

Abstract
Lysophosphatidic acid (LPA), a potent bioactive phospholipid, induces diverse cellular responses, including cell proliferation, migration, and cytokine release. LPA can be generated intracellular and extracellularly through multiple synthetic pathways by action of various enzymes, such as phospholipase A1/2 (PLA1/2), phospholipase D (PLD), acylglycerol kinase (AGK), and lysophospholipase D (lysoPLD). Metabolism of LPA is regulated by a family of lipid phosphate phosphatases (LPPs). Significant amounts of LPA have been detected in various biological fluids, including serum, saliva, and bronchoalveolar lavage fluid (BALF). The most significant effects of LPA appear to be through activation of the G-protein-coupled receptors (GPCRs), termed LPA1-6. LPA regulates gene expression through activation of several transcriptional factors, such as nuclear factor-κB (NF-κB), AP-1, and C/EBPβ. In addition to GPCRs, cross-talk between LPA receptors and receptor tyrosine kinases (RTKs) partly regulates LPA-induced intracellular signaling and cellular responses. Airway epithelial cells participate in innate immunity through the release of cytokines, chemokines, lipid mediators, other inflammatory mediators and an increase in barrier function in response to a variety of inhaled stimuli. Expression of LPA receptors have been demonstrated in airway epithelial cells. This review summarizes our recent observations of the role of LPA / LPA-Rs in regulation of airway epithelium, especially in relation to the secretion of pro- and anti-inflammatory mediators and regulation of airway barrier function.
Keywords: lysophosphatidic acid, airway epithelial cells, cytokine release, airway epithelium barrier function, G-protein-coupled receptors, RTKs, signal transduction, inflammation
1. Introduction
Phospholipids are major constituents of all biological membranes while lysophospholipids are present in relatively smaller proportion in tissues, biological fluids and cells. The prefix “Lyso” has been widely used by researchers in basic, translational and medical science to indicate loss or absence of a long chain fatty acid from the glycerol backbone of the glycerophospholipid. However, the prefix “Lyso” in the original context implied the ability of a phospholipid to induce lysis of cells, based on the observation that snake venom caused lysis of erythrocytes due to generation of a lipid from erythrocyte membranes, which was subsequently identified as lysophosphatidylcholine (LPC). Among the various lysoglycerophospholipids, lysophosphatidic acid (LPA) (1- or 2-radyl-sn-glycerol 3-phosphte) has the simple structure with either a long-chain saturated/monounsaturated fatty acid- or alkyl- or alk-1-enyl- moiety attached to sn-1 carbon or a polyunsaturated fatty acid group linked to sn-2, and a phosphate group at sn-3 position of the glycerol backbone. Similar to LPC, LPA is water soluble, present in nM to μM concentrations in plasma bound to either albumin or gelsolin, and plasma levels of LPA increase following activation of platelets and circulating monocytes/polymorphonuclear leukocytes [1-7]. In addition to its role as an intermediate in de novo biosynthesis of phospholipids in mammalian tissues/cells, LPA functions as a serum-derived growth factor, and also exhibits multiple pleiotropic effects as an inter- and intra-cellular lipid mediator of cellular functions such as proliferation [8-14], migration [11, 12, 15-18], and survival [19-21]. Many of these cellular effects of LPA are mediated via specific G protein-coupled LPA receptors [22-30], which are present on the cell surface, intra-cellular organelles and the nucleus. Additionally, the peroxosome proliferator-activated receptor-γ (PPARγ) has been identified as an intracellular receptor for LPA [31, 32]. LPA-Rs are coupled to multiple intracellular signaling pathways via heterotrimeric Gi, Gq, G12/13, and Gs regulating cell proliferation, migration and survival [22-29, 33-38]. While more than 60 reviews have dealt with the emerging role of LPA in proliferation, motility, and various diseases, there has been no mini- or comprehensive review that addresses the role of LPA in airway epithelium. Towes, M.L. et al. reviewed the effect of LPA on contraction, proliferation, and gene expression in airway smooth muscle cells in 2002 [39]. The present review focuses on LPA and its role in airway epithelial signaling, inflammatory responses, and remodeling with an emphasis on its pro- and anti-inflammatory effects in the airway.
2. Biosynthesis and catabolism of LPA
LPA is a natural constituent of all tissues, plasma [1-5, 7], saliva [40], bronchoalveolar lavage fluid (BALF) [41-43], follicular fluid [44], malignant effusions [45], and mildly oxidized LDL [46]. Plasma levels of LPA are low (< 100 nM). However, serum concentrations of LPA are much higher (> 1000 nM) and partly derived from activated platelets [1, 2, 7]. Furthermore, the fatty acid composition of LPA derived from plasma is different compared to serum LPA, which has more polyunsaturated fatty acids [1, 2, 7]. Plasma levels of LPA are normally low and regulated by production, degradation, and uptake by tissues and circulating cells. Mechanisms that regulate low LPA levels in plasma under normal conditions as well as enhanced LPA production during injury/pathophysiology states are not well understood, although plasma contains the necessary enzymes and substrates for LPA production. LPA in biological fluids could arise from at least two sources. First, LPA can be synthesized in the cells and then released, or LPA can be synthesized outside of cells. De novo synthesis of LPA is regulated by two key enzymes, glycerophosphate acyl transferase [47, 48] and acylglycerol kinase (AGK) [49, 50], which are predominantly localized in microsomes and mitochondria, respectively. Glycerophosphate acyl transferase catalyzes the transfer of long-chain fatty acid from fatty acyl CoA to glycerol-3-phosphate to biosynthesize LPA, while acylglycerol kinase phosphorylates monoacylglycerol to form LPA.
2.1. Intracellular generation of LPA
At least two pathways have been identified for intracellular LPA generation. In the first pathway, phosphatidic acid (PA) generated by phosphorylation of diacylglycerol (DAG) catalyzed by DAG kinase or agonist-stimulated phospholipase D (PLD) signal transduction is converted to LPA, a process mediated by phospholipase (PL) A1 or PLA2 type enzymes [7, 51-54]. While the specificity of PLA1 or PLA2 in using PA as a substrate in vivo is unclear, two membrane-bound PA-specific mPLA1 α and mPLA2 β, also called LIPH and LIPI belonging to the lipase gene family [54, 55] has been demonstrated, which may have specific role(s) in generating polyunsaturated 2-acyl-LPA. Interestingly, both mPLA1 α and mPLA2 β are located in lipid rafts indicating the possibility of LPA generation in the raft microdomains [54, 55]. However, the physiological implication of this distribution in lipid microdomain remains to be established. Although the role of the PLD/PA pathway in generation of LPA in airway cells has not been demonstrated, incubation of SK-OV-3 cells (ovarian cell line) with 1-butanol, which diverts PA formed by PLD to phosphatidylbutanol, caused a consistent reduction of 50 % of constitutively produced LPA and ~ 60 % of inducible LPA production by LPA treatment [51]. Further, Luquain et al. showed that overexpression of PLD2 (but not PLD1) resulted in LPA production by ovarian cancer cells in response to agonists stimulation [51]. The second pathway of intracellular generation of LPA is mediated by AGK, an enzyme that phosphorylates monoacylglycerol (MAG) to LPA [47-50] wherein MAG is derived either by the action of lipid phosphate phosphatases (LPPs) on LPA or lipase(s) on DAG [56-58]. In human bronchial epithelial cells (HBEpCs), overexpressed lentiviral V5-tagged AGK co-localized with MitoTracker Red [60], which was similar to mitochondrial localization of AGK in NIH 3T3 fibroblasts, HEK 293, and PC-3 cells [49, 50]. Furthermore, cell lysates of over-expressing V5-tagged AGK phosphorylated MAG and DAG, but not sphingosine or ceramide [60]; however, AGK expressed in bacteria phosphorylated MAG and DAG as well as sphingoid bases [49, 59]. AGK expression was up-regulated in prostate cancers compared to normal prostate tissues, and expression of AGK in PC-3 prostate cancer cells increased formation and secretion of LPA [50]. However, in HBEpCs, over-expression of AGK increased intracellular production of LPA, but the intracellularly generated LPA was not secreted [60], suggesting differences between the two cell types in the mechanism of action of intracellularly generated LPA signal transduction (Figure 1).
Figure 1. Biosynthesis and catabolism of LPA.

Activation of PLD generates PA, which is converted to LPA by the action of PLA1/PLA2. MAG is converted to LPA by AGK. PC in lipoproteins serves as a substrate of sPLA2 or LCAT, which converts PC to LPC. LPC serves as a substrate of lysoPLD for LPA generation. Levels of LPA are also regulated by LPPs and LPAAT.
2.2. Extracellular LPA generation
In this pathway, secretory PLA2 (sPLA2), phosphatidylserine (PS)-specific PLA1, and lysoPLD [also named as ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2) or autotaxin (ATX)] contribute to extracellular production of LPA. In activated platelets and other cell types, LPC, lysophsophatidylethanolamine (LPE), and lysophosphatidylserine (LPS) are generated by sPLA2 and PS-specific PLA1 in plasma [61, 62]. LPC is also formed from PC in lipoproteins by lecithin: cholesterol acyltransferase (LCAT) and PLA1 type enzymes [63, 64]. Lysophospholipids, thus generated, are acted upon by lysoPLD or autotaxin to LPA. LysoPLD activity, originally described in rat plasma [65], converts exogenously added LPC to LPA. Subsequent purification and characterization of lysoPLD from plasma showed that this was identical to autotaxin (ATX) [6]. ATX, in addition to generation of LPA from lysophospholipids, also produces cyclic phosphatidic acid (cPA), which contains a dioxaphospholane ring at sn-2 and sn-3 positions of the glycerol backbone [66-69]. cPA is also present in human serum [69-71] and its biological activities are distinct from that of LPA, exhibiting anti-proliferative and anti-metastatic properties in vivo and in vitro [66-68, 72]. In addition to hydrolysis of LPA, lysoPLD/ATX also possesses ecto-nucleotide pyrophosphatase/phosphodiesterase activity [73, 74]. Overexpression of lysoPLD mRNA in non-small cell lung cancer has been reported [75], however, the role of lysoPLD/autotaxin in airways and airway diseases is unknown. ATX deficiency is embryonically lethal and ATX (-/-) mice die at embryonic day 9.5 with profound vascular defects [76, 77]. Furthermore, sPLA2 activity was increased in BALF after inhaled antigen challenge in asthmatics [78, 79], and late-phase allergic reactions were characterized by increased phospholipids and lysophospholipids in BALF [80, 81].
2.3. LPA degradation
Three major pathways have been described for LPA degradation in mammalian systems. The first pathway involves dephosphorylation of LPA to MAG by phosphatases that belong to phosphatidate phosphatases type 2 (PAP-2), also known as lipid phosphate phosphohydrolase (LPP). There are three major isoforms of LPPs, LPP-1-3, that have been cloned and characterized in mammals [82-85]. While all the isoforms dephosphorylated, in vitro, a variety of lipid phosphates including LPA, PA, S1P, and ceramide-1-phosphate, LPA is a preferred substrate for LPP-1 compared to LPP-2 and LPP-3 [85]. Expression of LPP-1-3 was demonstrated in human bronchial epithelial cells (HBEpCs) by real time RT-PCR and Western blotting with LPP-specific antibodies [58]. Exogenous addition of [3H]oleoyl LPA was hydrolyzed to [3H]MAG, while over-expression of LPP-1 Wt enhanced LPA hydrolysis by ~2-3 fold compared to vector infected control cells [58]. As LPP-1 is an ecto-enzyme, one of its roles is to modulate LPA levels in milieu, which regulates LPA signaling and cell functions. The second pathway of LPA degradation involves conversion of LPA to PA catalyzed by a LPA acyltransferase (LPAAT) [86-90], which has not been well characterized in the airway. The third pathway responsible for LPA degradation involves hydrolysis of the long-chain fatty acyl group from LPA by the action of lysophospholipases. At least two distinct lysophospholipases with specificity towards LPC and LPA have been characterized [91-94]. The participation of lysophospholipases in LPA clearance from BALF and alveoli is unclear.
2.4. LPA levels in BALF
LPA is present in human BAL fluids as determined by tandem mass spectrometry (LC-MS/MS), and significantly increased following segmental allergen challenge [41]. Interestingly, analysis of the LPA by tandem MS/MS revealed that 20:4, 22:4, and 22:6 LPA molecular species were increased following segmental allergen challenge [41]. These increased accumulations of polyunsaturated LPA species, generated from complex phospholipids by activated phospholipases, are consistent with catabolic LPA production during lower airway inflammation. However, a role for lysoPLD/autotaxin in enhanced LPA production in the BALF after allergen challenge has not been established. In murine models of asthma, Schistosoma mansoni egg/soluble antigen or ovalbulmin challenge increased LPA levels in BALF by ~2-3 fold compared to control mice [43]. Recent studies by Tager et al. showed that LPA levels were increased in BALF in a murine model of bleomycin-induced pulmonary fibrosis, and in BALF from idiopathic pulmonary fibrosis (IPF) patients, compared to normal controls [42]. Furthermore, inhibition of LPA1 attenuated fibroblast chemotaxis induced by IPF BALF samples [42]. These studies from segmental allergen challenged patients [41] and IPF patients [42] show that elevated LPA levels in BALF may be a biomarker of airway inflammatory diseases. Further investigations on the mechanism(s) and cell type(s) involved in LPA production in normal and pathological conditions are necessary to understand the physiological role of LPA in airway inflammation.
3. LPA receptors
3.1. LPA receptors and G proteins
Discovery of LPA specific receptors in the plasma membrane of mammalian cells has led researchers to investigate LPA-Rs in LPA-induced intracellular signaling, and biological/physiological/pathological roles. Chun et al., for the first time, showed that LPA was a ligand for ventricular zone-1 (vzg-1) receptor, which is a member of the endothelial differentiation gene (EDG) family [23]. To date, six cell-surface LPA receptors, LPA1-6, have been cloned and described in mammals [22-29, 38, 95]. Based on sequence homology, three of these receptors belong to the EDG subfamily of the G-protein-coupled receptor (GPCR) superfamily. LPA1/EDG2, LPA2/EDG4, and LPA3/EDG7 share ~50% sequence homology [33-35, 95], while LPA4 /GPR23/P2Y9, and LPA5/GPR92, and LPA6/GPR87 are structurally distinct from the EDG family and share < 40% homology with conventional LPA1-3 [26-29]. The biological effects of LPA are mediated by ligation to specific LPA-Rs that are coupled to heterotrimeric G-protein families, the Gs, Gi, Gq, and G12/13. LPA1 and LPA2 are known to interact with Gi ,Gq, and G12/13; LPA3 interacts with Gi and Gq but not G12/13 [33, 36]; LPA4 appears to couple with all the G proteins [26, 37], and LPA5/GPR92 is likely coupled to Gs, G12/13, and Gq [27]. LPA signaling via LPA-Rs include: 1) the activation of phospholipase C (PLC) and calcium mobilization mediated by Gq [96-99]; 2) Gi-mediated inhibition of adenylate cyclase [100, 101], activation of the Ras-MAPK cascade [9, 15], and activation of PI3K-PKB/Akt signaling [10, 102], which promote cell motility and suppresses apoptosis; 3) G12/13-mediated activation of Rho/Rac GTPases signaling, which regulates cytoskeleton rearrangement [11, 12, 103]; and 4) G13-mediated membrane depolarization of chloride channels [5, 104]. LPA4 and LPA5 activate adenylate cyclase, resulting in the accumulation of cAMP and intracellular [Ca2+] [26, 27]. LPA also activates Src [105-107], PYK2 [105, 108], PKC [115-120] and PLD [115-120], transactivates growth factor receptors (EGF-R and PDGF-Rβ) [115, 120-129], induces COX-2 expression and PGE2 secretion [115, 127, 130-133], and regulates transcriptional factors such as C/EBPβ [115, 132], NF-κB [58, 114, 115, 132, 134, 135] and AP-1 [115, 132, 135-137].
3.2. LPA receptors in airway epithelium
The mRNA expression profile of LPA receptors in HBEpCs [41, 120, 135, 139], nasal polyp-derived epithelial cells [138], and airway epithelial cell lines [138] has been described. In HBEpCs, the relative abundance of mRNA detected was LPA1 > LPA3 > LPA2 > LPA4, while LPA-R5 was not detectable in HBEpCs [139]. LPA1-3 are not only localized at the plasma membrane, but are also distributed in the cytoplasm of the cells [120]. Studies from Georas et al. showed that LPA1 and LPA2 are expressed in airway epithelial cells of human lung tissue [41]. Furthermore, the expression of LPA1 on HBEpCs cell surface was determined by flow cytometry with an LPA1 receptor antibody directed against the conserved extracellular N-terminus [41]. The expression of LPA1 is not restricted to the plasma membrane and cytoplasmic organelles as nuclear localization of LPA1 has been shown in various cell types, including HBEpCs [30]; however, the biological role of nuclear LPA1 in airway epithelium remains to be investigated. In contrast to primary HBEpCs, only LPA1 and LPA2, but not LPA3 or LPA4, were expressed in epithelial cell lines A549 and BEAS-2B [138]. Nasal polyp epithelial cells exhibited expression of the LPA1, LPA2 and LPA4, but not LPA3 [138], suggesting differences in expression profile in different epithelial cell types. The regulation of LPA receptor expression is not well studied. The expression of LPA receptors are modulated differently in asthmatic HBEpCs compared to normal controls. In asthmatic HBEpCs, LPA1 mRNA was up-regulated ~ 3.2 fold, while LPA2 was down-regulated ~ 2.6 fold compared to normal HBEpCs [139]. Both IL-13 and IFN-γ significantly reduced LPA1 and LPA2 mRNA levels, while IL-4 had no effect on LPA receptor mRNA expression in the A549 epithelial cell line [138]. However, the effect of IL-13 and IFN-γ on LPA-induced signaling and biological responses remains to be investigated.
3.3 LPA receptors null mice in pulmonary/airway diseases
Given that LPA levels are elevated in BALF from segmental allergen challenged asthmatics [41], a bleomycin model of pulmonary fibrosis [42], and a Schistosoma mansoni egg/soluble antigen challenge murine model of inflammation [43], it becomes critical to determine the role of LPA/LPA-Rs underlying the pathobiology of airway/pulmonary diseases. Genetically engineered LPA1-3 null mice have been used to study a number of pathophysiological states including airway inflammation, asthma, and pulmonary fibrosis. LPA1 (+/-) mice or LPA2 (+/-) mice sensitized with inactivated Schistosoma mansoni eggs and challenged with soluble antigen Schistosoma mansoni eggs showed less infiltration of eosinophils, decreased PGE2 levels in BAL fluids, and lower COX-2 expression in the lung tissue [43]. Both LPA1&2 seem to be involved in the Schistosoma mansoni egg/soluble antigen challenge murine model of inflammation. Furthermore, 6 h after intratracheal administration of LPA (5 μM) to LPA1&2 (+/-) mice resulted in less infiltration of neutrophils into alveolar space compared to LPA-challenged Wild type mice [43]. LPA1 (-/-) mice showed significant increase in CD3/CD28-driven proliferation after ovalbumin challenge [140], suggesting an anti-inflammatory role for LPA1 in asthma. In the bleomycin model of lung injury mediated by fibroblast recruitment and vascular leak, mice lacking LPA1 were markedly protected from fibrosis and mortality suggesting a link between LPA1 and development of pulmonary fibrosis [42]. These studies indicate a potential role of LPA-Rs in lung injury, inflammation, and pulmonary fibrosis; however, the relative contribution of LPA-Rs present on the epithelial cells vs. endothelial vs. infiltrating cells to the inflammation or injury process is unclear that might require tissue targeted null knockdown of LPA-Rs.
3.4 LPA signaling via LPA-Rs in the airway epithelial cells
The role of LPA receptors in LPA signaling in HBEpCs has been investigated in the context of cytokine/chemokines secretion. Down-regulation of LPA1, or LPA2, or LPA3 by specific siRNA attenuated LPA-induced IL-8 secretion in HBEpCs, and LPA3 specific agonist increased IL-8 secretion in HBEpCs [135]. LPA signals via LPA receptors through different heterotrimeric G proteins, such as Gi, Gq, Gs, and G12/13, which couple to the C-terminus domain of LPA receptors. Pertussis toxin (PTx), which uncouples GPCR from its Gi/o and in turn reduces the affinity of GPCR toward its agonist, serves as a tool to study Gi-mediated signaling. In HBEpCs, PTx attenuated LPA-induced phosphorylation of EGF-R [128] and c-Met [113], as well as expression of IL-8 [114], COX-2 [115], and IL-13Rα2 [136]. To further investigate the involvement of G-proteins in LPA signaling, Cummings et al. transfected HBEpCs with minigenes that encode peptides that specifically block the respective G-protein/receptor interface [114]. Expression of G12/13 or Gi blocking peptides, but not Gq blocking peptide, resulted in attenuation of LPA-induced IL-8 secretion [114]. These studies suggest that LPA induced signaling in HBEpCs is through Gi and G12/13-coupled LPA receptors.
4. LPA regulates expression of pro- and anti-inflammatory genes
Airway epithelium is the first point of contact for inhaled substances such as cigarette smoke, microorganisms, allergens and environmental pollutants, and plays a key role in host defense and innate immune response [141-144]. The involvement of the airway epithelium in modulating immune responses is regulated by secretion of cytokines and other mediators of the airway epithelium in response to various stimuli. The earlier observations of LPA as the serum factor mediating serum-induced sensitization of cAMP responses in airway smooth muscle cells [39, 145], and a potential role of LPA in asthma [41, 43, 140] and IPF [42] have resulted in several investigations related to LPA in innate immunity, inflammation, repair and remodeling and gene expression in airway cells and animal models of airway diseases.
4.1. LPA modulates expression and release of cytokines and lipid mediators
4.1.1. IL-8 expression and secretion
IL-8, a major chemoattractant of neutrophils, plays a key role in innate immune responses [114, 146, 147]. Higher levels of IL-8 have been found in BALF of patients of airway inflammatory diseases including acute lung injury [114, 119]. LPA has been shown to be a strong stimulus for IL-8 secretion in various airway epithelial cell types, including HBEpCs, BEAS-2B, and A549 [58, 114, 128, 138, 139]. In HBEpCs, LPA mediated IL-8 gene expression at 1 h, and peaked at 2h, while IL-8 secretion by LPA reached its peak at 12 h [114]. LPA-induced IL-8 secretion was through Gi and G12/13-coupled LPA1-3 receptors [114, 135]. A similar increase in IL-6 and IL-8 production after LPA stimulation was observed in BEAS-2B cells [138]. Consistent with the in vitro studies, intratracheal administration of LPA in mice increased MIP-2 (the murine homolog of IL-8) levels at 3 h and neutrophils infiltration at 6 h. At later periods (>12 h), the levels of MIP-2 and neutrophils returned to near basal levels [114]. Hashimoto et al. also showed that inhalation induced histamine release and increased numbers of eosinophils and neutrophils in BAL of guinea pig [148, 149]. These results suggest that LPA regulates airway inflammation via stimulating cytokine and inflammatory mediator release and neutrophils or eosinophils infiltration in airway.
4.1.2. IL-13R alpha2 expression and release
The levels of interleukin-13 (IL-13), a Th2-type cytokine, are increased in BALF of asthma patients [150, 151] and ovalbumin challenged mice [152]. IL-13 induces phosphorylation of signal transducer and activator of transcription 6 (STAT6), which activates the transcription of many pro-inflammatory genes in epithelial cells and plays a critical role in the pathogenesis of bronchial asthma [153-155]. IL-13Rα1 binds to IL-13 and mediates IL-13-induced activation of Janus kinases (JAKs) and STAT6 [156, 157]. The human IL-13 decoy receptor, termed IL-13 Receptor α2 (IL-13Rα2), binds to IL-13 with much higher affinity (Kd=0.25-1.2 nM) compared to IL-13Rα1 (Kd=2-10 nM) [158-160], and overexpression of IL-13Rα2 selectively inhibited IL-13-induced response in airway epithelium and murine lung [161-163]. Elevated IL-13Rα2 mRNA expression has been detected in Schistosoma egg-induced liver fibrosis [164] and bleomycin-induced fibrosis [165]. In addition, interferon-γ (IFN-γ) or IL-13 treatment induced IL-13Rα2 mRNA expression that was localized to the cell surface in U937 [35] and bronchial epithelial cells [167]. While LPA induced IL-13 expression under conditions of semimaximal activation in T cells, in HBEpCs [168], LPA treatment induced IL-13Rα2 mRNA and protein expression and secretion through Gi-coupled LPA receptors, without altering IL-13Rα1 expression [136]. Further, elevated IL-13Rα2 expression by LPA in HBEpCs attenuated IL-13-mediated phosphorylation of STAT6 and eotaxin and SOCS-1 gene expression [136]. As LPA levels are elevated in BALF from segmental allergen challenged asthmatics [41] and IPF [42], and LPA modulates expression of IL-13Rα2 and IL-13-mediated STAT6 signaling [136], it is postulated that increased LPA levels may have a protective role in airway inflammation and remodeling of airways. Future studies related to LPA and its effect on production of Th2 cytokines, and airway remodeling and repair would increase our understanding of how LPA and its analogs ameliorate airway inflammation in asthma and fibrosis.
4.1.3. COX-2 expression and PGE2 release
Prostaglandin E2 (PGE2) is an autocrine lipid mediator derived from arachidonic acid (AA) metabolism by cyclooxygenase-1 or 2 (COX-1 or 2) [169]. PGE2 plays crucial roles in various biological events such as neuronal function [170], vascular hypertension [171], tumorigenesis [172-174], and inflammation [175]. Upregulation of COX-2 expression and PGE2 release plays a protective role in the innate immunity response and attenuated allergen-induced tissue repair process in airway inflammation [176, 177]. Inhaled PGE2 airway responses, hyperresponsiveness, and inflammation [178, 179]; however, the regulation of COX-2 expression and the physiological effect of PGE2 on airway epithelium has not been well examined. LPA challenge increased COX-2 expression and PGE2 production in HBEpCs [115]. Analyses of total RNA by Real-time RT-PCR showed that both COX-1 and COX-2 were expressed in HBEpCs and that the expression of COX-1 is ~ 2 fold higher than COX-2 [115]. Exposure of HBEpCs to LPA (1 μM) induced COX-2 gene and protein expression and PGE2, while such exposure had no effect on COX-1 gene and protein expression up to 24 h [115]. Further, PTx attenuated LPA-induced COX-2 mRNA and protein expression, suggesting the involvement of Gi-coupled LPA-Rs. COX-2 siRNA effectively down-regulated COX-2 protein expression and attenuated LPA-induced PGE2 release by ~ 50 % compared to scrambled siRNA cells exposed to LPA [115]. These results demonstrated that LPA induced PGE2 secretion was dependent on expression of COX-2 in HBEpCs. As PGE2 exhibits anti-inflammatory properties in the airway [176-179], enhanced COX-2 expression and PGE2 release induced by LPA may have a protective role in airway inflammation and remodeling.
4.2. LPA regulates transcriptional factors
4.2.1. Nuclear factor-κB (NF-κB)
NF-κB and AP-1 are transcriptional factors expressed in almost all mammalian cell types and play a key role in regulating the immune response to infection. NF-κB is a heterodimer composed of two Rel family members (RelA and B). In quiescent cells, NF-κB is present in cytosol in an inactive form bound to an inhibitory protein, I-κB, and after stimulation, I-κB is phosphorylated, and degraded by the 26S proteasome. NF-κB is released from NF-κB/I-κB complex, translocates from cytosol to nucleus, and activates gene transcription [180]. The activation of NF-κB by LPA in HBEpCs has been detected by several procedures including phosphorylation of I-κB, NF-κB nuclear translocation, NF-κB luciferase reporter assay, and electrophoretic motility-shift assay [58, 114, 115, 135]. Signaling pathways regulating LPA-induced NF-κB activation in HBEpCs have been partially defined. LPA induced NF-κB activation via p38 MAPK [139] and protein kinase C δ (PKCδ) [114]. The promoter region of human IL-8 and COX-2 have potential NF-κB binding elements [181, 182], however, the human IL-13Rα2 promoter region lacks NF-κB binding elements [183]. Inhibition of NF-κB activation attenuated LPA-induced IL-8 [114] and COX-2 [115] expression, suggesting that NF-κB plays a critical role in LPA-induced gene expression in HBEpCs. However, LPA-induced IL-13Rα2 expression was not dependent on the NF-κB pathway in HBEpCs [136].
4.2.2. C/EBPβ
The C/EBP family consists of six proteins that belong to the basic zipper transcriptional factors [184]. The C/EBPβ is important in the regulation of genes involved in immune and inflammatory responses and has been shown to bind to transcriptional regulatory regions of several acute-phase and cytokine genes. The human COX-2 promoter region has been sequenced and contains a putative transcriptional regulatory element of C/EBPβ [182]. The participation of C/EBPβ in COX-2 expression to various stimuli in different cell types has been described [185, 186]. In HBEpCs, LPA induced phosphorylation of C/EBPβ and downregulation of C/EBPβ expression by siRNA attenuated LPA-induced COX-2 expression [115], suggesting the involvement of C/EBPβ in LPA-induced gene expression in HBEpCs. Further evidence showed that LPA-induced activation of PLD2, PKCζ, and EGF-R contributed to C/EBP activation in HBEpCs [115]. The role of other C/EBP family members in LPA-mediated cytokine and lipid mediators release is still not clear.
4.2.3. JNK and p38 MAPK in LPA-induced NF-κB and AP-1 Activation
c-Jun N-terminal kinase (JNK) and p38 MAPK belong to MAP kinase family, which are important component of signal transduction pathways induced by growth factors, cytokines, and GPCR ligands. p38 MAPK and JNK1 and JNK2 isoforms are widely distributed in mammalian cells [187]. LPA treatment of HBEpCs induced phosphorylation of JNK1/2 [115, 135, 136] and c-Jun [135], induced c-Jun nuclear localization [135], and induced AP-1 activation [135]. The mechanism(s) of regulation of JNK activation by LPA is still not clear. Interestingly, overexpression of catalytic dominant negative PLD1 and 2 isoforms attenuated LPA-induced phosphorylation of JNK in HBEpCs [136], suggesting JNK activation may be regulated by PLD-generated phosphatidic acid (PA). Inhibition of JNK activation by JNK inhibitor (JNKiII) or downregulation of c-Jun by c-Jun siRNA attenuated LPA-induced activation of AP-1 [135], as well as IL-8 [135], COX-2 [115], and IL-13Rα2 [136] expression.
LPA-induced activation of NF-κB has been shown to be dependent on activation of p38 MAPK. An inhibitor of p38MAPK or p38 MAPK siRNA blocked phosphorylation of I-κB and NF-κB translocation to the nucleus without affecting JNK/ c-Jun phosphorylation and AP-1activation [135]. The role of LPA-induced activation of p38 MAPK in LPA-mediated IL-8 and COX-2 expression was investigated in HBEpCs. Inhibition of p38 MAPK activation attenuated LPA-induced IL-8 [135] through decreasing the NF-κB pathway, not the JNK/AP-1 pathway. Recent studies suggest that p38 MAPK also regulates COX-2 mRNA stability [188]. In HBEpCs, LPA induced COX-2 mRNA expression as well as COX-2 mRNA stability [189], while downregulation of p38 MAPK activation attenuated LPA-induced COX-2 mRNA stability and expression [189]. p38 MAPK regulates LPA-induced COX-2 expression through both transcriptional and post-transcriptional regulation. Further study will determine the role of p38 MAPK isoforms in the regulation of COX-2 mRNA levels in HBEpCs.
4.3. LPA activates PKC isoforms
PKC is a superfamily of kinases that phosphorylates protein substrates on serine and threonine residues and transduces the cellular signals. Eleven PKC isoforms have been identified in mammals and divided into three subgroups: classical PKCs (cPKCα, βI, βII, and γ), novel PKCs (nPKCδ, ε, η, μ and θ), and atypical PKCs (aPKCζ and γ) based on sequence homology. The cPKCs are calcium-dependent, and activated by diacylglycerol (DAG) and DAG-mimicking phorbol esters. The nPKCs are calcium-independent, activated by DAG and DAG-mimicking phorbol esters. The aPKCs are structurally divergent from other PKC family members and activated by acidic phospholipids such as phosphatidylserine (PS), polyphosphoinositides (PI) and phosphatidic acid (PA), but not by DAG or phorbol esters [190]. Expression of PKCα, δ, ι/λ, and ζ in HBEpCs were confirmed by Western blotting with specific antibodies to these isoforms [114]. LPA induced phosphorylation and plasma membrane translocation of PKCδ [114] and ζ [189]. The involvements of PKC isoforms in IL-8, IL-13Rα2, and COX-2 expression have been studied by using PKC isoform specific pharmaceutical inhibitors or overexpression of dominant negative PKC isoforms (dn-PKCs) in HBEpCs. PKCδ contributes to LPA-induced IL-8 [114], IL-13Rα2 [136], and COX-2 [189] expression. PKCζ regulates LPA-induced COX-2 expression [115] while PKCλ regulates LPA-induced IL-8 expression [114] in HBEpCs. It has been shown that PKC inhibitors attenuated activation of transcriptional factors in various cell types [191, 192]. Inhibition of PKCδ attenuated LPA-induced NF-κB activation [114] and inhibition of PKCζ modulated LPA-induced C/EBPβ activation in HBEpCs [115]. Regulation of PKCζ by PLD has been shown in human pulmonary arterial endothelial cells (HPAECs) [193] while the relationship between PKCζ and PLD will need to be further determined in human airway epithelial cells (Fig. 2).
Figure 2. LPA signaling in airway epithelial cells.
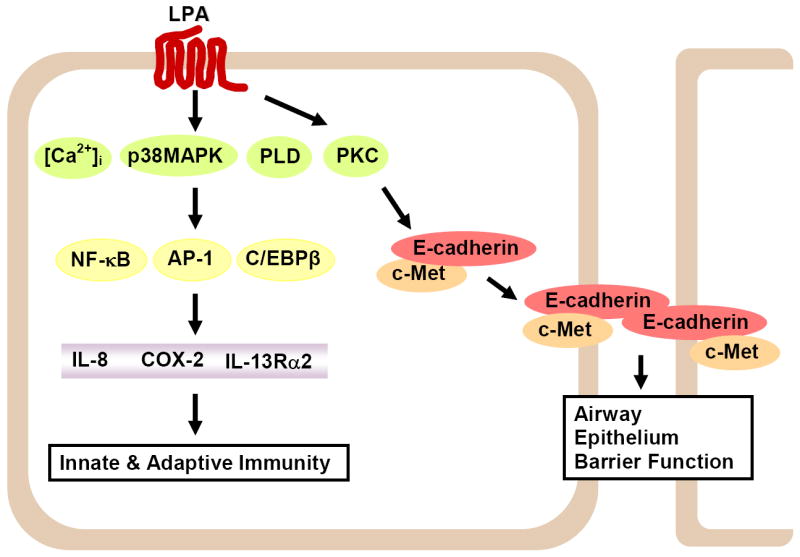
Ligation of LPA to LPA receptors increases intracellular calcium, activates p38 MAPK, PKCs and PLD, resulting in the activation of transcriptional factors and induction of cytokine(s) expression and release; thus regulating innate and adaptive immune responses. LPA induces E-cadherin and c-Met redistribution to cell periphery and regulates airway epithelium integrity.
4.4. Role of LPPs in regulation of LPA signaling
Recent studies have revealed that LPA degradation occurs through the LPPs family, which consists of three family members (LPP-1, LPP-2, LPP-3), attenuated LPA-induced intracellular signaling [82-85]. LPPs contain a novel conserved phosphatase sequence motif, K(X)6RP-(X12-54)-PSGH-(X31-54)-SR(X)5H(X)3D, that lies on the outside of the cell, or on the luminal surface of the endoplamic reticulum or Golgi [57, 85]. In the rat, the activity of LPPs in the lung is the highest among any tissue and mRNA expression for LPP-1 and LPP-3, but not LPP-2, was detected in rat lung [194]. LPPs activity is enriched in lipid-rich signaling platforms isolated from type II cell-mouse lung epithelial cell lines (MLE12 and MLE15) [195, 196], and using Western blotting and RT-PCR, expression of LPP-1, -2, and -3 were detected in HBEpCs [58]. In HBEpCs, exogenously added LPA was hydrolyzed by a LPP-like ecto-enzyme activity, which was inhibited by tyrosine kinase inhibitors, but not by serine-threonine inhibitors. Overexpression of LPP-1 Wild type (Wt) decreased extracellular LPA levels and attenuated LPA-induced intracellular Ca2+, ERK1/2 phosphorylation, activation of NF-κB pathway, secretion of IL-8, and expression of COX-2 [58, 189]. Downregulation of LPP-1 by LPP-1 siRNA enhanced LPA-induced COX-2 expression in HBEpCs [189]. These results suggest that LPP-1 regulated LPA-induced gene expression via degradation of extracellular LPA. Interestingly, overexpression of LPP-1 also attenuated the IL-8 secretion mediated by a non-hydrolyzed LPA analogue, suggesting an intracellular role of LPP-1 in regulation of LPA-induced signaling [58]. As LPA plays a critical role in innate immune response in airway diseases, and LPP-1 attenuates LPA function by its ecto-enzyme activity, targeting LPP-1 may regulate airway inflammation.
5. Cross-talk between LPA-Rs and Receptor Tyrosine Kinases (RTKs)
Several lines of investigation in a variety of cell types have shown that many of the growth promoting effects of GPCR agonists (LPA, sphingosine-1-phosphate (S1P), angiotensin, PGE2, and others) are mediated through activation of receptor tyrosine kinases (RTKs), a phenomenon termed as “Transactivation” [197-199]. The transactivation of RTKs by GPCR ligands has been demonstrated in various cell types and this cross-talk between GPCRs and RTKs provides additional non-canonical signaling pathways, which regulate cellular function. In addition, disruption of GPCR function via PTx or antagonist reduces the growth factor stimulated activation of ERK [200]. Both the GPCR (S1P1) and the βγ subunit of the activated G-protein form a functional signaling complex with the RTK, which may reprogram the mitogenic signal of the RTK to a migratory response [200]. Thus, potential signaling via “Transactivation” between GPCRs and RTKs will allow spatial regulation of effectors dictating nature of the biological responses.
5.1. LPA transactivates PDGF-Rβ
Among the various RTKs, GPCR mediated transactivation of platelet-derived growth factor receptor (PDGF-R) has been well described [198, 200-202]. The PDGF has been recognized as an important growth factor regulating cell proliferation and the development of many diseases including cancer [203-205]. PDGF-BB challenge stimulated phosphorylation of Erk1/2, suggesting that PDGF plays a proliferative role in HBEpCs [120]. In addition to PDGF-BB, LPA also induced tyrosine phosphorylation of PDGF-Rβ via a transactivation mechanism involving PLD2, but not PLD1, signaling [120]. Furthermore, transactivation of PDGF-Rβ by LPA regulated LPA-dependent phosphorylation of Erk1/2 [120] in HBEpCs. These results suggest that cross-talk between LPA-Rs and PDGF-Rβ may play a role in airway remodeling and repair. While the LPA1-PDGF-Rβ signaling complex could not be identified in HBEpCs [212], interaction between G-protein-coupled S1P1 and PDGF-Rβ has been identified and extensively studied in HEK293 and airway smooth muscle cells [201]. The formation of this signaling complex between S1P1 and PDGF-Rβ permits the usage of βγ-subunits of activated Gi by PDGF-R to enhance PDGF signaling [206]. The functional implications of the “Transactivation” between RTKs and GPCRs in health and disease warrant more investigation.
5.2. LPA transactivates EGF-R
EGF-R, similar to other growth factor receptors, is activated by EGF, heparin binding-EGF (HB-EGF) and tumor growth factor-α (TGF-α) [207] b Since the first report of transactivation of EGF-R by LPA in fibroblast [208], recent studies have established EGF-R transactivation by LPA in airway epithelial cells [123, 124, 128, 209, 210]. Two pathways of LPA-mediated transactivation of EGF-R have been described: the first involves activation of matrix metalloproteinases (MMPs), cleavage of pro-HB-EGF, and subsequent ligation and activation of EGF-R [125, 128, 209, 211], and the second pathway is HB-EGF shedding-independent involving the intracellular activation of EGF-R by Src kinases [209, 210]. In HBEpCs, LPA treatment induced tyrosine phosphorylation of EGF-R at different tyrosine residues, which were Gi-dependent [128, 212]. Furthermore, the LPA-induced EGF-R transactivation was regulated by PKCζ [115] and δ and Src kinases [128]. Among Src kinase family, using specific siRNA to down-regulate Src kinases, a role for Lyn kinase, but not Src or Yes kinase, in regulating LPA-induced EGF-R transactivation was established [128]. Similar to the mechanism of PDGF-Rβ transctivation by LPA, PLD2, but not PLD1, regulated the LPA-induced phosphorylation of EGF-R in HBEpCs [115]. Interestingly, LPA-mediated activation of Lyn was dependent on PKCδ signal transduction, with subsequent activation of MMP-2/9, and proHB-EGF shedding [128]. LPA induced PKCζ activation through PLD2 [115]. It is not clear if PKCδ is up-stream or down-stream of PLD2 in HBEpCs. Data on the physiological significance of EGF-R transactivation by LPA is limited. Inhibition of EGF-R tyrosine kinase with AG1478 attenuated LPA-induced phosphorylation of ERK 1/2 in PC12 [213], ovarian cancer cells [214], prostate cancer cells [50], and HBEpCs [212], suggesting a potential role in cell proliferation. Furthermore, He et al. demonstrated that though all NF-κB, AP-1, and C/EBPβ are involved in LPA-induced COX-2 expression and PGE2 release, however, only C/EBPβ is downstream signal molecule to EGF-R transactivation in HBEpCs [115] (Fig. 3). At present, it is unclear which of the LPA-Rs are involved in LPA-induced EGF-R transactivation. Another consequence of LPA-induced transactivation of EGF-R is a rapid and sustained decrease in EGF-R binding in human primary airway epithelial cells and BEAS-2B cell line [123, 124]. The rapid decrease in EGF-R binding via EGF-R transactivation mediated by LPA was dependent on MEK/ERK and PKC, whereas the sustained decrease was primarily regulated by the PKC signaling pathway in BEAS-2B cell line [123]. Further studies are necessary to delineate physiological significance(s) of EGF-R transactivation in airway inflammation, repair and remodeling.
Figure 3. Signaling pathways of LPA-induced transactivation of EGF-R.

Ligation of LPA to its G-protein-coupled receptors induces phosphorylation of EGF-R through PKCδ, Lyn kinase, MMP, and pro-HB-EGF pathway. PLD2 and PKCζ are involved in LPA-induced transactivation of EGF-R; however, the relationship between PLD2 and PKCδ or between PKCζ and Lyn kinase are unclear. Erk1/2 and C/EBPβ are downstream targets of EGF-R transactivation, which regulate cell proliferation and gene expression.
5.3. LPA transinactivates c-Met
c-Met, a proto-oncogene product, is a RTK, the receptor for hepatocyte growth factor (HGF), and predominantly expressed in various types of epithelium [215-216]. Increased expression and activation of c-Met is associated with tumor cell growth, scattering, invasion and metastasis [216-217]. Specific mutations of the tyrosine residues in the juxtamembrane and semaphorin domains of c-Met resulted in the regulation of the cytoskeleton through the focal adhesion protein paxillin [218]. LPA or S1P mediation enhanced tyrosine phosphorylation of c-Met via a transactivation pathway in human colon cancer cell lines and human gastric cancer cells [127, 219]; however, in HBEpCs, unlike LPA-mediated tyrosine phosphorylation of PDGF-Rβ and EGF-R [115, 120, 128], LPA-induced serine, but not tyrosine, phosphorylation, which was dependent on activation of PKCδ by LPA [113]. Further, serine phosphorylation of c-Met by LPA attenuated HGF-mediated activation of AKT and cell motility in HBEpCs, suggesting a regulatory role for c-Met serine phosphorylation in epithelial cell motility [113]. These results suggest that in the same type of cells, the same GPCR ligand treatment might induce “yin and yang” responses on different RTKs (Fig. 4). Recent study has demonstrated that the HGF-dependent tyrosine phosphorylation of c-Met was largely suppressed by PKCδ with a reciprocal relationship to Ser-985 phosphorylation in A549 cell line [220]. In HBEpCs, LPA induced c-Met relocalization from the cytoplasm to cell-cell contacts and this was dependent on activation of PKCδ and ζ and interaction with E-cadherin, suggesting a role of c-Met in cell adhesion and motility [113]. The role of c-Met in regulation of airway epithelium barrier function and cytokine release will be investigated.
Figure 4. Cross-talk between LPA receptors and RTKs.

LPA via its receptors induces tyrosine phosphorylation of EGF-R and PDGF-Rβ (termed as transactivation), and regulates cell proliferation and gene expression. Cross-talk between LPA receptors and c-Met induces serine phosphorylation of c-Met (termed as transinactivation), which enhances airway epithelial barrier function.
6. LPA regulates epithelial barrier via adherens junction proteins
The epithelial cell-cell junctional complex is composed of tight junctions, adherens junctions and desmosomes [221, 222]. These adherens junctions play a pivotal role in regulating the activity of the entire junctional complex since the formation of adherens junctions subsequently leads to the formation of other cell-cell junctions, including tight junction [221-223]. The major adhesion molecules in the adherens junctions are the cadherins [221-223]. E-cadherin is a member of the cadherin family that mediates calcium-dependent cell-cell adhesion [221-225]. The N-terminal ectodomain of E-cadherin contains homophilic-binding domain, and the cytoplasmic domain binds to catenins, which interact with actin [221-225]. The regulated expression and the plasma membrane localization of E-cadherin are critical for the maintenance of epithelial cell-cell junctions crucial to the functional integrity of the epithelial barrier [221-223]. The decrease of adhesive properties of E-cadherin has been shown to be related to the loss of differentiation and the subsequent acquisition of a higher motility and invasiveness of epithelial cells [226-228]. Dislocation or shedding of E-cadherin in the airway epithelium induces epithelial shedding and increases airway permeability in airway diseases [229-231]. However, the regulation and mechanism(s) of E-cadherin localization within the epithelium is not fully known, especially during pathological situations of inflammation and airway remodeling. LPA treatment tightened the airway epithelial cell barrier [41, 232]; however, the mechanism(s) regulating LPA-induced epithelial barrier function is yet to be fully defined. In HBEpCs, exposure to LPA enhanced accumulation of E-cadherin and c-Met complex at cell-cell contacts through activation of PKCδ, and E-cadherin relocalization to cell-cell contacts was critical for LPA-induced enhanced airway epithelial barrier function [232] (Fig. 2). The role of LPA in regulation of airway barrier function in airway inflammatory disease, such as acute lung injury, needs further investigation.
7. Conclusions and future directions
In this review, we summarized the mechanisms of LPA in regulating gene expression and barrier function in human airway epithelial cells. The study of the biological role of LPA in airway inflammatory diseases is an area yet to be well defined. The recent studies reveal that LPA regulates biological responses in airway epithelial, smooth muscle, fibroblast, and lymphocyte cells. Our focus is on the role of LPA in regulation of airway epithelial cytokine production, lipid mediator release and airway epithelial barrier function since airway epithelium is the first site that responds to the increase of LPA levels in BALF of asthmatic and IPF patents. LPA induces gene expression through LPA-Rs mediated intracellular signaling, such as changes of [Ca2+]i, activation of PKC isoforms, PLD, and transcriptional factors (NF-κB, AP-1, and C/EBPβ). Cross-talk between LPA-Rs and RTKs (EGF-R, PDGF-Rβ, and c-Met) regulate LPA-induced gene expression, cell proliferation, and airway epithelial barrier function. LPA upregulates Th1 type cytokine (such as IL-8) and PGE2 release and attenuates the effects of Th2 type cytokine (IL-13) by releasing of an IL-13 decoy receptor, IL-13Rα2, in airway epithelial cells. This indicates that LPA in the airway may play a protective role in inflammatory diseases. Furthermore, LPA increases airway epithelial integrity through PKC isoforms-mediated E-cadherin and c-Met protein complex assembly at cell-cell contacts, suggesting that LPA protects airway by regulation of cytokine releases and airway epithelial integrity (Fig. 2). While a majority of LPA receptors are coupled to heterotrimeric G protein, recently G-protein-independent signaling through GPCRs has been demonstrated [233]. Whether LPA induces part of intracellular signaling in the absence of G-proteins in airway epithelial cells will be further investigated. Furthermore, intravenous injection with LPA attenuated bacterial endotoxin-induced plasma TNFα production and myeloperioxidase activity in lung, suggesting an anti-inflammatory role of LPA in a murine model of acute lung injury [234]. To further understand the role of LPA from both plasma and BALF in airway inflammatory diseases, the lysoPLD transgenic mice and lung epithelial cells specific lysoPLD transgenic mice will serve as useful models. As there are six receptors for LPA, development of specific antagonists for each of the receptor subtype will allow therapeutic targeting of LPA-Rs that regulate inflammation and remodeling in airway diseases.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health HL79396 (to V.N.) and American Cancer Association Institutional Grant IRG-58-044-47 (to Y.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tigyi G, Miledi R. J Biol Chem. 1992;267(30):21360–21367. [PubMed] [Google Scholar]
- 2.Eichholtz T, Jalink K, Fahrenfort I, Moolenaar WH. Biochem J. 1993;291(Pt 3):677–680. doi: 10.1042/bj2910677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goetzl EJ, Lee H, Azuma T, Stossel TP, Turck CW, Karliner JS. J Biol Chem. 2000;275(19):14573–14578. doi: 10.1074/jbc.275.19.14573. [DOI] [PubMed] [Google Scholar]
- 4.Hosogaya S, Yatomi Y, Nakamura K, Ohkawa R, Okubo S, Yokota H, Ohta M, Yamazaki H, Koike T, Ozaki Y. Ann Clin Biochem. 2008;45(Pt 4):364–368. doi: 10.1258/acb.2008.007242. [DOI] [PubMed] [Google Scholar]
- 5.Postma FR, Jalink K, Hengeveld T, Bot AG, Alblas J, de Jonge HR, Moolenaar WH. Embo J. 1996;15(1):63–72. [PMC free article] [PubMed] [Google Scholar]
- 6.Tokumura A, Kanaya Y, Kitahara M, Miyake M, Yoshioka Y, Fukuzawa K. J Lipid Res. 2002;43(2):307–315. [PubMed] [Google Scholar]
- 7.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. J Biol Chem. 2002;277(50):48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 8.van Corven EJ, Groenink A, Jalink K, Eichholtz T, Moolenaar WH. Cell. 1989;59(1):45–54. doi: 10.1016/0092-8674(89)90868-4. [DOI] [PubMed] [Google Scholar]
- 9.Budnik LT, Brunswig-Spickenheier B, Mukhopadhyay AK. Mol Endocrinol. 2003;17(8):1593–1606. doi: 10.1210/me.2002-0371. [DOI] [PubMed] [Google Scholar]
- 10.Yart A, Chap H, Raynal P. Biochim Biophys Acta. 2002;1582(13):107–111. doi: 10.1016/s1388-1981(02)00144-0. [DOI] [PubMed] [Google Scholar]
- 11.Schmitz U, Thommes K, Beier I, Vetter H. Biochem Biophys Res Commun. 2002;291(3):687–691. doi: 10.1006/bbrc.2002.6493. [DOI] [PubMed] [Google Scholar]
- 12.van Leeuwen FN, Giepmans BN, van Meeteren LA, Moolenaar WH. Biochem Soc Trans. 2003;31(Pt 6):1209–1212. doi: 10.1042/bst0311209. [DOI] [PubMed] [Google Scholar]
- 13.Moolenaar WH, van Meeteren LA, Giepmans BN. Bioessays. 2004;26(8):870–881. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- 14.Mills GB, Moolenaar WH. Nat Rev Cancer. 2003;3(8):582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 15.Stahle M, Veit C, Bachfischer U, Schierling K, Skripczynski B, Hall A, Gierschik P, Giehl K. J Cell Sci. 2003;116(Pt 18):3835–3846. doi: 10.1242/jcs.00679. [DOI] [PubMed] [Google Scholar]
- 16.Manning TJ, Jr, Parker JC, Sontheimer H. Cell Motil Cytoskeleton. 2000;45(3):185–199. doi: 10.1002/(SICI)1097-0169(200003)45:3<185::AID-CM2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 17.Avraamides C, Bromberg ME, Gaughan JP, Thomas SM, Tsygankov AY, Panetti TS. Am J Physiol Heart Circ Physiol. 2007;293(1):H193–203. doi: 10.1152/ajpheart.00728.2006. [DOI] [PubMed] [Google Scholar]
- 18.Hao F, Tan M, Xu X, Han J, Miller DD, Tigyi G, Cui MZ. Biochim Biophys Acta. 2007;1771(7):883–892. doi: 10.1016/j.bbalip.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, Johnson LR. Gastroenterology. 2002;123(1):206–216. doi: 10.1053/gast.2002.34209. [DOI] [PubMed] [Google Scholar]
- 20.Ye X, Ishii I, Kingsbury MA, Chun J. Biochim Biophys Acta. 2002;1585(23):108–113. doi: 10.1016/s1388-1981(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 21.Rusovici R, Ghaleb A, Shim H, Yang VW, Yun CC. Biochim Biophys Acta. 2007;1770(8):1194–1203. doi: 10.1016/j.bbagen.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukushima N, Kimura Y, Chun J. Proc Natl Acad Sci U S A. 1998;95(11):6151–6156. doi: 10.1073/pnas.95.11.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hecht JH, Weiner JA, Post SR, Chun J. J Cell Biol. 1996;135(4):1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.An S, Dickens MA, Bleu T, Hallmark OG, Goetzl EJ. Biochem Biophys Res Commun. 1997;231(3):619–622. doi: 10.1006/bbrc.1997.6150. [DOI] [PubMed] [Google Scholar]
- 25.Chun J, Contos JJ, Munroe D. Cell Biochem Biophys. 1999;30(2):213–242. doi: 10.1007/BF02738068. [DOI] [PubMed] [Google Scholar]
- 26.Noguchi K, Ishii S, Shimizu T. J Biol Chem. 2003;278(28):25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 27.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. J Biol Chem. 2006;281(33):23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 28.Tabata K, Baba K, Shiraishi A, Ito M, Fujita N. Biochem Biophys Res Commun. 2007;363(3):861–866. doi: 10.1016/j.bbrc.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 29.Im DS, Heise CE, Harding MA, George SR, O’Dowd BF, Theodorescu D, Lynch KR. Mol Pharmacol. 2000;57(4):753–759. [PubMed] [Google Scholar]
- 30.Waters CM, Saatian B, Moughal NA, Zhao Y, Tigyi G, Natarajan V, Pyne S, Pyne NJ. Biochem J. 2006;398(1):55–62. doi: 10.1042/BJ20060155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Proc Natl Acad Sci U S A. 2003;100(1):131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Baker DL, Yasuda S, Makarova N, Balazs L, Johnson LR, Marathe GK, McIntyre TM, Xu Y, Prestwich GD, Byun HS, Bittman R, Tigyi G. J Exp Med. 2004;199(6):763–774. doi: 10.1084/jem.20031619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Contos JJ, Ishii I, Chun J. Mol Pharmacol. 2000;58(6):1188–1196. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- 34.Fukushima N, Chun J. Prostaglandins Other Lipid Mediat. 2001;64(14):21–32. doi: 10.1016/s0090-6980(01)00105-8. [DOI] [PubMed] [Google Scholar]
- 35.Graler MH, Goetzl EJ. Biochim Biophys Acta. 2002;1582(13):168–174. doi: 10.1016/s1388-1981(02)00152-x. [DOI] [PubMed] [Google Scholar]
- 36.Takuwa Y, Takuwa N, Sugimoto N. J Biochem. 2002;131(6):767–771. doi: 10.1093/oxfordjournals.jbchem.a003163. [DOI] [PubMed] [Google Scholar]
- 37.Yanagida K, Ishii S, Hamano F, Noguchi K, Shimizu T. J Biol Chem. 2007;282(8):5814–5824. doi: 10.1074/jbc.M610767200. [DOI] [PubMed] [Google Scholar]
- 38.An S, Bleu T, Hallmark OG, Goetzl EJ. J Biol Chem. 1998;273(14):7906–7910. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- 39.Toews ML, Ediger TL, Romberger DJ, Rennard SI. Biochim Biophys Acta. 2002;1582(13):240–250. doi: 10.1016/s1388-1981(02)00177-4. [DOI] [PubMed] [Google Scholar]
- 40.Sugiura T, Nakane S, Kishimoto S, Waku K, Yoshioka Y, Tokumura A. J Lipid Res. 2002;43(12):2049–2055. doi: 10.1194/jlr.m200242-jlr200. [DOI] [PubMed] [Google Scholar]
- 41.Georas SN, Berdyshev E, Hubbard W, Gorshkova IA, Usatyuk PV, Saatian B, Myers AC, Williams MA, Xiao HQ, Liu M, Natarajan V. Clin Exp Allergy. 2007;37(3):311–322. doi: 10.1111/j.1365-2222.2006.02626.x. [DOI] [PubMed] [Google Scholar]
- 42.Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, Hart WK, Pardo A, Blackwell TS, Xu Y, Chun J, Luster AD. Nat Med. 2008;14(1):45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y, Tong J, Gorshkova IA, He D, Stern R, Berdyshev EV, Pendyala S, Sperling AI, Chun J, Natarajan V. Am J Respir Crit Care Med. 2007;175:A926. [Google Scholar]
- 44.Tokumura A, Miyake M, Nishioka Y, Yamano S, Aono T, Fukuzawa K. Biol Reprod. 1999;61(1):195–199. doi: 10.1095/biolreprod61.1.195. [DOI] [PubMed] [Google Scholar]
- 45.Westermann AM, Havik E, Postma FR, Beijnen JH, Dalesio O, Moolenaar WH, Rodenhuis S. Ann Oncol. 1998;9(4):437–442. doi: 10.1023/a:1008217129273. [DOI] [PubMed] [Google Scholar]
- 46.Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, Bittman R, Tigyi G, Aepfelbacher M. Proc Natl Acad Sci U S A. 1999;96(12):6931–6936. doi: 10.1073/pnas.96.12.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vancura A, Haldar D. J Biol Chem. 1994;269(44):27209–27215. [PubMed] [Google Scholar]
- 48.Hammond LE, Gallagher PA, Wang S, Hiller S, Kluckman KD, Posey-Marcos EL, Maeda N, Coleman RA. Mol Cell Biol. 2002;22(23):8204–8214. doi: 10.1128/MCB.22.23.8204-8214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spiegel S, Milstien S. Biochem Soc Trans. 2005;33(Pt 6):1362–1365. doi: 10.1042/BST0331362. [DOI] [PubMed] [Google Scholar]
- 50.Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S. J Cell Biol. 2005;169(5):801–811. doi: 10.1083/jcb.200407123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luquain C, Singh A, Wang L, Natarajan V, Morris AJ. J Lipid Res. 2003;44(10):1963–1975. doi: 10.1194/jlr.M300188-JLR200. [DOI] [PubMed] [Google Scholar]
- 52.Xie Y, Meier KE. Cell Signal. 2004;16(9):975–981. doi: 10.1016/j.cellsig.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Sano T, Baker D, Virag T, Wada A, Yatomi Y, Kobayashi T, Igarashi Y, Tigyi G. J Biol Chem. 2002;277(24):21197–21206. doi: 10.1074/jbc.M201289200. [DOI] [PubMed] [Google Scholar]
- 54.Aoki J. Semin Cell Dev Biol. 2004;15(5):477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Hiramatsu T, Sonoda H, Takanezawa Y, Morikawa R, Ishida M, Kasahara K, Sanai Y, Taguchi R, Aoki J, Arai H. J Biol Chem. 2003;278(49):49438–49447. doi: 10.1074/jbc.M213018200. [DOI] [PubMed] [Google Scholar]
- 56.Pilquil C, Singh I, Zhang QX, Ling ZC, Buri K, Stromberg LM, Dewald J, Brindley DN. Prostaglandins Other Lipid Mediat. 2001;64(14):83–92. doi: 10.1016/s0090-6980(01)00101-0. [DOI] [PubMed] [Google Scholar]
- 57.Brindley DN, English D, Pilquil C, Buri K, Ling ZC. Biochim Biophys Acta. 2002;1582(13):33–44. doi: 10.1016/s1388-1981(02)00135-x. [DOI] [PubMed] [Google Scholar]
- 58.Zhao Y, Usatyuk PV, Cummings R, Saatian B, He D, Watkins T, Morris A, Spannhake EW, Brindley DN, Natarajan V. Biochem J. 2005;385(Pt 2):493–502. doi: 10.1042/BJ20041160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Waggoner DW, Johnson LB, Mann PC, Morris V, Guastella J, Bajjalieh SM. J Biol Chem. 2004;279(37):38228–38235. doi: 10.1074/jbc.M405932200. [DOI] [PubMed] [Google Scholar]
- 60.Kalari S, Zhao Y, Berdyshev E, Usatyuk PV, He D, Natarajan V. J Invest Medicine. 2007;55(2):S356. [Google Scholar]
- 61.Sato T, Aoki J, Nagai Y, Dohmae N, Takio K, Doi T, Arai H, Inoue K. J Biol Chem. 1997;272(4):2192–2198. doi: 10.1074/jbc.272.4.2192. [DOI] [PubMed] [Google Scholar]
- 62.Horigome K, Hayakawa M, Inoue K, Nojima S. J Biochem. 1987;101(3):625–631. doi: 10.1093/jb/101.3.625. [DOI] [PubMed] [Google Scholar]
- 63.Sekas G, Patton GM, Lincoln EC, Robins SJ. J Lab Clin Med. 1985;105(2):190–194. [PubMed] [Google Scholar]
- 64.Taniyama Y, Shibata S, Kita S, Horikoshi K, Fuse H, Shirafuji H, Sumino Y, Fujino M. Biochem Biophys Res Commun. 1999;257(1):50–56. doi: 10.1006/bbrc.1999.0411. [DOI] [PubMed] [Google Scholar]
- 65.Tokumura A. Biochim Biophys Acta. 2002;1582(13):18–25. doi: 10.1016/s1388-1981(02)00133-6. [DOI] [PubMed] [Google Scholar]
- 66.Fujiwara Y, Sebok A, Meakin S, Kobayashi T, Murakami-Murofushi K, Tigyi G. J Neurochem. 2003;87(5):1272–1283. doi: 10.1046/j.1471-4159.2003.02106.x. [DOI] [PubMed] [Google Scholar]
- 67.Fujiwara Y. Biochim Biophys Acta. 2008 [Google Scholar]
- 68.Gendaszewska-Darmach E. Acta Biochim Pol. 2008;55(2):227–240. [PubMed] [Google Scholar]
- 69.Tsuda S, Okudaira S, Moriya-Ito K, Shimamoto C, Tanaka M, Aoki J, Arai H, Murakami-Murofushi K, Kobayashi T. J Biol Chem. 2006;281(36):26081–26088. doi: 10.1074/jbc.M602925200. [DOI] [PubMed] [Google Scholar]
- 70.Kobayashi T, Tanaka-Ishii R, Taguchi R, Ikezawa H, Murakami-Murofushi K. Life Sci. 1999;65(21):2185–2191. doi: 10.1016/s0024-3205(99)00483-x. [DOI] [PubMed] [Google Scholar]
- 71.Shan L, Li S, Jaffe K, Davis L. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;862(12):161–167. doi: 10.1016/j.jchromb.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Murakami-Murofushi K, Mukai M, Kobayashi S, Kobayashi T, Tigyi G, Murofushi H. Ann N Y Acad Sci. 2000;905:319–321. doi: 10.1111/j.1749-6632.2000.tb06570.x. [DOI] [PubMed] [Google Scholar]
- 73.Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, Liotta LA. J Biol Chem. 1992;267(4):2524–2529. [PubMed] [Google Scholar]
- 74.Clair T, Lee HY, Liotta LA, Stracke ML. J Biol Chem. 1997;272(2):996–1001. doi: 10.1074/jbc.272.2.996. [DOI] [PubMed] [Google Scholar]
- 75.Yang Y, Mou L, Liu N, Tsao MS. Am J Respir Cell Mol Biol. 1999;21(2):216–222. doi: 10.1165/ajrcmb.21.2.3667. [DOI] [PubMed] [Google Scholar]
- 76.Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, Noji S, Yatomi Y, Aoki J, Arai H. J Biol Chem. 2006;281(35):25822–25830. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- 77.Ferry G, Giganti A, Coge F, Bertaux F, Thiam K, Boutin JA. FEBS Lett. 2007;581(18):3572–3578. doi: 10.1016/j.febslet.2007.06.064. [DOI] [PubMed] [Google Scholar]
- 78.Sane AC, Mendenhall T, Bass DA. J Leukoc Biol. 1996;60(6):704–709. doi: 10.1002/jlb.60.6.704. [DOI] [PubMed] [Google Scholar]
- 79.Bowton DL, Seeds MC, Fasano MB, Goldsmith B, Bass DA. Am J Respir Crit Care Med. 1997;155(2):421–425. doi: 10.1164/ajrccm.155.2.9032172. [DOI] [PubMed] [Google Scholar]
- 80.Chilton FH, Averill FJ, Hubbard WC, Fonteh AN, Triggiani M, Liu MC. J Exp Med. 1996;183(5):2235–2245. doi: 10.1084/jem.183.5.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Triggiani M, Oriente A, Seeds MC, Bass DA, Marone G, Chilton FH. J Exp Med. 1995;182(5):1181–1190. doi: 10.1084/jem.182.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kai M, Wada I, Imai S, Sakane F, Kanoh H. J Biol Chem. 1997;272(39):24572–24578. doi: 10.1074/jbc.272.39.24572. [DOI] [PubMed] [Google Scholar]
- 83.Leung DW, Tompkins CK, White T. DNA Cell Biol. 1998;17(4):377–385. doi: 10.1089/dna.1998.17.377. [DOI] [PubMed] [Google Scholar]
- 84.Roberts R, Sciorra VA, Morris AJ. J Biol Chem. 1998;273(34):22059–22067. doi: 10.1074/jbc.273.34.22059. [DOI] [PubMed] [Google Scholar]
- 85.Brindley DN. J Cell Biochem. 2004;92(5):900–912. doi: 10.1002/jcb.20126. [DOI] [PubMed] [Google Scholar]
- 86.West J, Tompkins CK, Balantac N, Nudelman E, Meengs B, White T, Bursten S, Coleman J, Kumar A, Singer JW, Leung DW. DNA Cell Biol. 1997;16(6):691–701. doi: 10.1089/dna.1997.16.691. [DOI] [PubMed] [Google Scholar]
- 87.Eberhardt C, Gray PW, Tjoelker LW. Adv Exp Med Biol. 1999;469:351–356. doi: 10.1007/978-1-4615-4793-8_51. [DOI] [PubMed] [Google Scholar]
- 88.Leung DW. Front Biosci. 2001;6:D944–953. doi: 10.2741/leung. [DOI] [PubMed] [Google Scholar]
- 89.Li D, Yu L, Wu H, Shan Y, Guo J, Dang Y, Wei Y, Zhao S. J Hum Genet. 2003;48(8):438–442. doi: 10.1007/s10038-003-0045-z. [DOI] [PubMed] [Google Scholar]
- 90.Tang W, Yuan J, Chen X, Gu X, Luo K, Li J, Wan B, Wang Y, Yu L. J Biochem Mol Biol. 2006;39(5):626–635. doi: 10.5483/bmbrep.2006.39.5.626. [DOI] [PubMed] [Google Scholar]
- 91.Thompson FJ, Clark MA. Biochem J. 1994;300(Pt 2):457–461. doi: 10.1042/bj3000457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang A, Dennis EA. Biochim Biophys Acta. 1999;1439(1):1–16. doi: 10.1016/s1388-1981(99)00063-3. [DOI] [PubMed] [Google Scholar]
- 93.Wang A, Yang HC, Friedman P, Johnson CA, Dennis EA. Biochim Biophys Acta. 1999;1437(2):157–169. doi: 10.1016/s1388-1981(99)00012-8. [DOI] [PubMed] [Google Scholar]
- 94.Baker RR, Chang HY. Biochim Biophys Acta. 1999;1438(2):253–263. doi: 10.1016/s1388-1981(99)00057-8. [DOI] [PubMed] [Google Scholar]
- 95.Chun J. Crit Rev Neurobiol. 1999;13(2):151–168. doi: 10.1615/critrevneurobiol.v13.i2.20. [DOI] [PubMed] [Google Scholar]
- 96.Ohata H, Aizawa H, Momose K. Life Sci. 1997;60(15):1287–1295. doi: 10.1016/s0024-3205(97)00072-6. [DOI] [PubMed] [Google Scholar]
- 97.Okajima F, Tomura H, Sho K, Kimura T, Sato K, Im DS, Akbar M, Kondo Y. Endocrinology. 1997;138(1):220–229. doi: 10.1210/endo.138.1.4883. [DOI] [PubMed] [Google Scholar]
- 98.Zhou WL, Sugioka M, Yamashita M. J Neurobiol. 1999;41(4):495–504. doi: 10.1002/(sici)1097-4695(199912)41:4<495::aid-neu5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 99.Xu YJ, Tappia PS, Goyal RK, Dhalla NS. J Cell Mol Med. 2008;12(3):942–954. doi: 10.1111/j.1582-4934.2008.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pietruck F, Busch S, Virchow S, Brockmeyer N, Siffert W. Naunyn Schmiedebergs Arch Pharmacol. 1997;355(1):1–7. doi: 10.1007/pl00004906. [DOI] [PubMed] [Google Scholar]
- 101.Thoreson WB, Ryan JS, Shi C, Kelly ME, Bryson EJ, Toews ML, Ediger TL, Chacko DM. Invest Ophthalmol Vis Sci. 2002;43(7):2450–2461. [PubMed] [Google Scholar]
- 102.Peres C, Yart A, Perret B, Salles JP, Raynal P. FEBS Lett. 2003;534(13):164–168. doi: 10.1016/s0014-5793(02)03832-2. [DOI] [PubMed] [Google Scholar]
- 103.Hahn A, Barth H, Kress M, Mertens PR, Goppelt-Struebe M. Biochem J. 2002;362(Pt 1):33–40. doi: 10.1042/0264-6021:3620033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang J, Carbone LD, Watsky MA. Invest Ophthalmol Vis Sci. 2002;43(10):3202–3208. [PubMed] [Google Scholar]
- 105.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. Nature. 1996;383(6600):547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 106.He Q, LaPointe MC. Hypertension. 2001;37(2 Part 2):478–484. doi: 10.1161/01.hyp.37.2.478. [DOI] [PubMed] [Google Scholar]
- 107.Takeda H, Matozaki T, Fujioka Y, Takada T, Noguchi T, Yamao T, Tsuda M, Ochi F, Fukunaga K, Narumiya S, Yamamoto T, Kasuga M. Oncogene. 1998;16(23):3019–3027. doi: 10.1038/sj.onc.1201839. [DOI] [PubMed] [Google Scholar]
- 108.Wu SS, Chiu T, Rozengurt E. Am J Physiol Cell Physiol. 2002;282(6):C1432–1444. doi: 10.1152/ajpcell.00323.2001. [DOI] [PubMed] [Google Scholar]
- 109.Bassa BV, Roh DD, Vaziri ND, Kirschenbaum MA, Kamanna VS. Am J Physiol. 1999;277(3 Pt 2):F328–337. doi: 10.1152/ajprenal.1999.277.3.F328. [DOI] [PubMed] [Google Scholar]
- 110.Lin WW, Chang SH, Wang SM. Br J Pharmacol. 1999;128(6):1189–1198. doi: 10.1038/sj.bjp.0702906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rhim JH, Jang IS, Yeo EJ, Song KY, Park SC. Aging Cell. 2006;5(6):451–461. doi: 10.1111/j.1474-9726.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- 112.Seewald S, Schmitz U, Seul C, Ko Y, Sachinidis A, Vetter H. Am J Hypertens. 1999;12(5):532–537. doi: 10.1016/s0895-7061(98)00269-6. [DOI] [PubMed] [Google Scholar]
- 113.Zhao Y, He D, Stern R, Usatyuk PV, Spannhake EW, Salgia R, Natarajan V. Cell Signal. 2007;19(11):2329–2338. doi: 10.1016/j.cellsig.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JG, Watkins T, He D, Saatian B, Natarajan V. J Biol Chem. 2004;279(39):41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 115.He D, Natarajan V, Stern R, Gorshkova IA, Solway J, Spannhake EW, Zhao Y. Biochem J. 2008;412(1):153–162. doi: 10.1042/BJ20071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hong JH, Oh SO, Lee M, Kim YR, Kim DU, Hur GM, Lee JH, Lim K, Hwang BD, Park SK. Biochem Biophys Res Commun. 2001;281(5):1337–1342. doi: 10.1006/bbrc.2001.4517. [DOI] [PubMed] [Google Scholar]
- 117.Qi C, Park JH, Gibbs TC, Shirley DW, Bradshaw CD, Ella KM, Meier KE. J Cell Physiol. 1998;174(2):261–272. doi: 10.1002/(SICI)1097-4652(199802)174:2<261::AID-JCP13>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 118.Shin I, Kweon SM, Lee ZW, Kim SI, Joe CO, Kim JH, Park YM, Ha KS. Mol Cells. 1999;9(3):292–299. [PubMed] [Google Scholar]
- 119.Tou JS, Gill JS. Cell Signal. 2005;17(1):77–82. doi: 10.1016/j.cellsig.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 120.Wang L, Cummings R, Zhao Y, Kazlauskas A, Sham JK, Morris A, Georas S, Brindley DN, Natarajan V. J Biol Chem. 2003;278(41):39931–39940. doi: 10.1074/jbc.M302896200. [DOI] [PubMed] [Google Scholar]
- 121.Gohla A, Harhammer R, Schultz G. J Biol Chem. 1998;273(8):4653–4659. doi: 10.1074/jbc.273.8.4653. [DOI] [PubMed] [Google Scholar]
- 122.Gschwind A, Prenzel N, Ullrich A. Cancer Res. 2002;62(21):6329–6336. [PubMed] [Google Scholar]
- 123.Kassel KM, Dodmane PR, Schulte NA, Toews ML. J Pharmacol Exp Ther. 2008;325(3):809–817. doi: 10.1124/jpet.107.133736. [DOI] [PubMed] [Google Scholar]
- 124.Kassel KM, Schulte NA, Parker SM, Lanik AD, Toews ML. J Pharmacol Exp Ther. 2007;323(1):109–118. doi: 10.1124/jpet.107.120584. [DOI] [PubMed] [Google Scholar]
- 125.Mori K, Kitayama J, Shida D, Yamashita H, Watanabe T, Nagawa H. J Surg Res. 2006;132(1):56–61. doi: 10.1016/j.jss.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 126.Shida D, Fang X, Kordula T, Takabe K, Lepine S, Alvarez SE, Milstien S, Spiegel S. Cancer Res. 2008;68(16):6569–6577. doi: 10.1158/0008-5472.CAN-08-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, Nagawa H. World J Gastroenterol. 2005;11(36):5638–5643. doi: 10.3748/wjg.v11.i36.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. J Biol Chem. 2006;281(28):19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Goppelt-Struebe M, Fickel S, Reiser CO. Biochem J. 2000;345(Pt 2):217–224. [PMC free article] [PubMed] [Google Scholar]
- 130.Kang S, Luo R, Smicun Y, Fishman DA, Meng Y. FEBS Lett. 2006;580(2):443–449. doi: 10.1016/j.febslet.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 131.Lin CI, Chen CN, Huang MT, Lee SJ, Lin CH, Chang CC, Lee H. Cell Signal. 2008;20(10):1804–1814. doi: 10.1016/j.cellsig.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 132.Oyesanya RA, Lee ZP, Wu J, Chen J, Song Y, Mukherjee A, Dent P, Kordula T, Zhou H, Fang X. Faseb J. 2008 doi: 10.1096/fj.07-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Symowicz J, Adley BP, Woo MM, Auersperg N, Hudson LG, Stack MS. Cancer Res. 2005;65(6):2234–2242. doi: 10.1158/0008.5472.CAN-04-2781. [DOI] [PubMed] [Google Scholar]
- 134.Shahrestanifar M, Fan X, Manning DR. J Biol Chem. 1999;274(6):3828–3833. doi: 10.1074/jbc.274.6.3828. [DOI] [PubMed] [Google Scholar]
- 135.Saatian B, Zhao Y, He D, Georas SN, Watkins T, Spannhake EW, Natarajan V. Biochem J. 2006;393(Pt 3):657–668. doi: 10.1042/BJ20050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhao Y, He D, Zhao J, Wang L, Leff AR, Spannhake EW, Georas S, Natarajan V. J Biol Chem. 2007;282(14):10172–10179. doi: 10.1074/jbc.M611210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fang X, Yu S, Bast RC, Liu S, Xu HJ, Hu SX, LaPushin R, Claret FX, Aggarwal BB, Lu Y, Mills GB. J Biol Chem. 2004;279(10):9653–9661. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- 138.Barekzi E, Roman J, Hise K, Georas S, Steinke JW. Prostaglandins Leukot Essent Fatty Acids. 2006;74(6):357–363. doi: 10.1016/j.plefa.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 139.Saatian B, zhao Y, watkins T, He D, Spannhake EWm, Natarajan V. Proc Am Thorac Soc. 2005;2:A756. [Google Scholar]
- 140.Chen R, Roman JM, Belmote K, Guo J, Georas SN. Proc Am Thorac Soc. 2005;2:A366. [Google Scholar]
- 141.Martin LD, Rochelle LG, Fischer BM, Krunkosky TM, Adler KB. Eur Respir J. 1997;10(9):2139–2146. doi: 10.1183/09031936.97.10092139. [DOI] [PubMed] [Google Scholar]
- 142.Raeburn D, Webber SE. Eur Respir J. 1994;7(12):2226–2233. doi: 10.1183/09031936.94.07122226. [DOI] [PubMed] [Google Scholar]
- 143.Message SD, Johnston SL. J Leukoc Biol. 2004;75(1):5–17. doi: 10.1189/jlb.0703315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Elias JA. Am J Respir Crit Care Med. 2000;161(3 Pt 2):S168–171. doi: 10.1164/ajrccm.161.supplement_2.a1q4-4. [DOI] [PubMed] [Google Scholar]
- 145.Toews ML, Ustinova EE, Schultz HD. J Appl Physiol. 1997;83(4):1216–1222. doi: 10.1152/jappl.1997.83.4.1216. [DOI] [PubMed] [Google Scholar]
- 146.Aggarwal A, Baker CS, Evans TW, Haslam PL. Eur Respir J. 2000;15(5):895–901. doi: 10.1034/j.1399-3003.2000.15e14.x. [DOI] [PubMed] [Google Scholar]
- 147.Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Am J Respir Crit Care Med. 1996;154(3 Pt 1):602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 148.Hashimoto T, Yamashita M, Ohata H, Momose K. J Pharmacol Sci. 2003;91(1):8–14. doi: 10.1254/jphs.91.8. [DOI] [PubMed] [Google Scholar]
- 149.Hashimoto T, Nakano Y, Yamashita M, Fang YI, Ohata H, Momose K. Jpn J Pharmacol. 2002;88(3):256–261. doi: 10.1254/jjp.88.256. [DOI] [PubMed] [Google Scholar]
- 150.Huang SK, Xiao HQ, Kleine-Tebbe J, Paciotti G, Marsh DG, Lichtenstein LM, Liu MC. J Immunol. 1995;155(5):2688–2694. [PubMed] [Google Scholar]
- 151.Bodey KJ, Semper AE, Redington AE, Madden J, Teran LM, Holgate ST, Frew AJ. Allergy. 1999;54(10):1083–1093. doi: 10.1034/j.1398-9995.1999.00889.x. [DOI] [PubMed] [Google Scholar]
- 152.Henderson WR, Jr, Tang LO, Chu SJ, Tsao SM, Chiang GK, Jones F, Jonas M, Pae C, Wang H, Chi EY. Am J Respir Crit Care Med. 2002;165(1):108–116. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- 153.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Science. 1998;282(5397):2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 154.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. J Clin Invest. 1999;103(6):779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Taube C, Duez C, Cui ZH, Takeda K, Rha YH, Park JW, Balhorn A, Donaldson DD, Dakhama A, Gelfand EW. J Immunol. 2002;169(11):6482–6489. doi: 10.4049/jimmunol.169.11.6482. [DOI] [PubMed] [Google Scholar]
- 156.Zurawski SM, Chomarat P, Djossou O, Bidaud C, McKenzie AN, Miossec P, Banchereau J, Zurawski G. J Biol Chem. 1995;270(23):13869–13878. doi: 10.1074/jbc.270.23.13869. [DOI] [PubMed] [Google Scholar]
- 157.Kotowicz K, Callard RE, Friedrich K, Matthews DJ, Klein N. Int Immunol. 1996;8(12):1915–1925. doi: 10.1093/intimm/8.12.1915. [DOI] [PubMed] [Google Scholar]
- 158.Zhang JG, Hilton DJ, Willson TA, McFarlane C, Roberts BA, Moritz RL, Simpson RJ, Alexander WS, Metcalf D, Nicola NA. J Biol Chem. 1997;272(14):9474–9480. doi: 10.1074/jbc.272.14.9474. [DOI] [PubMed] [Google Scholar]
- 159.Donaldson DD, Whitters MJ, Fitz LJ, Neben TY, Finnerty H, Henderson SL, O’Hara RM, Jr, Beier DR, Turner KJ, Wood CR, Collins M. J Immunol. 1998;161(5):2317–2324. [PubMed] [Google Scholar]
- 160.Kawakami K, Taguchi J, Murata T, Puri RK. Blood. 2001;97(9):2673–2679. doi: 10.1182/blood.v97.9.2673. [DOI] [PubMed] [Google Scholar]
- 161.Rahaman SO, Sharma P, Harbor PC, Aman MJ, Vogelbaum MA, Haque SJ. Cancer Res. 2002;62(4):1103–1109. [PubMed] [Google Scholar]
- 162.Tanabe T, Fujimoto K, Yasuo M, Tsushima K, Yoshida K, Ise H, Yamaya M. Clin Exp Allergy. 2008;38(1):122–134. doi: 10.1111/j.1365-2222.2007.02871.x. [DOI] [PubMed] [Google Scholar]
- 163.Zheng T, Liu W, Oh SY, Zhu Z, Hu B, Homer RJ, Cohn L, Grusby MJ, Elias JA. J Immunol. 2008;180(1):522–529. doi: 10.4049/jimmunol.180.1.522. [DOI] [PubMed] [Google Scholar]
- 164.Chiaramonte MG, Mentink-Kane M, Jacobson BA, Cheever AW, Whitters MJ, Goad ME, Wong A, Collins M, Donaldson DD, Grusby MJ, Wynn TA. J Exp Med. 2003;197(6):687–701. doi: 10.1084/jem.20020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. Nat Med. 2006;12(1):99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 166.Daines MO, Hershey GK. J Biol Chem. 2002;277(12):10387–10393. doi: 10.1074/jbc.M108109200. [DOI] [PubMed] [Google Scholar]
- 167.Heller NM, Matsukura S, Georas SN, Boothby MR, Rothman PB, Stellato C, Schleimer RP. Am J Respir Cell Mol Biol. 2004;31(5):573–582. doi: 10.1165/rcmb.2004-0195OC. [DOI] [PubMed] [Google Scholar]
- 168.Rubenfeld J, Guo J, Sookrung N, Chen R, Chaicumpa W, Casolaro V, Zhao Y, Natarajan V, Georas S. Am J Physiol Lung Cell Mol Physiol. 2006;290(1):L66–74. doi: 10.1152/ajplung.00473.2004. [DOI] [PubMed] [Google Scholar]
- 169.Ghosh M, Stewart A, Tucker DE, Bonventre JV, Murphy RC, Leslie CC. Am J Respir Cell Mol Biol. 2004;30(1):91–100. doi: 10.1165/rcmb.2003-0005OC. [DOI] [PubMed] [Google Scholar]
- 170.Uchida K, Kumihashi K, Kurosawa S, Kobayashi T, Itoi K, Machida T. Zoolog Sci. 2002;19(11):1211–1216. doi: 10.2108/zsj.19.1211. [DOI] [PubMed] [Google Scholar]
- 171.Blanco-Rivero J, Cachofeiro V, Lahera V, Aras-Lopez R, Marquez-Rodas I, Salaices M, Xavier FE, Ferrer M, Balfagon G. Hypertension. 2005;46(1):107–112. doi: 10.1161/01.HYP.0000171479.36880.17. [DOI] [PubMed] [Google Scholar]
- 172.Dempke W, Rie C, Grothey A, Schmoll HJ. J Cancer Res Clin Oncol. 2001;127(7):411–417. doi: 10.1007/s004320000225. [DOI] [PubMed] [Google Scholar]
- 173.Gately S, Li WW. Semin Oncol. 2004;31(2 Suppl 7):2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 174.Krysan K, Reckamp KL, Sharma S, Dubinett SM. Anticancer Agents Med Chem. 2006;6(3):209–220. doi: 10.2174/187152006776930882. [DOI] [PubMed] [Google Scholar]
- 175.Inoue W, Matsumura K, Yamagata K, Takemiya T, Shiraki T, Kobayashi S. Neurosci Res. 2002;44(1):51–61. doi: 10.1016/s0168-0102(02)00083-4. [DOI] [PubMed] [Google Scholar]
- 176.Oguma T, Asano K, Shiomi T, Fukunaga K, Suzuki Y, Nakamura M, Matsubara H, Sheldon HK, Haley KJ, Lilly CM, Drazen JM, Yamaguchi K. Am J Respir Crit Care Med. 2002;165(3):382–386. doi: 10.1164/ajrccm.165.3.2103093. [DOI] [PubMed] [Google Scholar]
- 177.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. Trends Immunol. 2004;25(1):40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 178.Gauvreau GM, Watson RM, O’Byrne PM. Am J Respir Crit Care Med. 1999;159(1):31–36. doi: 10.1164/ajrccm.159.1.9804030. [DOI] [PubMed] [Google Scholar]
- 179.Walters EH, Bevan C, Parrish RW, Davies BH, Smith AP. Thorax. 1982;37(6):438–442. doi: 10.1136/thx.37.6.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ghosh S, May MJ, Kopp EB. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 181.Mastronarde JG, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. J Infect Dis. 1998;177(5):1275–1281. doi: 10.1086/515279. [DOI] [PubMed] [Google Scholar]
- 182.Wu KK, Liou JY, Cieslik K. Arterioscler Thromb Vasc Biol. 2005;25(4):679–685. doi: 10.1161/01.ATV.0000157899.35660.61. [DOI] [PubMed] [Google Scholar]
- 183.Wu AH, Low WC. Neuro Oncol. 2003;5(3):179–187. doi: 10.1215/S1152-8517-02-00051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wedel A, Ziegler-Heitbrock HW. Immunobiology. 1995;193(24):171–185. doi: 10.1016/s0171-2985(11)80541-3. [DOI] [PubMed] [Google Scholar]
- 185.Caivano M, Gorgoni B, Cohen P, Poli V. J Biol Chem. 2001;276(52):48693–48701. doi: 10.1074/jbc.M108282200. [DOI] [PubMed] [Google Scholar]
- 186.Gorgoni B, Caivano M, Arizmendi C, Poli V. J Biol Chem. 2001;276(44):40769–40777. doi: 10.1074/jbc.M106865200. [DOI] [PubMed] [Google Scholar]
- 187.Hazzalin CA, Mahadevan LC. Nat Rev Mol Cell Biol. 2002;3(1):30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- 188.Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Mol Cell Biol. 2000;20(12):4265–4274. doi: 10.1128/mcb.20.12.4265-4274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.He D, Zhao Y, Stern R, Gorshkova IA, Kalari S, Spannhake EWm, Natarajan V. Am J Respir Crit Care Med. 2007;175:A279. [Google Scholar]
- 190.Mellor H, Parker PJ. Biochem J. 1998;332(Pt 2):281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Manji HK, Lenox RH. Synapse. 1994;16(1):11–28. doi: 10.1002/syn.890160103. [DOI] [PubMed] [Google Scholar]
- 192.Wooten MW. J Neurosci Res. 1999;58(5):607–611. doi: 10.1002/(sici)1097-4547(19991201)58:5<607::aid-jnr1>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 193.Gorshkova I, He D, Berdyshev E, Usatuyk P, Burns M, Kalari S, Zhao Y, Pendyala S, Garcia JG, Pyne NJ, Brindley DN, Natarajan V. J Biol Chem. 2008;283(17):11794–11806. doi: 10.1074/jbc.M800250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nanjundan M, Possmayer F. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1484–1493. doi: 10.1152/ajplung.2001.281.6.L1484. [DOI] [PubMed] [Google Scholar]
- 195.Nanjundan M, Possmayer F. Biochem J. 2001;358(Pt 3):637–646. doi: 10.1042/0264-6021:3580637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nanjundan M, Possmayer F. Am J Physiol Lung Cell Mol Physiol. 2003;284(1):L1–23. doi: 10.1152/ajplung.00029.2002. [DOI] [PubMed] [Google Scholar]
- 197.Natarajan K, Berk BC. Methods Mol Biol. 2006;332:51–77. doi: 10.1385/1-59745-048-0:51. [DOI] [PubMed] [Google Scholar]
- 198.Pyne NJ, Waters CM, Long JS, Moughal NA, Tigyi G, Pyne S. Adv Enzyme Regul. 2007;47:271–280. doi: 10.1016/j.advenzreg.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liebmann C, Bohmer FD. Curr Med Chem. 2000;7(9):911–943. doi: 10.2174/0929867003374589. [DOI] [PubMed] [Google Scholar]
- 200.Pyne NJ, Pyne S. Biochim Biophys Acta. 2008 [Google Scholar]
- 201.Waters C, Sambi B, Kong KC, Thompson D, Pitson SM, Pyne S, Pyne NJ. J Biol Chem. 2003;278(8):6282–6290. doi: 10.1074/jbc.M208560200. [DOI] [PubMed] [Google Scholar]
- 202.Wang C, Wu LL, Liu J, Zhang ZG, Fan D, Li L. Chin Med J (Engl) 2008;121(3):236–240. [PubMed] [Google Scholar]
- 203.Bonner JC. Cytokine Growth Factor Rev. 2004;15(4):255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 204.Ingram JL, Bonner JC. Curr Mol Med. 2006;6(4):409–421. doi: 10.2174/156652406777435426. [DOI] [PubMed] [Google Scholar]
- 205.Szabo S, Sandor Z. Baillieres Clin Gastroenterol. 1996;10(1):97–112. doi: 10.1016/s0950-3528(96)90042-1. [DOI] [PubMed] [Google Scholar]
- 206.Kluk MJ, Colmont C, Wu MT, Hla T. FEBS Lett. 2003;533(13):25–28. doi: 10.1016/s0014-5793(02)03742-0. [DOI] [PubMed] [Google Scholar]
- 207.Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA. Atherosclerosis. 2006;186(1):38–53. doi: 10.1016/j.atherosclerosis.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 208.Laffargue M, Raynal P, Yart A, Peres C, Wetzker R, Roche S, Payrastre B, Chap H. J Biol Chem. 1999;274(46):32835–32841. doi: 10.1074/jbc.274.46.32835. [DOI] [PubMed] [Google Scholar]
- 209.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. J Embo. 1997;16(23):7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shah BH, Baukal AJ, Shah FB, Catt KJ. Mol Endocrinol. 2005;19(10):2535–2548. doi: 10.1210/me.2005-0082. [DOI] [PubMed] [Google Scholar]
- 211.Umata T, Hirata M, Takahashi T, Ryu F, Shida S, Takahashi Y, Tsuneoka M, Miura Y, Masuda M, Horiguchi Y, Mekada E. J Biol Chem. 2001;276(32):30475–30482. doi: 10.1074/jbc.M103673200. [DOI] [PubMed] [Google Scholar]
- 212.Zhao Y, He D, Saatian B, Watkins T, Spannhake EWm, Natarajan V. Proc Am Thorac Soc. 2005;2:A25. [Google Scholar]
- 213.Kim SN, Park JG, Lee EB, Kim SS, Yoo YS. J Cell Biochem. 2000;76(3):386–393. [PubMed] [Google Scholar]
- 214.Miyamoto S, Hirata M, Yamazaki A, Kageyama T, Hasuwa H, Mizushima H, Tanaka Y, Yagi H, Sonoda K, Kai M, Kanoh H, Nakano H, Mekada E. Cancer Res. 2004;64(16):5720–5727. doi: 10.1158/0008-5472.CAN-04-0811. [DOI] [PubMed] [Google Scholar]
- 215.Rubin JS, Bottaro DP, Aaronson SA. Biochim Biophys Acta. 1993;1155(3):357–371. doi: 10.1016/0304-419x(93)90015-5. [DOI] [PubMed] [Google Scholar]
- 216.Rosen EM, Nigam SK, Goldberg ID. J Cell Biol. 1994;127(6 Pt 2):1783–1787. doi: 10.1083/jcb.127.6.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Jafri NF, Ma PC, Maulik G, Salgia R. J Environ Pathol Toxicol Oncol. 2003;22(3):147–165. doi: 10.1615/jenvpathtoxoncol.v22.i3.10. [DOI] [PubMed] [Google Scholar]
- 218.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, Johnson BE, Salgia R. Cancer Res. 2003;63(19):6272–6281. [PubMed] [Google Scholar]
- 219.Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, Yatomi Y, Nagawa H. FEBS Lett. 2004;577(3):333–338. doi: 10.1016/j.febslet.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 220.Hashigasako A, Machide M, Nakamura T, Matsumoto K, Nakamura T. J Biol Chem. 2004;279(25):26445–26452. doi: 10.1074/jbc.M314254200. [DOI] [PubMed] [Google Scholar]
- 221.Bush KT, Tsukamoto T, Nigam SK. Am J Physiol Renal Physiol. 2000;278(5):F847–852. doi: 10.1152/ajprenal.2000.278.5.F847. [DOI] [PubMed] [Google Scholar]
- 222.Chen X, Gumbiner BM. Curr Opin Cell Biol. 2006;18(5):572–578. doi: 10.1016/j.ceb.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 223.Miyoshi J, Takai Y. Adv Drug Deliv Rev. 2005;57(6):815–855. doi: 10.1016/j.addr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 224.Huntley GW, Gil O, Bozdagi O. Neuroscientist. 2002;8(3):221–233. doi: 10.1177/1073858402008003008. [DOI] [PubMed] [Google Scholar]
- 225.Wheelock MJ, Johnson KR. Curr Opin Cell Biol. 2003;15(5):509–514. doi: 10.1016/s0955-0674(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 226.Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W. J Cell Biol. 1993;120(3):757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Otto T, Rembrink K, Goepel M, Meyer-Schwickerath M, Rubben H. Urol Res. 1993;21(5):359–362. doi: 10.1007/BF00296837. [DOI] [PubMed] [Google Scholar]
- 228.Yang JY, Zong CS, Xia W, Wei Y, Ali-Seyed M, Li Z, Broglio K, Berry DA, Hung MC. Mol Cell Biol. 2006;26(19):7269–7282. doi: 10.1128/MCB.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Goto Y, Uchida Y, Nomura A, Sakamoto T, Ishii Y, Morishima Y, Masuyama K, Sekizawa K. Am J Respir Cell Mol Biol. 2000;23(6):712–718. doi: 10.1165/ajrcmb.23.6.4031. [DOI] [PubMed] [Google Scholar]
- 230.Evans SM, Blyth DI, Wong T, Sanjar S, West MR. Am J Respir Cell Mol Biol. 2002;27(4):446–454. doi: 10.1165/rcmb.4776. [DOI] [PubMed] [Google Scholar]
- 231.Zabner J, Winter M, Excoffon KJ, Stoltz D, Ries D, Shasby S, Shasby M. J Appl Physiol. 2003;95(1):394–401. doi: 10.1152/japplphysiol.01134.2002. [DOI] [PubMed] [Google Scholar]
- 232.He D, Usatyuk PV, Spannhake EWm, Natarajan V, Zhao Y. Am J Respir Crit Care Med. 2008;175:A726. [Google Scholar]
- 233.Sun Y, McGarrigle D, Huang X-Y. Mol BioSyst. 2007;3:849–854. doi: 10.1039/b706343a. [DOI] [PubMed] [Google Scholar]
- 234.Fan H, Zingarelli B, Harris V, Tempel GE, Halushka PV, Cook JA. Mol Med. 2008;14(78):422–428. doi: 10.2119/2007-00106.Fan. [DOI] [PMC free article] [PubMed] [Google Scholar]