

Abstract
Fibrin impairs surfactant function in vitro, and inhibition of fibrinolysis by plasminogen activator inhibitor (PAI-1) is thought to promote fibrin accumulation in acute lung injury (ALI). This has led to speculation that impaired PAI-1 and fibrin accumulation should protect lung function in ALI. We tested this hypothesis by investigating ALI severity in fibrinogen-deficient (Fgn−/−) and PAI-1-deficient (PAI-1−/−) mice. PAI-1−/−, C57BL/6, Fgn−/−, and Fgn+/− females were anesthetized and allowed to aspirate 4 μl/g of hydrochloric acid (pH 1.0) and then reanesthetized and connected to a ventilator 48 h later. Naive C57BL/6 and Fgn+/− females served as controls. Following deep inflation (DI), forced oscillations were delivered periodically over 8 min to measure changes in elastance (H) as a surrogate of lung derecruitment, at positive end-expiratory pressures (PEEP) of 6, 3, and 1 cmH2O. Increases in H following DI in acid-injured mice were greater than naive strain-matched controls. Increases in H were no different between injured PAI-1−/− and C57BL/6, or between injured Fgn−/− and +/− mice, at any PEEP. Pressure-volume curves were no different between injured groups. Total lung fibrin was lower in injured PAI-1−/− and Fgn−/− mice relative to injured C57BL/6 and Fgn+/− mice, respectively, but indices of permeability were no different between strains. Unexpectedly, neither fibrin nor PAI-1 deficiency protects lung mechanical function in mice with acid-induced ALI. We speculate that in vivo lung function may be more closely tied to permeability and alveolar protein in general, rather than being linked specifically to fibrin.
Keywords: lung mechanics, respiratory impedance, acid aspiration, coagulation
acute lung injury (ALI) is a severe form of noncardiogenic pulmonary edema and hypoxemic respiratory failure stemming from numerous causes (60). Current treatment of ALI rests largely on supportive care with mechanical ventilation, and its prognosis remains poor with a mortality of 30–40% in the general population, and higher in the elderly (49). The pathology of ALI typically progresses through an initial exudative phase characterized by neutrophil infiltration, edema, and accumulation of hyaline membranes, the latter consisting primarily of necrotic debris and fibrin (58). In this regard, the coagulation pathway and its end product fibrin have excited particular interest, in part due to the ability of fibrin to inhibit surfactant function in vitro (52, 54), and the increasingly recognized interplay between coagulation and innate immunity (17, 63). Fibrin formation and clearance in the lung are governed by the relative quantity and activity of fibrinolysis promoters such as plasminogen activators and fibrinolysis inhibitors such as plasminogen activator inhibitor-1 (PAI-1) (28). The importance of PAI-1 in ALI pathogenesis is suggested by its upregulation in various ALI models (5, 7) and by the finding that PAI-1-deficient mice fail to accumulate alveolar fibrin and die less quickly in response to injury (8). The importance of PAI-1 in ALI is further underscored by the finding that elevated plasma and edema fluid levels of PAI-1 are associated with higher mortality in ALI patients (45, 61). In agreement with the widely speculated role for PAI-1 and fibrin in the impairment of lung function in ALI, we demonstrated that progressive derangement in lung mechanics over 48 h in mice with acid aspiration injury corresponds with an increase in air space PAI-1 and fibrin (5). Although implied by our findings, the direct roles of fibrin accumulation and its clearance, as governed by PAI-1, in the derangement of in vivo lung mechanical function has yet to be firmly established. We thus set out to ascertain the direct roles of PAI-1 and air space fibrin in the disruption of lung mechanical function in an acid-aspiration model of ALI among varying strains of mice with different capacities to generate fibrin or PAI-1. Among these different strains, and their respective controls, we examined the effects of acid aspiration on lung mechanical derangement, various markers of injury, and on the accumulation of fibrin and fibrin breakdown products within the lung.
METHODS
Animals and injury protocol.
Female, 8- to 10-wk-old PAI-1-deficient (PAI-1−/−) mice (B6.129S2-Serpine1tm1Mlg; 18.9 ± 0.4 g; n = 7) were obtained from Jackson Labs (Bar Harbor, ME) and then bred and housed in a pathogen-free facility at the University of Vermont. Weight-matched and background-matched C57BL/6 controls (19.1 ± 0.4 g; n = 8) were purchased from Jackson Labs and housed in the same facility. Female, 10- to 12-wk-old fibrinogen-deficient (Fgn−/−) mice (24.3 ± 0.6 g; n = 9) and age-/weight-matched heterozygous (Fgn+/−) littermate controls (23.4 ± 1.1 g; n = 9) were bred and genotyped at the University of Vermont. The Fgn−/− and Fgn+/− mice were generously supplied by Jay Degen (Children's Hospital Medical Center, Cincinnati, OH) through the Trudeau Institute Breeding Facility in Saranac Lake, NY. Fgn−/− mice have a targeted deletion within the gene coding for the Aα-chain of fibrinogen and thus have no circulating fibrinogen (56). The heterozygous mice (Fgn+/−) have a plasma fibrinogen level that is 70% that of the wild type (26). For the induction of lung injury, mice were anesthetized with 3% inhaled isoflurane, positioned vertically upright, their tongues retracted, and their deep oropharynx slowly instilled with hydrochloric acid (HCl) (pH 1.0, 4 μl/g) using a plastic pipette tip. The tongue was held retracted until the mice fully aspirated the fluid. Then the mice were briefly rotated to the left and right lateral decubitus position, allowed to recover from anesthesia, and monitored periodically thereafter. Lung mechanics were measured 48 h following acid aspiration in each strain of mouse with ALI. At this point, the mice were anesthetized with intraperitoneal pentobarbital (90 mg/kg), underwent surgical tracheal intubation with an 18-gauge metal cannula, and were ventilated in a quasi-sinusoidal fashion with tidal volumes of 10 ml/kg at 180 breaths/min on a flexiVent (SCIREQ, Montreal, Canada) small animal ventilator. Weight-matched female C57BL/6 mice (n = 7) were used as naive controls for the injured C57BL/6 and PAI-1−/− mice, and weight-matched female Fgn+/− mice (n = 7) were used as naive controls for the injured Fgn−/− and Fgn+/− mice. The mice were paralyzed with an intraperitoneal injection of pancuronium bromide (0.5 ml/kg) and allowed 5 min for full drug effect while ventilating at a positive end-expiratory pressure (PEEP) of 3 cmH2O. The entire protocol was reviewed and approved by the Institutional Animal Care and Use Committee at the University of Vermont.
Experimental protocol.
Following a 5-min stabilization period, the level of PEEP was set at 6 cmH2O, and a 1.0-ml deep inflation (DI) was delivered twice over 4 s at constant flow and a pressure limit of 25 cmH2O. The mice were then returned to quasi-sinusoidal ventilation at 180 breaths/min. Respiratory system input impedance (Zrs) was measured via a forced-oscillation technique (described later) immediately following the two DIs and then subsequently every 20 s for 8 min. The post-DI measurement period was then repeated at a PEEP of 3 and 1 cmH2O. At the end of each post-DI measurement period, a quasistatic pressure-volume curve was obtained from resting residual capacity by dropping PEEP to 0 cmH2O and then immediately delivering seven steps of inspiratory volume to a total volume of 40 ml/kg, followed by seven equal expiratory steps, pausing at each step for 1 s. Plateau cylinder pressure was measured during each pause and plotted against piston displacement (corrected for gas compression and slow leak).
Specimen collection.
At the end of the timed ventilator protocol, the mice were euthanized with pentobarbital, followed by thoracotomy. Immediately thereafter, bronchoalveolar lavage fluid (BALF) was obtained by instilling 1 ml of PBS into the lungs via the tracheal cannula and slowly suctioning back for a return of ∼0.8 ml. The left atrium was then cut, and 10 ml of PBS with heparin (5 U/ml) was slowly perfused through the right ventricular outflow tract to blanch the lungs of intravascular blood and prevent further coagulation. Blanched lungs were then surgically removed, and the left lung was tied off with suture, dissected away, flash frozen in liquid nitrogen, and stored at −80°C. The right lung was instilled with 10% buffered formalin to a pressure of 30 cmH2O, fixed with 70% ethanol, and later embedded in paraffin, cut, and mounted for staining.
BALF analysis.
Immediately following collection, BALF was centrifuged, and the supernatant was stored at −80°C. The cell pellet was resuspended, and the total cell count was determined by an Advia 120 hematology analyzer (Bayer, Tarrytown, NY). Cytospun slides were stained with hematoxylin and eosin for differential count determination. Protein content was calculated using a colorimetric assay (Bio-Rad Laboratories, Hercules, CA) standardized to graded concentrations of BSA. ELISA kits for D-dimer (Diagnostica Stago, France) and murine total PAI-1 (Molecular Innovations, Southfield, MI) were used according to the manufacturer's protocol for BALF measurements.
Permeability index.
Immediately before anesthesia, mice received a tail vein injection with a 2.5 mg/ml solution of FITC-labeled dextran (4,000 Da; Sigma Chemical, St. Louis, MO) at a dose of 25 mg/kg, according to a protocol previously described (5, 24). Fifty microliters of BALF was later loaded on a microwell plate, excited at 485 nm, read at 528 nm, and the concentration determined from a linear dilution standard curve. This concentration was used as an index of combined endothelial and epithelial permeability.
Tissue fibrin immunohistochemical staining.
Formalin-fixed, cut, and mounted slides from the right lung were deparaffinized with xylene and graded ethanol series, and rinsed with water. Antigen was unmasked with sodium citrate, and slides were treated with an M.O.M. immunodetection kit (Vector Labs, Burlingame, CA) to block unwanted background staining. Fibrin and fibrinogen were stained using a mouse anti-fibrin(ogen) β-chain antibody (Accurate Chemical & Scientific, Westbury, NY), followed by a biotinylated anti-mouse antibody, a Vectastain avidin-based alkaline phosphatase solution, and a Vector Red Alkaline Phosphatase substrate. Following fibrin(ogen) staining, hematoxylin was used for background staining. The most injured appearing section out of four different specimens was chosen for imaging to represent each of the injured strains. Images were obtained using a Zeiss AxioCam HR digital camera (Oberkochen, Germany) adapted to an Olympus (Center Valley, PA) BH-2 microscope. Images were processed using Zeiss AxioVision software (version 3.1). All images were obtained using the same exposure and color setting and optimized using an automated brightness and contrast adjustment before being converted to tagged information file format (TIFF) and then combined into a final figure using Adobe Photoshop CS3, version 10.0.
Fibrin Western blot.
Frozen left lung tissue specimens were individually crushed, weighed, and suspended in an extraction buffer composed of sodium phosphate (10 mM), EDTA (5 mM), ε-aminocaproic acid (100 mM), aprotinin (10 U/ml), heparin (10 U/ml), and PMSF (2 mM). Then each sample was homogenized at 2,600 rpm (Polytron, Kinematica) for 1 min and spun at 10,000 g for 10 min at 4°C, after which the supernatant was removed and the pellet resuspended in sodium phosphate (10 mM) and EDTA (5 mM). Samples were spun again at 10,000 g for 10 min, the supernatant was removed, and the pellet was resuspended in 3 M urea, spun at 14,000 g, and the supernatant was again discarded. The pellet was then resuspended in SDS buffer at a final concentration of 6 mg of tissue (original weight) per milliliter of SDS and then incubated at 65°C for 1.5 h before adding β-mercaptoethanol and loading into the gel. A fibrin standard was generated by mixing 5 mg of murine fibrinogen (Sigma Chemical, St. Louis, MO) with 5 units of bovine thrombin (Sigma Chemical), incubated at 37°C for 10 min, solubilized in SDS buffer and β-ME, and diluted to a concentration such that the greatest total amount of fibrin loaded into the well was 2,000 ng. Following gel electrophoresis, each gel was first blocked with 1% BSA on TBS/Tween (TBST) and incubated overnight at 4°C with a 1:500 dilution of monoclonal mouse anti-β-chain fibrin antibody (MAb350; American Diagnostica, Stamford, CT), which specifically binds to a heptapeptide sequence on the β-chain of fibrin after cleavage of fibrinopeptide B by thrombin, thus binding specifically to previously polymerized fibrin monomers and not to fibrinogen (55). The gel was then incubated for 1 h with a horseradish peroxidase-conjugated sheep ECL anti-mouse IgG antibody (GE Healthcare, UK) for 1 h, washed five times in TBST, and exposed with Super Signal West Pico chemiluminescent substrate (Pierce, Rockford, IL) for 5 min before chemiluminescence measurements. Nonsaturated chemiluminescence measurements were converted to nanograms using a standard curve generated from the same gel as their respective samples and then normalized to total tissue protein.
Impedance data analysis.
Respiratory impedance, Zrs, was determined by measuring piston volume displacement and cylinder pressure while delivering 2-s oscillatory volume perturbations to the airway opening in a method described previously (3). These perturbations were composed of 13 superimposed sine waves with frequencies ranging from 1.0 to 20.5 Hz, all mutually prime to reduce harmonic distortion that can occur in nonlinear systems (25). Initial dynamic calibration signals were obtained to correct for the physical characteristics of the ventilator and tubing in subsequent measurements of Zrs (27, 51). Zrs itself was determined via Fourier transform from the signals of ventilator piston volume and cylinder pressure as described previously (19, 27). Zrs was interpreted by being fit with the model
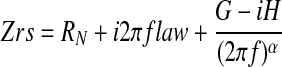 |
(1) |
where
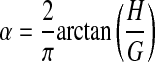 |
(2) |
and f is frequency, and i is the square root of −1. The parameters RN and Iaw largely characterize the resistive and inertive properties, respectively, of the airways, whereas G and H characterize the dissipative and elastic properties of the lung tissues (25). In particular, the parameter H is equal to respiratory elastance at an oscillation frequency of 1/2π Hz. Hysteresivity (η) is the quotient G/H. Increases in η are believed to reflect changes in intrinsic tissue properties and/or increased regional heterogeneity in lung function (36, 37). The relative contribution of Iaw to Zrs in a mouse has been determined to be negligible (19).
Lung mechanics measurements were excluded from the analysis in one PAI-1−/− and one Fgn+/− mouse due to measurement error or poor model fit. All graphing and statistical analyses were performed using Origin software (version 7.5, Northampton, MA). ANOVA was used to compare values among all groups, followed by post hoc Bonferonni tests for mean comparison between groups.
RESULTS
Lung mechanical function.
One PAI-1−/− mouse and one Fgn+/− mouse died before the end of the protocol, and data from these mice were excluded. The total increase in H over time following DI, a surrogate of lung derecruitment (6), was significantly greater in acid-injured PAI-1−/− and C57BL/6 mice (Fig. 1A) compared with their naive, weight-matched C57BL/6 controls (ANOVA, P < 0.05). Likewise, the total increase in H following DI was significantly greater in acid-injured Fgn−/− and Fgn+/− mice (Fig. 1D) compared with their naive Fgn+/− controls (ANOVA, P < 0.05). The increases in H following DI in the PAI-1−/− and C57BL/6 acid-injured mice nearly overlap one another (Fig. 1A), and the total increases in H over time (difference from DI to end) were not significantly different. Likewise, the total increases in H following DI in the Fgn−/− and Fgn+/− were also not significantly different, and curves nearly overlap one another (Fig. 1D). The increased rises in H over time in the injured animals relative to naive controls were not accompanied by any significant differences in hysteresivity (η) between groups, (Fig. 1, B and E), signifying active temporal derecruitment of lung volume from air space instability as opposed to any changes in heterogeneity or intrinsic tissue properties (6, 9).
Fig. 1.

Mean values (±SE) for elastance (H) and hysteresivity (η) are plotted against time at positive end-expiratory pressure (PEEP) levels of 6, 3, and 1 cmH2O in A and B for naive C57BL/6 (white circles), injured C57BL6 (gray circles), and injured PAI-1−/− mice (black circles). Mean values (±SE) for H and η are plotted in D and E for naive Fgn+/− (white circles), injured Fgn+/− (gray circles), and injured Fgn−/− mice (black circles). Averaged pressure-volume curves (±SE) obtained following PEEP 1 cmH2O are plotted in C for C57BL/6 and PAI-1−/− mice and in F for Fgn+/− and Fgn−/− mice. *Significant change in the post-DI (deep inflation) rise in H relative to naive controls (P < 0.05). #Significant increase in pressure-volume area (product) relative to naive controls (P < 0.05). Differences in η between groups were nonsignificant.
Pressure-volume (PV) curves of PAI-1−/− and C57BL/6 acid-injured mice demonstrated greater hysteresis and rightward shift of the inflation limb compared with those from naive C57BL/6 controls (Fig. 1C). PV curves of PAI-1−/− and C57BL/6 acid-injured mice overlapped considerably with no significant difference in total integrated area at all levels of PEEP. Likewise, PV curves of Fgn−/− and Fgn+/− acid-injured mice demonstrated greater hysteresis and rightward shift of the inflation limb compared with those from naive Fgn+/− controls (Fig. 1E), and PV curves of Fgn−/− and Fgn+/− acid-injured mice overlapped considerably with no significant difference in total integrated area at all levels of PEEP.
Inflammatory markers.
BALF total cell and neutrophil counts (Fig. 2) were significantly greater in the injured PAI-1−/− (11.98 ± 3.36 × 105 and 8.80 ± 2.81 × 105 cells/ml) and C57BL/6 mice (5.50 ± 1.37 and 3.07 ± 0.71 × 105 cells/ml) compared with naive C57BL/6 controls (9.71 ± 1.67 and 0.26 ± 0.19 × 104 cells/ml) (ANOVA, P < 0.05). Total cell counts were higher (ANOVA, P < 0.05) and neutrophil counts trended higher (P = 0.06) in injured PAI-1−/− mice compared with injured C57BL/6 mice. Total cell and neutrophil counts were significantly greater in the injured Fgn−/− mice (2.23 ± 0.38 × 105 and 0.54 ± 0.12 × 105 cells/ml) and Fgn+/− mice (2.36 ± 0.19 and 0.84 ± 0.07 × 105 cells/ml) compared with naive Fgn+/− controls (6.94 ± 1.17 and 0.056 ± 0.037 × 104 cells/ml) (ANOVA, P < 0.05) but were no different between injured Fgn−/− and Fgn+/− mice.
Fig. 2.

Mean bronchoalveolar lavage (BAL) fluid cell counts (±SE) are plotted for naive and acid-injured (ALI) strains. Total cell counts (black bars), macrophage counts (gray bars), and neutrophil counts (white bars) are significantly greater in injured C57BL/6 and PAI-1−/− mice relative to naive C57BL/6 controls (*P < 0.05). Total cell, macrophage, and neutrophil counts are significantly greater in injured Fgn+/− and Fgn−/− mice relative to naive Fgn+/− controls (#P < 0.05). Total cell counts were higher (^P < 0.05) and neutrophil counts trended higher (^^P = 0.06) in injured PAI-1−/− mice compared with injured C57BL/6 mice.
BALF protein levels (Fig. 3) were higher in injured PAI-1−/− (1,017 ± 201 μg/ml) and C57BL/6 mice (948 ± 208 μg/ml) compared with naive C57BL/6 mice (139 ± 11 μg/ml) and were likewise higher in the injured Fgn−/− (1,568 ± 276 μg/ml) and Fgn+/− (1,449 ± 127 μg/ml) mice compared with naive Fgn+/− controls (223 ± 19 μg/ml) (ANOVA, P < 0.05). As an index of permeability, BALF levels of FITC-dextran (Fig. 3) were higher in injured PAI-1−/− (2,826 ± 941 ng/ml) and C57BL/6 mice (2,021 ± 972 ng/ml) compared with naive C57BL/6 mice (216 ± 86 ng/ml) and were likewise higher in the injured Fgn−/− (2,013 ± 427 ng/ml) and Fgn+/− (2,005 ± 214 ng/ml) mice compared with naive Fgn+/− mice (682 ± 104 ng/ml) (ANOVA, P < 0.05). BALF protein and FITC-dextran levels were not statistically different between injured PAI-1−/− and C57BL/6 mice or between injured Fgn−/− and Fgn+/− mice.
Fig. 3.

Mean BAL fluid (BALF) protein (left axis) levels (±SE) are plotted as gray bars, and mean BALF FITC-labeled dextran (right axis) levels (±SE) are plotted as hatched bars for naive and acid-injured (ALI) strains. BALF protein and FITC-dextran levels were significantly elevated in injured C57BL/6 and PAI-1−/− mice relative to naive C57BL/6 controls (*P < 0.05) and significantly elevated in injured Fgn+/− and Fgn−/− mice relative to naive Fgn+/− controls (#P < 0.05).
PAI-1 and D-dimer.
BALF D-dimer levels (Fig. 4A) were higher in the injured PAI-1−/− (799.7 ± 388.8 ng/ml) and C57BL/6 mice (257.0 ± 89.3 ng/ml) compared with naive C57BL/6 controls (9.9 ± 1.7 ng/ml) and higher in the injured Fgn+/− mice (246.0 ± 68.3 ng/ml) compared with naive Fgn+/− mice (9.5 ± 1.4 ng/ml) (ANOVA, P < 0.05). D-dimer trended nonsignificantly higher in the injured PAI-1−/− compared with injured C57BL/6 mice (P = 0.14). D-dimer was not detectable in the BALF of Fgn−/− mice. BALF PAI-1 levels (Fig. 4B) were higher in the injured Fgn−/− (144.7 ± 31.1 ng/ml) and Fgn+/− (94.5 ± 14.1 ng/ml) mice compared with naive Fgn+/− mice (2.7 ± 0.2 ng/ml) and higher in the injured C57BL/6 mice (47.9 ± 15.9 ng/ml) compared with naive C57BL/6 controls (0.8 ± 0.2 ng/ml) (ANOVA, P < 0.05). PAI-1 was not detectable in the BALF of PAI-1−/− mice.
Fig. 4.

Mean BALF D-dimer levels (±SE) are plotted in A for naive and acid-injured strains. D-dimer levels were significantly elevated in injured C57BL/6 and PAI-1−/− mice relative to naive C57BL/6 controls (*P < 0.05) and significantly elevated in injured Fgn+/− relative to naive Fgn+/− controls (#P < 0.05). D-dimer was not detectable (ND) in the BALF of injured Fgn−/− mice. Mean BALF PAI-1 levels (±SE) are plotted in B for naive and acid-injured strains. PAI-1 levels were significantly elevated in injured C57BL/6 mice relative to naive C57BL/6 controls (*P < 0.05) and significantly elevated in injured Fgn+/− and Fgn−/− mice relative to naive Fgn+/− controls (#P < 0.05). PAI-1 was ND in the BALF of injured PAI-1−/− mice.
Lung fibrin staining and Western blot determination.
Histological lung specimens from acid-injured lungs of C57BL/6, PAI-1−/−, and Fgn+/− mice demonstrated patchy but large accumulations of fibrin(ogen) within the distal alveolar compartment (Fig. 5, A–C). In contrast, fibrin(ogen) was entirely absent in the immunostained lung specimens from acid-injured Fgn−/− mice (Fig. 5D). Fibrin is undetectable by Western blot in perfused lung specimens of naive uninjured mice (5). Mean whole lung fibrin levels (Fig. 6) were higher in injured C57BL/6 mice (49.7 ± 16.2 ng/mg tissue) compared with injured PAI-1−/− mice (7.6 ± 4.4 ng/mg tissue) (P < 0.05) and were higher in injured Fgn+/− mice (29.7 ± 15.8 ng/mg tissue) than in the injured Fgn−/− mice (not detectable) (P < 0.05).
Fig. 5.

Representative images of lung specimens following fibrin(ogen) immunostaining, marked with Vector Red, shown at ×100 and ×400 (inset) magnification, in A (injured C57BL/6), B (injured PAI-1−/−), C (injured Fgn+/−), and D (injured Fgn−/−). Background staining was done with hematoxylin. Primary antibody unstained slides (not shown) demonstrated negligible nonspecific staining with secondary antibody and Vector Red.
Fig. 6.

A: representative image of an immunoblot for the β-chain of fibrin in mouse fibrin standards (top) and tissue extractions (bottom), visualized at a level corresponding to a 57-kDa protein size. Bottom blot shows sample bands from 2 separate representative mice of each acid-injured mouse strain. B: mean values for total lung fibrin, normalized to lung tissue (ng/mg tissue), are plotted for injured C57BL/6, PAI-1−/−, Fgn+/−, and Fgn−/− groups (±SE). Fgn−/− and naive mice (not shown) have undetectable levels of lung fibrin. Lung fibrin levels are significantly lower in injured PAI-1−/− mice relative to injured C57BL/6 mice (*P < 0.05) and are significantly different between injured Fgn+/− and Fgn−/− mice (#P < 0.05).
DISCUSSION
Polymerized fibrin is a chief component of the alveolar hyaline membranes that are pathognomonic for the diffuse alveolar damage seen histologically in ALI (58, 60). Historically, this fibrin has been credited with possessing potent surfactant-inhibiting properties at the air-liquid interface (53, 54), thus being in part responsible for the increased lung stiffness that classically manifests in ALI (42). Contrary to popular theory and our own hypothesis, in an acid aspiration model of ALI yielding a significant disruption in lung mechanics, the diminution of distal air space and alveolar fibrin did not translate into improved lung mechanical function, either in response to DI recruitment and PEEP or in quasistatic lung compliance. This was the case both in mice possessing an augmented ability to breakdown fibrin (PAI-1−/− phenotype) and in mice lacking the ability to form fibrin (Fgn−/− phenotype). This was also found to be the case at 24 h following injury (4) (data not shown) in an earlier model of acid ALI in which acid injury was greater compared with the present study (5) but was ultimately discovered to be enhanced by the presence of transition metals, which we were careful to exclude in the present model. In the present study, the 48-h time point was ultimately fully explored since it is the experimental time point when air space fibrin and PAI-1 are known to be more significantly elevated in our model. This is the first study to our knowledge that demonstrates evidence challenging the widely espoused view of fibrin being dominant over other plasma proteins in disrupting air space surface tension in vivo. We should acknowledge that because of the near equivalence in mean rise in H between injured PAI-1−/− and Fgn−/− mice and their respective injured controls, the results are weakly powered to demonstrate a significant difference between the groups, thus leaving a chance for type II error of declaring no significant difference when in fact one exists. However, we assert that even if we were to study the projected 256 Fgn−/− and Fgn+/− mice needed for the 1.3 cmH2O·ml−1 difference in delta H at PEEP 1 cmH2O to reach statistical significance, such a small difference would be of negligible clinical relevance. Nevertheless, the conclusions of this study by no means discount the yet many potential roles of both alveolar fibrin and PAI-1 in ALI's pathogenesis, its fibroproliferative progression, or its recovery.
Through 1-to-1 inhibitory binding of urokinase-type plasminogen activator (uPA) and tissue-type PA (tPA), PAI-1 is a potent inhibitor of fibrinolysis (59), and elevated levels of PAI-1 are believed to contribute to disrupted fibrinolysis within the lungs of patients with ALI (10, 23, 29). Furthermore, air space PAI-1 levels are markedly increased in several models of lung injury, including hyperoxia (8), endotoxin (7), pneumonia (47), and acid aspiration (5). Certainly, other PA inhibitors such as PAI-2 and α2-antiplasmin can be elevated during injury and potentially compensate for the absence of PAI-1 (23, 33), particularly in mice constitutively deficient in PAI-1. However, earlier studies in PAI-1−/− mice have confirmed an increase in fibrinolysis through increased (less inhibited) activity of uPA and tPA (11, 59). This likely explains the increased levels of the fibrin degradation product, D-dimer (Fig. 4A), and the reduced levels of lung fibrin (Fig. 6) in the acid-injured PAI-1−/− mice relative to injured C57BL/6 mice in this study.
Investigators have demonstrated a protective role of PAI-1 deletion in other animal models of lung injury (8, 15). Results from a hyperoxia model of lung injury demonstrated improved survival and reduced alveolar fibrin deposits in PAI-1−/− mice compared with wild type (8). Other investigators have demonstrated a reduction in air space neutrophils following pharmacological interference with PAI-1 following endotoxin exposure (7). This reduction in neutrophil recruitment was also seen in PAI-1−/− mice in a model of Klebsiella pneumoniae, but ultimately led to decreased organism clearance and survival, implying a vital role of PAI-1 in host defenses to gram-negative infections (46). However, in a model of Streptococcal pneumoniae, investigators could demonstrate no significant difference in the host immune response or in acute mortality between PAI-1−/− mice and controls (47). Protection incurred by PAI-1 deletion thus appears to be dependent on the model of injury and the type of infection. Furthermore, lung mechanics may be affected differently by PAI-1 deletion in our model in the setting of an additional insult in which PAI-1 is known to be important, such as long-term injurious mechanical ventilation (12, 13).
Our finding of increased neutrophil counts in the injured PAI-1−/− mice relative to C57BL/6 controls is difficult to explain in lieu of other investigators having demonstrated just the opposite under PAI-1-deficient conditions (7, 46). At this point, we can only speculate whether this may be due to the known neutrophil chemoattractant capacity of D-dimer in vivo (20, 34) or simply the timing of our model. Nevertheless, the reduced levels of alveolar fibrin in acid-injured PAI-1−/− mice are compatible with the findings in hyperoxia injury (8), and the increased levels of D-dimer in acid-injured PAI-1−/− mice are compatible with findings in a K. pneumoniae model. With respect to later endpoints of ALI, Eitzman and colleagues (15) demonstrated a reduction in bleomycin-induced pulmonary fibrosis in PAI-1−/− mice at 2 wk compared with normal PAI-1-expressing mice and an increase in bleomycin-induced fibrosis in PAI-1-overexpressing mice. However, none of the previously cited studies specifically assessed lung mechanics to discern whether such decreases in inflammation and fibrin accumulation ever translated into improved lung function during the earlier exudative phase of injury.
With regard to fibrin, several studies have demonstrated the surfactant-inhibiting properties of fibrinogen and fibrin in vitro (52, 54), and other studies have suggested an improvement in lung function with the administration of fibrinolytic or anti-coagulant therapies (16, 22, 50). Although fibrin is known to interfere with surfactant function in vitro, other plasma proteins could sufficiently interfere with surfactant in the absence of fibrin (52) in vivo. Furthermore, aside from surfactant function, disrupted permeability and lung edema accumulation also contribute to the derangement of lung function in ALI (64). Although in vitro studies demonstrate a greater inhibitory function of polymerized fibrin compared with other plasma proteins (52, 54), these studies employ highly artificial conditions, and it is hard to determine the relative importance of the competing effects of different plasma proteins on surfactant in vivo, especially in the setting of what may be an overwhelming amount of alveolar edema and total protein. In other words, in a region where several factors are contributing to increased surface tension within the injured alveolus, it may be that total protein and fluid “trump” fibrin. However, it is also important to note that more recent evidence suggests that plasma proteins may have less effect on the surface tension lowering capacity of surfactant (35, 41) than originally believed. In fact cholesterols, elevated to the levels measured in the airways of subjects with ALI (14, 44), more prominently inhibit the surface tension lowering effects of surfactant in vitro than do serum proteins (21), a disparity that could also apply in vivo. With this in mind, it is curious to note that most studies reporting improved lung function with the use of fibrinolytic agents in ALI have focused predominantly on ventilation-perfusion matching and oxygenation (2, 16, 50, 57), with limited reporting on changes in lung mechanical function. The one study reporting a reduction in peak airway pressures following aerosolized tPA used a sheep model of burn and smoke inhalation injury (16), a model characterized by extensive obstruction of large and small airways with fibrinous debris (16). This smoke inhalation model differs significantly from our model of distal air space edema and fibrin and is a model we would expect to demonstrate a reduction in peak airway pressures with successful reductions in central airway occlusion.
Even if fibrin and its breakdown products play a negligible role in the acute derangement of surface tension and lung function, they could still have significant effects on other important ALI parameters such as vascular permeability (18, 48), inflammatory cell recruitment (20, 34), and remodeling/fibrosis (28, 38). Furthermore, we fully acknowledge that our results in no way directly challenge the widely touted role for tissue factor and thrombin activation in ALI pathogenesis and progression (17, 63). In fact, it may be that fibrin and PAI-1 are simply markers of thrombin activation and hence ALI severity, and the ancillary effects of thrombin on epithelial and endothelial barrier integrity (32) are what drive the generation of protein-rich edema and subsequent disruption of lung mechanical function. This alone could explain the prognostic role of elevated serum and edema fluid levels of PAI-1 in human ALI (45, 61), which itself is upregulated by thrombin (65). However, PAI-1 might also play a role in the downstream promotion of fibroproliferative remodeling (15, 59), a phenomenon known to promote unfavorable outcomes in ALI (39, 40). Thus, although not linked to changes in lung function (3), the increased air space cell counts in injured PAI-1−/− mice relative to C57BL/6 mice (Fig. 2) in the current study might portend a worsening ultimate outcome in our model. These many and varied potential effects of fibrin and PAI-1 may explain why so many studies of anti-coagulation agents in sepsis and ALI have yielded conflicting and often disappointing results (1, 30, 62) and why we still do not fully understand the role of fibrin in protecting the lung from vascular leak, hemorrhage, or ensuing infection (31, 43).
In conclusion, we have demonstrated in an acid-induced model of ALI with substantial fibrinous exudate formation that impaired distal air space fibrin accumulation does not help preserve lung mechanical function. Quasistatic lung compliance and dynamic changes in lung elastance following DI recruitment were similar between injured strains despite significant differences in total lung fibrin, a finding most conclusively demonstrated in Fgn+/− and Fgn−/− mice. This has led us to propose that although fibrin has been shown to have potent surfactant-inhibiting properties in vitro (52, 54), the influences in vivo from several other variables such as fluid, other plasma proteins, and perhaps even air space cholesterols (21), are likely to overwhelm any effect from fibrin alone, at least during the early phase of injury when alveolar edema and protein are elevated. Our findings thus present an important challenge to the popular notion that alveolar fibrin is critical to the in vivo disruption of alveolar surface tension and lung function in ALI.
GRANTS
This work was supported by National Institutes of Health Grants K08-HL-074107, R01-HL-75593, and COBRE P20RR15557.
Acknowledgments
Fibrinogen-deficient mice were generously supplied by Jay Degen (Children's Hospital Medical Center, Cincinnati, OH) through the Trudeau Institute Breeding Facility (Saranac Lake, NY).
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, Beale R, Svoboda P, Laterre PF, Simon S, Light B, Spapen H, Stone J, Seibert A, Peckelsen C, De Deyne C, Postier R, Pettila V, Artigas A, Percell SR, Shu V, Zwingelstein C, Tobias J, Poole L, Stolzenbach JC, Creasey AA. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA 290: 238–247, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Abubakar K, Schmidt B, Monkman S, Webber C, deSA D, Roberts R. Heparin improves gas exchange during experimental acute lung injury in newborn piglets. Am J Respir Crit Care Med 158: 1620–1625, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Allen G, Bates JH. Dynamic mechanical consequences of deep inflation in mice depend on type and degree of lung injury. J Appl Physiol 96: 293–300, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Allen GB, Cloutier ME, Larrabee YC, Smiley ST, Bates JHT. Neither fibrinogen nor PAI-1 deficiency confers protection of lung mechanical function in a mouse model of acute lung injury (Abstract). Am J Respir Crit Care Med 177: A62, 2008. [Google Scholar]
- 5.Allen GB, Leclair T, Cloutier M, Thompson-Figueroa J, Bates JH. The response to recruitment worsens with progression of lung injury and fibrin accumulation in a mouse model of acid aspiration. Am J Physiol Lung Cell Mol Physiol 292: L1580–L1589, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Allen GB, Pavone LA, DiRocco JD, Bates JHT, Nieman GF. Pulmonary impedance and alveolar instability during injurious ventilation in rats. J Appl Physiol 99: 723–730, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Arndt PG, Young SK, Poch KR, Nick JA, Falk S, Schrier RW, Worthen GS. Systemic inhibition of the angiotensin-converting enzyme limits lipopolysaccharide-induced lung neutrophil recruitment through both bradykinin and angiotensin II-regulated pathways. J Immunol 177: 7233–7241, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Barazzone C, Belin D, Piguet PF, Vassalli JD, Sappino AP. Plasminogen activator inhibitor-1 in acute hyperoxic mouse lung injury. J Clin Invest 98: 2666–2673, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bates JH, Allen GB. The estimation of lung mechanics parameters in the presence of pathology: a theoretical analysis. Ann Biomed Eng 34: 384–392, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Bertozzi P, Astedt B, Zenzius L, Lynch K, LeMaire F, Zapol W, Chapman HA Jr. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med 322: 890–897, 1990. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Stassen JM, Schoonjans L, Ream B, van den Oord JJ, De Mol M, Mulligan RC, Collen D. Plasminogen activator inhibitor-1 gene-deficient mice. II. Effects on hemostasis, thrombosis, and thrombolysis. J Clin Invest 92: 2756–2760, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahlem P, Bos AP, Haitsma JJ, Schultz MJ, Meijers JC, Lachmann B. Alveolar fibrinolytic capacity suppressed by injurious mechanical ventilation. Intensive Care Med 31: 724–732, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Dahlem P, Bos AP, Haitsma JJ, Schultz MJ, Wolthuis EK, Meijers JC, Lachmann B. Mechanical ventilation affects alveolar fibrinolysis in LPS-induced lung injury. Eur Respir J 28: 992–998, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Davidson KG, Bersten AD, Barr HA, Dowling KD, Nicholas TE, Doyle IR. Lung function, permeability, and surfactant composition in oleic acid-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 279: L1091–L1102, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 97: 232–237, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enkhbaatar P, Murakami K, Cox R, Westphal M, Morita N, Brantley K, Burke A, Hawkins H, Schmalstieg F, Traber L, Herndon D, Traber D. Aerosolized tissue plasminogen inhibitor improves pulmonary function in sheep with burn and smoke inhalation. Shock 22: 70–75, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Esmon CT Interactions between the innate immune and blood coagulation systems. Trends Immunol 25: 536–542, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge M, Ryan TJ, Lum H, Malik AB. Fibrinogen degradation product fragment D increases endothelial monolayer permeability. Am J Physiol Lung Cell Mol Physiol 261: L283–L289, 1991. [DOI] [PubMed] [Google Scholar]
- 19.Gomes RF, Shen X, Ramchandani R, Tepper RS, Bates JH. Comparative respiratory system mechanics in rodents. J Appl Physiol 89: 908–916, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Gross TJ, Leavell KJ, Peterson MW. CD11b/CD18 mediates the neutrophil chemotactic activity of fibrin degradation product D domain. Thromb Haemost 77: 894–900, 1997. [PubMed] [Google Scholar]
- 21.Gunasekara L, Schurch S, Schoel WM, Nag K, Leonenko Z, Haufs M, Amrein M. Pulmonary surfactant function is abolished by an elevated proportion of cholesterol. Biochim Biophys Acta 1737: 27–35, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Gunther A, Lubke N, Ermert M, Schermuly RT, Weissmann N, Breithecker A, Markart P, Ruppert C, Quanz K, Ermert L, Grimminger F, Seeger W. Prevention of bleomycin-induced lung fibrosis by aerosolization of heparin or urokinase in rabbits. Am J Respir Crit Care Med 168: 1358–1365, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Gunther A, Mosavi P, Heinemann S, Ruppert C, Muth H, Markart P, Grimminger F, Walmrath D, Temmesfeld-Wollbruck B, Seeger W. Alveolar fibrin formation caused by enhanced procoagulant and depressed fibrinolytic capacities in severe pneumonia. Comparison with the acute respiratory distress syndrome. Am J Respir Crit Care Med 161: 454–462, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Han X, Fink MP, Uchiyama T, Yang R, Delude RL. Increased iNOS activity is essential for pulmonary epithelial tight junction dysfunction in endotoxemic mice. Am J Physiol Lung Cell Mol Physiol 286: L259–L267, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol 72: 168–178, 1992. [DOI] [PubMed] [Google Scholar]
- 26.Hattori N, Degen JL, Sisson TH, Liu H, Moore BB, Pandrangi RG, Simon RH, Drew AF. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest 106: 1341–1350, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirai T, McKeown KA, Gomes RF, Bates JH. Effects of lung volume on lung and chest wall mechanics in rats. J Appl Physiol 86: 16–21, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Idell S Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med 31: S213–S220, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest 84: 695–705, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jian MY, Koizumi T, Tsushima K, Fujimoto K, Kubo K. Activated protein C attenuates acid-aspiration lung injury in rats. Pulm Pharmacol Ther 18: 291–296, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Johnson LL, Berggren KN, Szaba FM, Chen W, Smiley ST. Fibrin-mediated protection against infection-stimulated immunopathology. J Exp Med 197: 801–806, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawkitinarong K, Linz-McGillem L, Birukov KG, Garcia JG. Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am J Respir Cell Mol Biol 31: 517–527, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Laterre PF, Wittebole X, Dhainaut JF. Anticoagulant therapy in acute lung injury. Crit Care Med 31: S329–S336, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Leavell KJ, Peterson MW, Gross TJ. The role of fibrin degradation products in neutrophil recruitment to the lung. Am J Respir Cell Mol Biol 14: 53–60, 1996. [DOI] [PubMed] [Google Scholar]
- 35.Lee MM, Green FH, Schurch S, Cheng S, Bjarnason SG, Leonard S, Wallace W, Possmayer F, Vallyathan V. Comparison of inhibitory effects of oxygen radicals and calf serum protein on surfactant activity. Mol Cell Biochem 259: 15–22, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Lutchen KR, Greenstein JL, Suki B. How inhomogeneities and airway walls affect frequency dependence and separation of airway and tissue properties. J Appl Physiol 80: 1696–1707, 1996. [DOI] [PubMed] [Google Scholar]
- 37.Lutchen KR, Hantos Z, Petak F, Adamicza A, Suki B. Airway inhomogeneities contribute to apparent lung tissue mechanics during constriction. J Appl Physiol 80: 1841–1849, 1996. [DOI] [PubMed] [Google Scholar]
- 38.Marshall R, Bellingan G, Laurent G. The acute respiratory distress syndrome: fibrosis in the fast lane. Thorax 53: 815–817, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall RP, Bellingan G, Webb S, Puddicombe A, Goldsack N, McAnulty RJ, Laurent GJ. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med 162: 1783–1788, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Martin C, Papazian L, Payan MJ, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest 107: 196–200, 1995. [DOI] [PubMed] [Google Scholar]
- 41.Marzan Y, Mora R, Butler A, Butler M, Ingenito EP. Effects of simultaneous exposure of surfactant to serum proteins and free radicals. Exp Lung Res 28: 99–121, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Matamis D, Lemaire F, Harf A, Brun-Buisson C, Ansquer JC, Atlan G. Total respiratory pressure-volume curves in the adult respiratory distress syndrome. Chest 86: 58–66, 1984. [DOI] [PubMed] [Google Scholar]
- 43.Mullarky IK, Szaba FM, Berggren KN, Parent MA, Kummer LW, Chen W, Johnson LL, Smiley ST. Infection-stimulated fibrin deposition controls hemorrhage and limits hepatic bacterial growth during listeriosis. Infect Immun 73: 3888–3895, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panda AK, Nag K, Harbottle RR, Rodriguez-Capote K, Veldhuizen RA, Petersen NO, Possmayer F. Effect of acute lung injury on structure and function of pulmonary surfactant films. Am J Respir Cell Mol Biol 30: 641–650, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol 285: L20–L28, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Renckens R, Roelofs JJ, Bonta PI, Florquin S, de Vries CJ, Levi M, Carmeliet P, van't Veer C, van der Poll T. Plasminogen activator inhibitor type 1 is protective during severe Gram-negative pneumonia. Blood 109: 1593–1601, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Rijneveld AW, Florquin S, Bresser P, Levi M, De Waard V, Lijnen R, Van Der Zee JS, Speelman P, Carmeliet P, Van Der Poll T. Plasminogen activator inhibitor type-1 deficiency does not influence the outcome of murine pneumococcal pneumonia. Blood 102: 934–939, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Rowland FN, Donovan MJ, Picciano PT, Kreutzer DL. Fibrin-mediated vascular injury: demonstration of vascular endothelial cell retraction in response to soluble fibrin-associated factors. J Exp Pathol 1: 217–240, 1984. [PubMed] [Google Scholar]
- 49.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Schermuly RT, Gunther A, Weissmann N, Ghofrani HA, Seeger W, Grimminger F, Walmrath D. Differential impact of ultrasonically nebulized versus tracheal-instilled surfactant on ventilation-perfusion (VA/Q) mismatch in a model of acute lung injury. Am J Respir Crit Care Med 161: 152–159, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Schuessler TF, Bates JH. A computer-controlled research ventilator for small animals: design and evaluation. IEEE Trans Biomed Eng 42: 860–866, 1995. [DOI] [PubMed] [Google Scholar]
- 52.Seeger W, Grube C, Gunther A, Schmidt R. Surfactant inhibition by plasma proteins: differential sensitivity of various surfactant preparations. Eur Respir J 6: 971–977, 1993. [PubMed] [Google Scholar]
- 53.Seeger W, Gunther A, Thede C. Differential sensitivity to fibrinogen inhibition of SP-C- vs. SP-B-based surfactants. Am J Physiol Lung Cell Mol Physiol 262: L286–L291, 1992. [DOI] [PubMed] [Google Scholar]
- 54.Seeger W, Stohr G, Wolf HR, Neuhof H. Alteration of surfactant function due to protein leakage: special interaction with fibrin monomer. J Appl Physiol 58: 326–338, 1985. [DOI] [PubMed] [Google Scholar]
- 55.Smyth SS, Reis ED, Vaananen H, Zhang W, Coller BS. Variable protection of beta 3-integrin-deficient mice from thrombosis initiated by different mechanisms. Blood 98: 1055–1062, 2001. [DOI] [PubMed] [Google Scholar]
- 56.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 9: 2020–2033, 1995. [DOI] [PubMed] [Google Scholar]
- 57.Tasaki O, Mozingo DW, Dubick MA, Goodwin CW, Yantis LD, Pruitt BA Jr. Effects of heparin and lisofylline on pulmonary function after smoke inhalation injury in an ovine model. Crit Care Med 30: 637–643, 2002. [DOI] [PubMed] [Google Scholar]
- 58.Tomashefski JF Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med 21: 435–466, 2000. [DOI] [PubMed] [Google Scholar]
- 59.Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest 88: 1067–1072, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. [DOI] [PubMed] [Google Scholar]
- 61.Ware LB, Matthay MA, Parsons PE, Thompson BT, Januzzi JL, Eisner MD. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit Care Med 35: 1821–1828, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Penzes I, Kubler A, Knaub S, Keinecke HO, Heinrichs H, Schindel F, Juers M, Bone RC, Opal SM. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA 286: 1869–1878, 2001. [DOI] [PubMed] [Google Scholar]
- 63.Welty-Wolf KE, Carraway MS, Ortel TL, Piantadosi CA. Coagulation and inflammation in acute lung injury. Thromb Haemost 88: 17–25, 2002. [PubMed] [Google Scholar]
- 64.Wilson TA, Anafi RC, Hubmayr RD. Mechanics of edematous lungs. J Appl Physiol 90: 2088–2093, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Wojta J, Gallicchio M, Zoellner H, Hufnagl P, Last K, Filonzi EL, Binder BR, Hamilton JA, McGrath K. Thrombin stimulates expression of tissue-type plasminogen activator and plasminogen activator inhibitor type 1 in cultured human vascular smooth muscle cells. Thromb Haemost 70: 469–474, 1993. [PubMed] [Google Scholar]