garnered little attention in PKD. To address the role of 20-HETE in PKD, we recently designed experiments in two animal models of ARPKD, the BPK mouse (26) and the PCK rat. The BPK mouse has been extensively characterized in our laboratory (23, 24, 26). The BPK mouse is a nonorthologous model of ARPKD, since the specific molecular mutation responsible for the cystic phenotype has been identified as the bicaudal C1 (bicc1) gene, which is not the gene associated with human ARPKD, the polycystic kidney and hepatic gene 1 (PKHD1) gene. Regardless, the time course and phenotypic characteristics of the cystic disease in the BPK mouse consistently mimic the effects observed in human ARPKD. It is important to note that homozygous BPK mice develop massively enlarged kidneys, leading to renal failure and death within 28 days after birth. This makes the BPK mouse an ideal model to study medicinal interventions related to cystic kidney disease, since the effects on the kidney can be rapidly screened.
Therefore, we used the BPK mouse in our proof-of-concept study to determine whether 20-HETE played an important role as a mitogen in ARPKD (26). Our laboratory reported that 20-HETE production was significantly higher in BPK kidneys compared with its genetically compatible noncystic Balb/c kidneys. In addition, we found chronic blockade of 20-HETE synthesis with HET-0016, which is a specific inhibitor of CYP4A and 4F, resulted in a significant reduction in cyst size and number (26). The chronic administration of HET-0016 was found to be able to prolong the longevity of the BPK mice to nearly double the normal lifespan (∼46 days) compared with the vehicle-treated BPK mice, which, as explained earlier, normally perish before PN day 28 (26).
To extend our previous work in the BPK mouse, we studied the genetically orthologous rodent model of ARPKD, the PCK rat (13). The PCK rat is a model of human ARPKD caused by a splicing mutation (IVS35-2A3T) that leads to a frameshift in the ortholog Pkhd1 as a result of skipping of exon 36 (40). The PCK is currently the only available orthologous model of human ARPKD that develops the dual-organ pathology characteristic of human ARPKD. Principal manifestations of human ARPKD involve the fusiform dilation of renal collecting tubules or ducts (CT) and biliary dysgenesis due to ductal plate abnormalities (6). The progressive CT cystogenesis in the PCK rat (40) is relatively slow compared with the BPK mouse. For this reason, the BPK (26) and PCK animal models (in the current study) were treated at different stages of cystic disease progression. By PN day 21, the BPK mouse is premorbid and in severe renal failure, emphasizing the value of the aggressive pace of disease in this model for preclinical screening. Conversely, by PN day 56, the PCK rat is still at an early stage of disease with mild renal insufficiency as determined by BUN and serum creatinine (Ref. 13 and unpublished observations in our laboratory) and has a lifespan that extends for at least 1 yr despite progressive cystic disease. It is known that changes in renal function will be preceded by altered cell proliferation rates. Even with the slower onset of functional renal disease manifestations in the PCK rat, it does not diminish its value as an effective model for screening antiproliferative therapies since KW/BW ratios, cystic indices, and proliferation rates can be used effectively to assess efficacy on proliferation.
This illustrates the importance of the PCK rat model to study the antiproliferative effects of HET-0016 for the treatment of ARPKD, and, consistent with our findings in the murine BPK model of ARPKD, we found that there was a significant reduction in the CT CI in the PCK rat kidneys (Table 1). The dose of HET-0016 (10 mg/kg) was found to be predominantly inhibitory to 20-HETE synthesis, as shown in Fig. 1A, without altering the production of other eicosanoids. We also measured the 20-HETE synthesis using the kidneys chronically treated with HET-0016 (10 mg/kg), where the last dose of the drug was administered 24 h before death. As shown in Fig. 1B, there was a mild, but significant reduction in the 20-HETE synthesis, which correlated with the reduced kidney size and cystic indices (Table 1). The reason that we did not detect a larger inhibition of 20-HETE synthesis from the chronic HET-0016-treated kidneys is attributed to the relatively short half-life of HET-0016 (∼50 min in vivo) as measured by Poloyac et al. (29). It is likely that all of the HET-0016 was metabolized within the 24-h window before each subsequent injection. It is likely that improved inhibition of cyst formation would occur using a multiple-dosing regimen with HET-0016 rather than a single daily dose as we used in the current study.
On a molecular level, the PCK rat kidney was found to have increased steady-state levels of CYP4A8 mRNA, the rat homolog of the murine Cyp4a12, which we previously found was elevated in the kidney of the BPK mouse (26). We also demonstrated that the increased renal expression of CYP4504A mRNA isoforms in the PCK rat kidney was normalized in the PCK rats treated with HET-0016 to expression levels detected in the noncystic SD rats. This may reflect the marked decrease in the number of cystic cells that are actively undergoing proliferation in the PCK rats treated with HET-0016. On the protein level, we did not observe any marked difference in CYP4A expression, although there was a tendency for the protein to be higher by densitometry in the PCK vs. SD rats. The lack of a reduction in the CYP4A protein in the HET-0016-treated kidneys may be attributed to a positive feedback loop in which reduced levels of the end product, 20-HETE, resulting in increased translation and/or stability of the CYP4A protein. This may have occurred even under conditions in which we measured lower steady-state levels of the CYP4A mRNA isoforms, and this would indicate that there is a more complex regulatory system in the PCK rat that regulates the production of 20-HETE, such as the substrate (i.e., arachidonic acid) levels and availability, phospholipase A2 or D activity, CYP enzyme stability, etc. In all, the present phenotypic results are entirely consistent with our previous findings in the BPK mouse model of ARPKD, but more molecular and cellular analyses are needed to dissect out the mechanisms involved in promoting 20-HETE activity in both models of ARPKD.
To further explore the role of 20-HETE relative to other metabolites of arachidonic acid that are normally produced in the kidney, additional experiments were performed to determine the effects of chronically blocking the formation of EETs and DiHETEs using selective epoxygenase inhibitors MS-PPOH and FS-PPOH (38). EETs are known to be synthesized in the proximal convoluted tubules, thick ascending limbs, and collecting ducts through the activity of numerous cytochrome P-450 isoforms, including enzymes of the CYP1A, CYP2B, CYP2C, CYP2D, CYP2G, CYP2J, and CYP2N families. Although each of these CYP450 isozymes is involved in EET production, it is well established that CYP2C and CYP2J are the major enzymes for EET production (10). Moreover, EETs have been reported to increase mitogenesis in the proximal tubule. In the present study, we found that chronic administration of MS-PPOH did not significantly reduce kidney size, but it did have a mild effect in reducing the CT CI. The reduction with MS-PPOH was not as large as the decrease calculated for the kidneys treated with HET-0016. These results are consistent with our previous work in the BPK mouse and indicate that 20-HETE rather that EETs acts as the primary proliferative eicosanoids in the cystic kidneys. However, it is likely that EETs could play a minor role in mitigating the epithelial cell cystic response. Because of our findings, it is plausible that HET-0016 or drugs related to inhibiting 20-HETE function may become an important therapeutic to attenuate epithelial cell proliferation and cystogenesis in PKD.
Very little is known about the mitogenic signaling pathway(s) by which 20-HETE promotes cystogenesis. Previous studies have shown other cystic hormones, such as epidermal growth factor (EGF) (4, 15) and angiotensin II, increase the formation of 20-HETE in both renal- and non-renal-derived cells. Other cystic hormones, including arginine vasopressin (AVP), which are known to activate cAMP-dependent proliferative pathways, have yet to be studied with regard to their potential interaction with 20-HETE and/or other eicosanoids. One major proliferative signaling cascade that has emerged to play a pivotal role in mediating the epithelial cell proliferation and cytogenesis in animal models and humans with PKD, including the PCK rat, is the Raf/MEK/ERK pathway (5, 21, 32, 39). Previous in vitro studies using non-PKD cell types demonstrated 20-HETE as a proliferative mediator through the Raf/MEK/ERK pathway (15, 17–19, 36), which was consistent with an interactive role between 20-HETE synthesis and the ERK1/2 pathway in our PCK rat model of ARPKD. We found less phosphorylation (activation) of ERK1/2 following chronic inhibition with HET-0016 in the PCK rats, and this was associated with the reduction in the number and size of renal cysts. Previous studies have shown that various potential cystic hormones can mediate the production and release of 20-HETE (17, 18), resulting in increased cell cycle progression as measured by [3H]thymidine incorporation in vascular smooth muscle cells (19) and renal proximal tubular epithelial cells (15) through a MAPK-dependent pathway (17, 19, 36). More recent studies by Guo et al. (7, 8) have shown significant reduction in the active rate of cell proliferation in human U251 glioma (7) and rat 9L gliosarcoma cells (8) in vitro following blockade of 20-HETE synthesis using HET-0016. In our laboratory, we have found that another important procystic hormonal system, EGF and its cognate receptor ErbB1 (EGFR), may also be involved in the regulation of the ERK1/2 pathway, since BPK mouse kidneys had decreased activity (phosphorylation) following chronic HET-0016 treatment compared with vehicle-treated BPK mice (26). Guo et al. (7) found that 20-HETE synthesis inhibition using HET-0016 could block the EGF-mediated cell proliferation of U251 glioma cells, documenting the potential interactions of 20-HETE and EGF signaling.
In addition to the reduced activation of the ERK1/2 pathway, we found that the chronically treated PCK rat kidneys with HET-0016 had significantly reduced levels of the second messenger cAMP. It is well known that an important second messenger in PKD is cAMP, and the regulation of cAMP production appears to be a critical regulator of Ras/MEK/ERK activation and cell proliferation depending on the genotype of the cell. In PKD-affected kidneys, cAMP stimulates epithelial cell proliferation whereas cAMP inhibits proliferation in normal human kidney cells (2, 9, 42). Although our data correlate the reduced cAMP levels with the decreased kidney mass and cystic index, it does not provide any direct insight into an interaction of 20-HETE production and/or function with the cAMP pathway. The decreased cAMP levels are likely attributed to the reduced number of proliferating epithelial cells and cysts, but further in vitro experiments will be needed to address a direct causative role by 20-HETE activity with cAMP. There is strong evidence showing a functional interaction with cAMP-dependent pathways with the ERK1/2 signaling cascade, and so 20-HETE may be produced similar to the mechanisms previously shown in vascular smooth muscle cells through phospholipase A2 and activation of proliferative pathways, which were blocked in the current study with HET-0016 (12).
In conclusion, this is the first study to investigate the potential role of 20-HETE as a mediator of renal cyst formation in an orthologous rodent model of ARPKD. In the PCK rat, we demonstrated that chronic blockade with a specific inhibitor to the CYP4A and CYP4F enzyme family prevented the formation of 20-HETE, resulting in a significant decrease in renal cyst formation. These new findings provide strong evidence that eicosanoids, specifically 20-HETE, play a role in the epithelial cell proliferation and cyst formation in ARPKD, and inhibitors to 20-HETE synthesis and/or function could prove to be a future therapeutic target for ARPKD and possibly, the more prevalent genetic form of PKD, ADPKD.
GRANTS
This work was funded through grants from the Polycystic Kidney Disease (PKD) Foundation (F. Park), Advancing a Wealthier Wisconsin (F. Park), National Institutes of Health Grants HL-36279 (R. J. Roman) and P50-DK-057306 (E. D. Avner), and GM-31278 (J. R. Falck), and the Robert A. Welch Foundation (J. R. Falck).
Acknowledgments
The authors thank Averia Steinman and Lisa Henderson for help with the cytochrome P-450 assay, Jenifer Goepfert for performing the blood analysis, and Dr. Noriyuki Miyata (Taisho Pharmaceuticals, Tokyo, Japan) for generously providing the HET-0016 for this study.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Avner ED, Sweeney WE Jr. Renal cystic disease: new insights for the clinician. Pediatr Clin North Am 53: 889–909, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM Jr, Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66: 964–973, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Chang MY, Parker E, Ibrahim S, Shortland JR, Nahas ME, Haylor JL, Ong AC. Haploinsufficiency of Pkd2 is associated with increased tubular cell proliferation and interstitial fibrosis in two murine Pkd2 models. Nephrol Dial Transplant 21: 2078–2084, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Chen P, Guo M, Wygle D, Edwards PA, Falck JR, Roman RJ, Scicli AG. Inhibitors of cytochrome P450 4A suppress angiogenic responses. Am J Pathol 166: 615–624, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cowley BD Jr, Chadwick LJ, Grantham JJ, Calvet JP. Elevated proto-oncogene expression in polycystic kidneys of the C57BL/6J (cpk) mouse. J Am Soc Nephrol 1: 1048–1053, 1991 [DOI] [PubMed] [Google Scholar]
- 6.Dell KM, Avner ED. Autosomal recessive polycystic kidney disease. In: GeneClinics: Clinical Genetic Information Resource (Online). University of Washington, Seattle, WA. http://www.geneclinics.org July 2001[August 2008].
- 7.Guo M, Roman RJ, Falck JR, Edwards PA, Scicli AG. Human U251 glioma cell proliferation is suppressed by HET0016 [N-hydroxy-N′-(4-butyl-2-methylphenyl)formamidine], a selective inhibitor of CYP4A. J Pharmacol Exp Ther 315: 526–533, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Guo M, Roman RJ, Fenstermacher JD, Brown SL, Falck JR, Arbab AS, Edwards PA, Scicli AG. 9L gliosarcoma cell proliferation and tumor growth in rats are suppressed by N-hydroxy-N′-(4-butyl-2-methylphenol) formamidine (HET0016), a selective inhibitor of CYP4A. J Pharmacol Exp Ther 317: 97–108, 2006 [DOI] [PubMed] [Google Scholar]
- 9.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol 11: 1179–1187, 2000 [DOI] [PubMed] [Google Scholar]
- 10.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol 289: F496–F503, 2005 [DOI] [PubMed] [Google Scholar]
- 11.Ito O, Omata K, Ito S, Hoagland KM, Roman RJ. Effects of converting enzyme inhibitors on renal P-450 metabolism of arachidonic acid. Am J Physiol Regul Integr Comp Physiol 280: R822–R830, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Kalyankrishna S, Malik KU. Norepinephrine-induced stimulation of p38 mitogen-activated protein kinase is mediated by arachidonic acid metabolites generated by activation of cytosolic phospholipase A2 in vascular smooth muscle cells. J Pharmacol Exp Ther 304: 761–772, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Lager DJ, Qian Q, Bengal RJ, Ishibashi M, Torres VE. The pck rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int 59: 126–136, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Lanoix J, D'Agati V, Szabolcs M, Trudel M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 13: 1153–1160, 1996 [PubMed] [Google Scholar]
- 15.Lin F, Rios A, Falck JR, Belosludtsev Y, Schwartzman ML. 20-Hydroxyeicosatetraenoic acid is formed in response to EGF and is a mitogen in rat proximal tubule. Am J Physiol Renal Fluid Electrolyte Physiol 269: F806–F816, 1995 [DOI] [PubMed] [Google Scholar]
- 16.MacRae Dell K, Nemo R, Sweeney WE Jr, Avner ED. EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int 65: 2018–2029, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Muthalif MM, Benter IF, Karzoun N, Fatima S, Harper J, Uddin MR, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci USA 95: 12701–12706, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muthalif MM, Benter IF, Uddin MR, Malik KU. Calcium/calmodulin-dependent protein kinase IIalpha mediates activation of mitogen-activated protein kinase and cytosolic phospholipase A2 in norepinephrine-induced arachidonic acid release in rabbit aortic smooth muscle cells. J Biol Chem 271: 30149–30157, 1996 [DOI] [PubMed] [Google Scholar]
- 19.Muthalif MM, Karzoun NA, Gaber L, Khandekar Z, Benter IF, Saeed AE, Parmentier JH, Estes A, Malik KU. Angiotensin II-induced hypertension: contribution of Ras GTPase/mitogen-activated protein kinase and cytochrome P450 metabolites. Hypertension 36: 604–609, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG. Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol 5: 1462–1468, 1995 [DOI] [PubMed] [Google Scholar]
- 21.Nagao S, Yamaguchi T, Kusaka M, Maser RL, Takahashi H, Cowley BD, Grantham JJ. Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int 63: 427–437, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Nakanishi K, Sweeney WE Jr, Macrae Dell K, Cotton CU, Avner ED. Role of CFTR in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 12: 719–725, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Nauta J, Ozawa Y, Sweeney WE Jr, Rutledge JC, Avner ED. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr Nephrol 7: 163–172, 1993 [DOI] [PubMed] [Google Scholar]
- 24.Nauta J, Sweeney WE, Rutledge JC, Avner ED. Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr Res 37: 755–763, 1995 [DOI] [PubMed] [Google Scholar]
- 26.Park F, Sweeney WE, Jia G, Roman RJ, Avner ED. 20-HETE mediates proliferation of renal epithelial cells in polycystic kidney disease. J Am Soc Nephrol 19: 1929–1939, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J Cereb Blood Flow Metab 26: 1551–1561, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Richards WG, Sweeney WE, Yoder BK, Wilkinson JE, Woychik RP, Avner ED. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J Clin Invest 101: 935–939, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Sorenson CM, Sheibani N. Sustained activation of MAPK/ERKs signaling pathway in cystic kidneys from bcl-2 −/− mice. Am J Physiol Renal Physiol 283: F1085–F1090, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int 57: 33–40, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Sweeney WE Jr, Avner ED. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res 326: 671–685, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Sweeney WE Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int 64: 1310–1319, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Takahashi T, Kawahara Y, Okuda M, Ueno H, Takeshita A, Yokoyama M. Angiotensin II stimulates mitogen-activated protein kinases and protein synthesis by a Ras-independent pathway in vascular smooth muscle cells. J Biol Chem 272: 16018–16022, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Wang MH, Brand-Schieber E, Zand BA, Nguyen X, Falck JR, Balu N, Schwartzman ML. Cytochrome P450-derived arachidonic acid metabolism in the rat kidney: characterization of selective inhibitors. J Pharmacol Exp Ther 284: 966–973, 1998 [PubMed] [Google Scholar]
- 39.Wang X, Gattone V, 2nd Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 16: 846–851, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30: 259–269, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Williams JM, Sharma M, Anjaiahh S, Falck JR, Roman RJ. Role of endogenous CYP450 metabolites of arachidonic acid in maintaining the glomerular protein permeability barrier. Am J Physiol Renal Physiol 293: F501–F505, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 63: 1983–1994, 2003 [DOI] [PubMed] [Google Scholar]