

Abstract
Tafazzin is a conserved mitochondrial protein that is required to maintain normal content and composition of cardiolipin. We used electron tomography to investigate the effect of tafazzin deletion on mitochondrial structure and found that cellular differentiation plays a crucial role in the manifestation of abnormalities. This conclusion was reached by comparing differentiated cardiomyocytes with embryonic stem cells from mouse and by comparing different tissues from Drosophila melanogaster. The data suggest that tafazzin deficiency affects cardiolipin in all mitochondria, but significant alterations of the ultrastrcture, such as remodeling and aggregation of inner membranes, will only occur after specific differentiation.
Keywords: Tafazzin, Cardiolipin, Mitochondria, Barth Syndrome, Electron Microscopy, Electron Tomography
1. Introduction
Tafazzin is a phospholipid-lysophospholipid transacylase that is essential for the synthesis of the remodeled molecular species of mitochondrial cardiolipin (Xu et al. 2006a). Although the cardiolipin composition is heterogeneous, it is normally dominated by one or two types of fatty acids. Deletion or mutation of tafazzin increases this heterogeneity, increases the proportion of monolysocardiolipin, and reduces the overall abundance of cardiolipin. This specific combination of changes has been observed in yeast and fruit flies (Gu et al. 2004; Xu et al. 2006b), and humans (Vreken et al. 2000; Schlame et al. 2002; Valianpour et al. 2002; Schlame et al. 2003; Valianpour et al. 2005), although the exact concentration and submitochondrial localization of monolysocardiolipin has not been determined.
Not surprisingly, the changes in cardiolipin content and composition have been associated with a number of alterations in mitochondrial structure and function. Early on, abnormal mitochondria have been described in tissue biopsies from patients with tafazzin mutations (Barth et al. 1983; Ino et al. 1988; Hug et al. 1990; Christodoulou et al. 1994). In lymphoblast cultures from those patients, we found mitochondrial hyperproliferation and abnormal size distribution, as well as adhesion zones between mitochondrial inner membranes (Xu et al. 2005; Acehan et al. 2007). Mitochondria with tafazzin deletion show reduced coupling, reduced membrane potential, and a blunted swelling-shrinking response (Ma et al. 2004; Xu et al. 2005). The bioenergetic changes may be a consequence of impaired complex assembly and poor supercomplex stability, two features that have been described in tafazzin-deficient mitochondria (Brandner et al. 2005; McKenzie et al. 2006). Thus, the emerging view is that cardiolipin remodeling is essential for the assembly of protein complexes and supercomplexes responsible for oxidative phosphorylation and perhaps for creating the domain architecture of the inner membrane.
In humans, tafazzin mutations cause a disease known as Barth syndrome (Barth et al. 1999; Spencer et al. 2006). In its most characteristic expression, Barth syndrome includes cardiomyopathy, skeletal myopathy, neutropenia, chronic fatigue, and growth disturbances. However, there is considerable variability in the clinical presentation, i.e. some patients may have only one or two of the cardinal symptoms while others may be largely unaffected. Furthermore, episodes of severe illness may be followed by extended periods of remission, in which patients are close to normal. Despite this clinical variability, similar changes in the cardiolipin composition have been found in all Barth patients studied to date (Schlame et al. 2002; Schlame et al. 2003; van Werkhoven et al. 2006; Kulik et al. 2008). Thus it is surprising that a disease, consistently affecting a crucial component of the mitochondrial inner membrane, has consequences only in selected tissues and only at certain times. This raises the question as to what makes mitochondria susceptible to tafazzin deficiency and how does this susceptibility change as mitochondria develop and differentiate.
2. Materials and methods
2.1. Generation of a tafazzin knockout mutation in mouse embryonic stem cells
The tafazzin (Taz) gene targeting vector was constructed by the recombineering-based method (Liu et al. 2003). Electroporation of targeting constructs, manipulation of embryonic stem cells, and clone selection were performed as described (Matise et al. 2003). Cre-recombinase expressing plasmid pCAG-Cre (Matsuda et al. 2007) was electroporated into the Tazflox embryonic stem cells and the cells were plated on top of a feeder layer of inactivated mouse embryonic fibroblasts. Cultures were grown until isolated stem cell colonies appeared. Stem cell colonies were screened by PCR with primers P1 (5′-GAGGTAGGCTTGCTCATTCTTTGGC-3′) and P2 (5′-CTTCTACCCTTCTGACATTCTCTAAC-3′). We isolated and expanded clones, in which the first four exons of the tafazzin gene were deleted and verified the deletion by RT-PCR analysis. To this end, total RNA was extracted using Trizol reagent (Invitrogen) and 4 μg were used for oligo-dT primed reverse transcription. An aliquot of the cDNA product was used for PCR amplification with the primers P3 (5′-CCCTCCATGTGAAGTGGCCATTCC-3′) and P4 (5′-CCTCTTGAATGAAGTCTGTGAGGGCTT-3′).
2.2. Differentiation of stem cells into cardiomyocytes
Embryonic stem cells were cultured as 3-dimensional aggregates in suspension to form embryoid bodies. After 3 days, the embryoid bodies were transferred to gelatin-coated 6 well dishes (tissue culture grade) or gelatin-coated glass cover slips, to form attached cultures. Cells were grown for an additional two weeks in the presence of Noggin to promote differentiation into cardiomyocytes (Yuasa et al. 2005). For further analysis, cells were washed 3 times with phosphate-buffered saline and mechanically detached from tissue culture plates. Cells were collected by centrifugation.
2.3. Drosophila Culture
We created a tafazzin mutant of Drosophila melanogaster by imprecise excision of a P-element inserted upstream of the coding region of the gene, which resulted in deletion of the full-length isoform of tafazzin (isoform A) and cardiolipin deficiency (Xu et al. 2006b). Mutants and precise-excision controls were grown in 7.5 cm culture vials at 22°C in a standard cornmeal-sucrose-yeast medium. Flies were examined during the second week of their adult life.
2.4. Preparation of cultured cells for electron microscopy
Cells were treated for 20 minutes with primary fixative (2% glutaraldehyde in 0.1M Pipes buffer, pH 7.0). The prefixed cells were collected into centrifuge tubes and pelleted at 3000 r.p.m. in a benchtop centrifuge. The supernatant was replaced with fresh primary fixative and cells were incubated overnight at 4°C. The cell pellets were then incubated for 20 minutes on ice in secondary fixative (2% glutaraldehyde and 0.2% tannic acid in 0.1 M Pipes buffer, pH 7.0). Fixed cells were washed extensively and treated with 1% OsO4 in 0.1 M Pipes buffer for 60 minutes at 4°C. This was followed by three washes for 10 minutes in 0.1 M Pipes buffer, two rinses in water, and en bloc staining in 1% aqueous uranyl acetate.
2.5. Preparation of Drosophila for electron microscopy
Head, wings and hind tip of the flies were removed to facilitate penetration of fixatives (see above). The remaining parts of the flies were incubated in a series of solutions, including 4% paraformaldehyde/2% glutaraldehyde for 1 hour, 4% glutaraldehyde/0.5% tannic acid for 1 hour, 4% glutaraldehyde overnight at 4°C, 1.5% OsO4 for 30 minutes at room temperature, 1.5% OsO4 for 30 minutes at 4°C, and 2% aqueous uranyl acetate for 1 hour, with repeated washes in between steps.
2.6. Sectioning and staining
For all samples, conventional chemical fixation was followed by a series of ethanol substitutions from 10% to 100% ethanol, using incrementally decreasing temperatures from 20°C to −35°C. After the samples were fully dehydrated, they were infiltrated with LX-112 epoxy resin over a period of 18 hours, slowly increasing the resin content to 100%. Samples were then heat cured in resin at 60°C and sectioned to a thickness of 50–100 nm. Sections were collected on plastic coated copper EM grids and stained with 2% aqueous uranyl acetate in water and Sato Lead Solution. Stained sample grids were screened for well preserved areas using a Phillips CM12 electron microscope. The major Drosophila flight muscles can be easily identified in the thorax. Drosophila heart is attached to the dorsal wall and runs from the last abdominal segment to the cerebral hemispheres with chambers that can be located with respect to other features such as abdominal segments (Wasserthal 2007). The identities of heart and flight muscle cells were further verified by the presence of striated fibers. Twenty well preserved sections were analyzed for each tissue.
2.7. Electron microscopic tomography
Selected EM grids with good sections were coated with a thin layer of carbon and colloidal gold markers were randomly attached via poly-l-lysine. Samples were tilted between −70° and +70° at one-degree intervals and electron micrographs were recorded at 9,000 to 25,000 fold magnifications with a Tecnai TF20 microscope (FEI Corporation, Hillsboro, OR, USA) equipped with a 4k × 4k CCD camera (TVIPS, Gauting, Germany). For this procedure, we used a high tilt sample holder (Fischione, Export, PA, USA) and the SerialEM program for automated data collection (Mastronarde 2005). A second tilt series of the same area was collected after manually rotating the specimen support by ninety degrees. Dual-axis tomographic reconstructions were calculated using colloidal gold as fiducial marker for alignment in IMOD (Kremer et al. 1996). Features of interest within the tomogram volumes were segmented manually using the software AMIRA (Mercury Computer Systems, San Diego, CA, USA).
2.8. Evaluation of heart and muscle function in Drosophila
The cardiac function of Drosophila strains was evaluated by optical coherence tomography as described previously (Wolf et al. 2006). M-mode images were obtained in the transverse plane of awake immobilized flies at room temperature. Cardiac chamber dimensions were measured from the inner edge of the superior and inferior walls during mid-diastole for end-diastolic dimension (EDD) and mid-systole for end-systolic dimension (ESD). Fractional shortening was calculated as (EDD-ESD)/EDD. Heart rate was calculated from 2.5 seconds M-mode tracings. Muscle function was evaluated by locomotor assays as described (Xu et al. 2006b). The flying score was calculated from the distance an animal crossed in its first attempt to fly on a 75×75 cm airfield. Flies scored one point for each unit of 6 cm up to a maximal score of 6. The climbing score was measured by countercurrent distribution (Benzer 1967). Flies, placed at the bottom of an 11 cm tube, were given 20 seconds to climb into an inverted tube placed on top. Flies that successfully climbed into the upper tube were given another chance to climb and so forth. Flies scored one point for every tube they climbed out of.
2.9. Isolation of mitochondria and submitochondrial fractions
Mitochondria were isolated from rat liver and whole flies by differential centrifugation. Homogenates were prepared in isolation buffer (0.15 M sucrose, 0.05 M KCl, 0.02 M Hepes, 1 mM EDTA, 2 mM 2-mercaptoethanol, pH 7.4) at 4°C, using a tight-fitting Teflon-glass homogenizer. Nuclei and debris were removed by centrifugation at 750 × g for 10 minutes. Mitochondria were spun at 17,000 × g for 10 minutes, washed and then re-collected. Submitochondrial fractions were prepared according to Greenawalt (Greenawalt 1974). Briefly, mitochondria (protein concentration of about 50 g/l) were treated with digitonin for 12 minutes on ice, using a digitonin/protein mass ratio of 1:10. The treatment was stopped by 3-fold dilution with cold isolation buffer. Mitoplasts (inner membranes plus matrix) were collected by centrifugation at 15,000 × g for 5 minutes and outer membranes were collected from the post-mitoplast supernatant by centrifugation at 100,000 × g for 60 minutes.
2.10. Analysis of cardiolipin and monolysocardiolipin in cell cultures
Phospholipids were extracted from lyophilized embryonic stem cells or cardiomyocytes. The cells were resuspended in 1 ml demineralized water, followed by the addition of 3 ml chloroform-methanol (2:1, v/v) and internal standard (0.4 nmol of tetramyristoyl-cardiolipin, Avanti Polar Lipids, Alabaster, AL). After 20 minutes of sonication in a sonicator bath, the mixture was cooled on ice for 15 minutes and centrifuged for 10 minutes at 1,000 × g. The lower phase was transferred to another tube and the upper phase was re-extracted with 3 ml chloroform-methanol (2:1, v/v). The combined organic phases were dried and dissolved in 150 μl chloroform/methanol/water (50:45:5, by volume), containing 0.01% ammonia. An aliquot of 10 μl was used for high-performance-liquid-chromatography (HPLC) mass-spectrometry exactly as described previously (Houtkooper et al. 2006). The ratio of monolysocardiolipin over cardiolipin was calculated from the areas under the curve of the HPLC profiles of the complete mass spectra (Kulik et al. 2008).
2.11. Analysis of cardiolipin and monolysocardiolipin in mitochondrial membranes
To measure actual concentrations of cardiolipin and monolysocardiolipin in mitochondria and submitochondrial membranes, we used HPLC with fluorescence detection. Lipids were extracted, derivatized with 1-naphthylacetic anhydride, and purified by solid-phase extraction as decribed (Schlame et al. 1999; Schlame 2007). The subsequent HPLC method was modified in order to include monolysocardiolipin in the analysis. Derivatized samples were injected via a 20-μl-loop into a Hypersil ODS column (150 mm × 4.6 mm, particle size 5 μm). A linear gradient was run from 100% acetonitrile to 100% 2-propanol in 100 minutes at a flow rate of 2 ml/minute. Fluorescence was recorded at an excitation wavelength of 280 nm and an emission wavelength of 360 nm. We showed that monolysocardiolipin gave the same fluorescence yield per nanomole as cardiolipin. Quantification was performed with the internal standard oleoyl-tristearoyl-cardiolipin, which was produced from bovine heart cardiolipin by catalytic hydrogenation (Schlame 2007).
3. Results
3.1. Generation of tafazzin-deficient mouse embryonic stem cells and differentiation into cardiomyocytes
Mouse embryonic stem cells with loxP flanked tafazzin allele (Tazflox) were generated employing standard technology of homologous recombination (Fig. 1 A–C). Correct targeting of the tafazzin gene was verified by Southern blot analysis (Fig. 1 D). Tafazzin-deficient stem cells (ΔTaz) were generated by excision of the first four exons of the tafazzin gene by transient expression of Cre-recombinase (Fig. 1 E). Inactivation of the tafazzin gene was demonstrated by RT-PCR analysis (Fig. 1 F). Wild-type and ΔTaz embryonic stem cells were differentiated into contracting cardiomyocytes in the presence of Noggin. Cardiomyocytes were identified by rhythmic contractions and by immunofluorescent staining for cardiac specific myosin. About twenty to thirty percent of cells were cardiomyocytes after Noggin treatment.
Fig. 1. Generation of tafazzin-deficient mouse embryonic stem cells.

A, The normal mouse tafazzin gene is shown with exons (boxes) and untranslated regions (half-height boxes). B, The targeting vector contains the neomycin-resistant cassette (Neor) and the thymidine kinase gene (TK), which are used as positive and negative selectors, respectively. Shaded areas depict regions homologous to the sequence of the mouse tafazzin gene. C, The linearized targeting vector was inserted into embryonic stem cells by electroporation followed by selection for neomycin and ganciclovir resistance. The panel shows the correctly targeted tafazzin gene (Tazflox). D, Clones were screened for homologous recombination by Southern blot analysis with a 535 bp DNA probe (marked sp on panel C). Clones containing wild (WT) type and targeted tafazzin alleles (Tazflox) produce 6.8 kb and 1.9 kb bgl II fragments, respectively. E, The tafazzin gene was inactivated in Tazflox cells by transient expression of Cre-recombinase. Cell colonies were screened by PCR with primers P1 and P2. F, Inactivation of the tafazzin gene was verified by RT-PCR analysis of total RNA from differentiated wild type (WT) and Taz-deficient (ΔTaz) cells with mRNA-specific primers P3 and P4 (see text). Glyceroaldehydephosphate dehydrogenase (GAPDH) mRNA was amplified as a control.
3.2. Cardiolipin analysis in stem cells and cardiomyocytes
Cardiolipin was analyzed in cell cultures by mass spectrometry, revealing molecular species with acyl moieties that have 66 (C66, m/z ~ 685–690) to 74 (C74, m/z ~ 730–740) carbon atoms (Fig. 2). The abundance of the species clusters ranked C74~C66<C68<C70<C72 in both embryonic stem cells and differentiated cardiomyocytes. Tafazzin deletion changed the pattern of cardiolipin species in embryonic stem cells and it increased the ratio of monolysocardiolipin to cardiolipin, consistent with the results in other tafazzin-deficient models (Vreken et al. 2000; Schlame et al. 2002; Valianpour et al. 2002; Schlame et al. 2003; Gu et al. 2004; Valianpour et al. 2005; Xu et al. 2006b). Upon differentiation, the cardiolipin changes persisted and it became obvious that each cardiolipin cluster was slightly shifted towards higher m/z values, indicating a higher level of saturation (Fig. 2).
Fig. 2. Cardiolipin and monolysocardiolipin in mouse embryonic stem cells (SC) and differentiated cardiomyocytes (CM).

Lipid extracts of controls and tafazzin-deficient cells (ΔTAZ) were analyzed by mass spectrometry. A, Mass spectra. B, Ratio of monolysocardiolipin (MLCL) to cardiolipin (CL). Tafazzin deletion caused an increase in MLCL relative to CL and a change in the pattern of molecular species. CL i.s., internal standard (tetramyristoyl-cardiolipin).
3.3. Electron microscopy of stem cells and cardiomyocytes
We assessed the effect of tafazzin deletion on the ultrastructure of mitochondria in differentiated and undifferentiated cells by electron microscopic tomography. Mitochondria of embryonic stem cells had a unique morphology, characterized by large tubular or vesicular cristae with only sparse connections to the inner boundary membrane (Fig. 3A). The cristae morphology underwent profound changes upon differentiation of the stem cells into cardiomyocytes. Specifically, we observed re-orientation of the inner membranes to parallel sheets and narrowing of the intermembrane space, which gave the cristae a lamellar appearance (Fig. 3B). After deletion of tafazzin, we found little change in the morphology of stem cell mitochondria (Fig. 3C). Morphometric analysis confirmed that mitochondria maintained their size (Fig. 4A) and their predominantly tubular cristae structure (Fig. 4B) in the absence of tafazzin. However, tafazzin deficiency inhibited the mitochondrial differentiation process because the characteristic lamellar-type mitochondria were less abundant in tafazzin-deficient cardiomyocytes (Fig. 4B). In these “under-differentiated” mitochondria, we found distorted cristae with an irregular pattern nearly twice as frequent as in controls (Fig. 4C). Aberrant cristae lost their parallel orientation and instead formed networks of branching lamellae, in which there were inverted inner membrane vesicles (Fig. 3D). No characteristic patterns were noted in the spatial distribution of mitochondria in stem cells and cardiomyocytes both in controls and tafazzin-deficient cells.
Fig. 3. Electron microscopic tomograms of mitochondria from mouse embryonic stem cells and differentiated cardiomyocytes.

The left panels show a representative slice of the tomogram; the other panels show 3-D models of mitochondria in different tilt angles. White arrowheads point to the mitochondria for which models were created. Inner boundary membranes are shown in yellow and cristae membranes are shown in shades of blue. A, undifferentiated control stem cell; B, differentiated control cardiomyocyte; C, undifferentiated stem cell with tafazzin deletion; D, differentiated cardiomyocyte with tafazzin deletion. Embryonic stem cells contain mainly tubular mitochondria that are not altered by tafazzin deletion. Differentiated cardiomyocytes contain mainly lamellar mitochondria that show significant abnormalities in the absence of tafazzin. Bars: 500 nm.
Fig. 4. Morphometry of mitochondria in mouse embryonic stem cells (SC) and differentiatedcardiomyocytes (CM).

Control cells and tafazzin-deficient cells (ΔTAZ) were analyzed. A, Average size of mitochondrial cross sections. B, Proportion of mitochondria with tubular (T), lamellar (L), and mixed (L/T) cristae morphology. C, Proportion of mitochondria with abnormal cristae. Embryonic stem cells contain mainly tubular mitochondria that are not altered by tafazzin deletion. Differentiated cardiomyocytes contain mainly lamellar mitochondria that show significant abnormalities in the absence of tafazzin. Data are means with s.e.m. of two experiments, in each of which we evaluated hundred mitochondrial cross sections. *Significant difference between control SC and control CM (p<0.01); **Significant difference between control CM and ΔTAZ CM (p<0.05).
3.4. Cardiolipin analysis in Drosophila mitochondria
Previously, we created a tafazzin mutant of Drosophila melanogaster, in which aberrant cardiolipin was associated with mitochondrial myopathy (Xu et al. 2006b). To further characterize the metabolic changes in this mutant, we determined the concentration and submitochondrial localization of cardiolipin and monolysocardiolipin by HPLC with fluorescence detection. Tafazzin deletion decreased the concentration of cardiolipin in Drosophila mitochondria from 17 to 4 nmol/mg protein and it increased the concentration of monolysocardiolipin from 0.5 to 7 nmol/mg protein (Fig. 5A). The submitochondrial localization of monolysocardiolipin was identical to the submitochondrial localization of cardiolipin, i.e. both lipids were found primarily in the inner mitochondrial membrane (Fig. 5B). Likewise, monolysocardiolipin was found primarily in the inner membrane of rat liver mitochondria, although only as a very minor component (Fig. 5 A, B).The data demonstrate that monolysocardiolipin is a trace phospholipid of the inner membrane of normal mitochondria, whereas similar amounts of monolysocardiolipin and cardiolipin are present in the inner membrane of Drosophila mitochondria with tafazzin deficiency.
Fig. 5. Quantification and submitochondrial localization of cardiolipin and monolysocardiolipin.
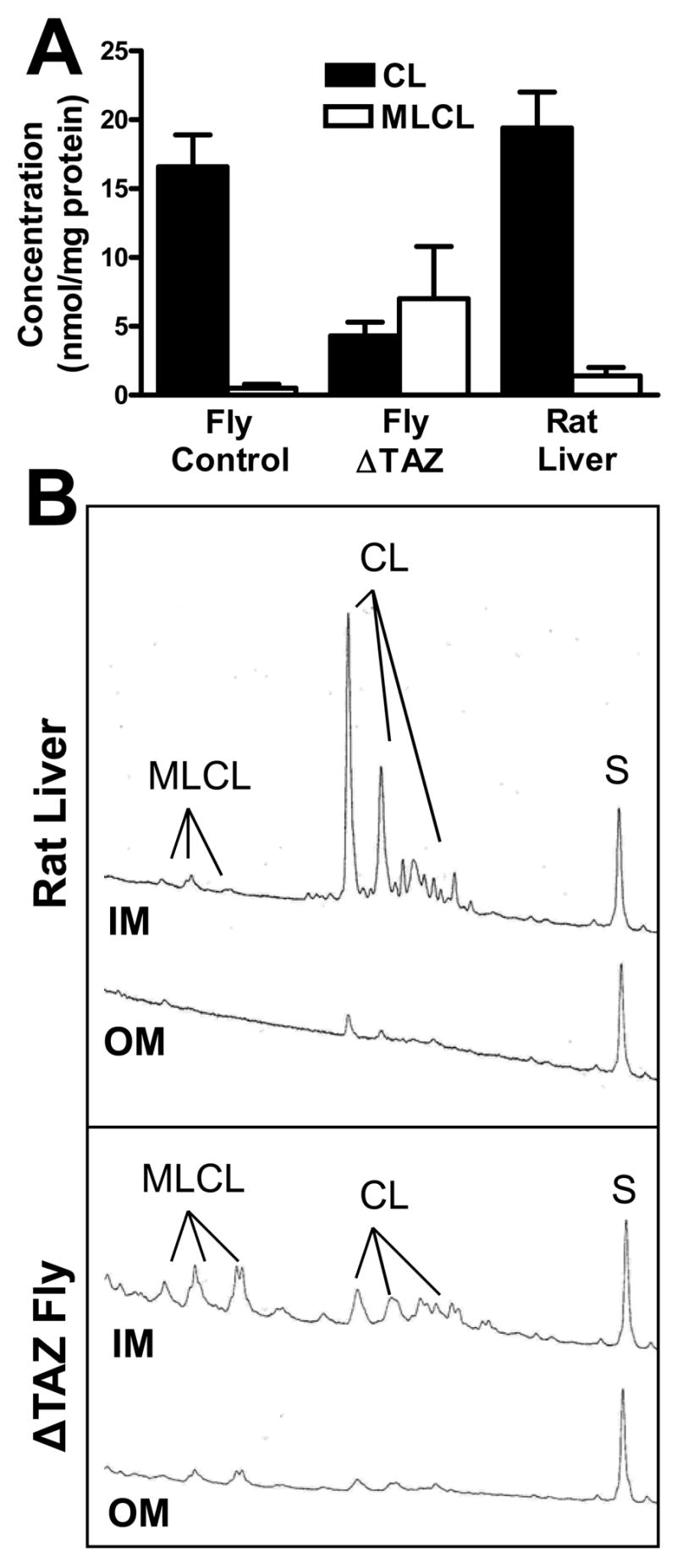
Cardiolipin (CL) and monolysocardiolipin (MLCL) were quantified in isolated mitochondria and submitochondrial membranes from Drosophila and rat liver by fluorescence-HPLC with pre-column derivatization. A, Concentration in whole mitochondria. Monolysocardiolipin is a minor component of normal mitochondria but it increases significantly in tafazzin-deficient mitochondria (ΔTAZ). B, Distribution between inner (IM) and outer (OM) membrane (membrane aliquots corresponding to 1.5 mg protein). The fluorescence yield is plotted against the retention time between 15 and 60 min. CL and MLCL were quantified by the magnitude of their peak integrals relative to the integral of the internal standard peak (S, oleoyl-tristearoyl-cardiolipin). The data show that both CL and MLCL are mainly present in the inner membrane.
3.5. Functional evaluation of Drosophila heart and flight muscles
We compared the effect of tafazzin deletion on the physiologic activity of two types of muscle tissue in Drosophila, namely heart muscle and flight muscle (indirect wing muscle). Consistent with our previous report (Xu et al. 2006b), we observed that tafazzin deletion caused a significant decline in indirect flight muscle function, which we determined by the ability to fly and to climb against gravity (Table 1). In contrast, we did not observe any change in cardiac function when we measured heart rate, heart chamber size, and fractional shortening by optical coherence tomography. Thus, it appeared that the Drosophila heart maintained normal contractility, compliance, and chronotropic properties in the absence of tafazzin (Table 1). The data suggest that mitochondria from Drosophila wing muscles are more susceptible to tafazzin deficiency than mitochondria from Drosophila heart.
TABLE 1.
Heart and flight muscle function of adult flies
| Physiologic variable | Control | ΔTAZ | t-test |
|---|---|---|---|
| Cardiac function | (n=21) | (n=84) | |
| End-diastolic dimension (μm) | 70±4 | 76±2 | NS |
| End-systolic dimension (μm) | 7±3 | 11±2 | NS |
| Fractional shortening (%) | 91±3 | 87±2 | NS |
| Heart rate (beats/min) | 326±9 | 357±4 | NS |
|
| |||
| Flight muscle function | |||
| Flying score (n=360) | 3.53±0.12 | 2.16±0.13 | p<0.0001 |
| Climbing score (n=220) | 4.41±0.13 | 2.50±0.14 | p<0.0001 |
Data are means with standard error of mean of n measurements.
3.6. Electron microscopy of Drosophila heart and flight muscle
In parallel to our physiologic studies, we compared the effect of tafazzin deletion on the ultrastructure of mitochondria from heart and flight muscle. As reported previously (Xu et al. 2006b), tafazzin deletion caused in flight muscle an increase of mitochondria with significant crista abnormalities from 2 to 14 per 300 μm2 as well as an increase in large mitochondria (>3.5 μm2 cross section) from 1 to 3 per 300 μm2. In contrast, we were not able to identify any structural abnormalities in heart mitochondria in the same flies that contained altered flight muscle mitochondria (Fig. 6). The two most striking morphologic differences between heart and flight muscle mitochondria were found in size and cristae density, both of which were greater in flight muscle mitochondria (Fig. 6). To characterize in more detail the structural changes induced by tafazzin deficiency in flight muscle mitochondria, we used electron microscopic tomography. 3-D models of normal wing muscle mitochondria showed that they are highly organized structures, in which cristae form a reticular network of densely packed lamellae aligned in parallel fashion. The model also showed that the reticular appearance resulted from frequent interruptions of the lamellar continuity by cristae fenestrations that were filled with matrix space (Fig. 7A). Tafazzin deficiency resulted in the regional formation of enlarged, round-shaped cristae, which virtually eliminated the presence of fenestrations and high-curvature membrane folds (Fig. 7B). In addition, we frequently observed hyperdense bodies in the center of mitochondria, which tomography revealed to be compact multi-layered blocks of tubules and sheets, apparently formed from stacks of aggregated inner membranes (Fig. 7C).
Fig. 6. Electron micrographs of flight muscle and heart muscle of Drosophila.

The images show mitochondria (marked by arrowheads) and myofibers (MF) in flight muscle and heart from the same fly preparations. Flight muscle mitochondria are larger and have a higher cristae density than heart mitochondria. Tafazzin deficiency (ΔTAZ) causes characteristic structural abnormalities in flight muscle mitochondria but not in heart mitochondria. Bars: 500 nm.
Fig. 7. Electron microscopic tomograms of mitochondria from Drosophila flight muscles.

The images show 3-D models of mitochondrial compartments in different tilt angles. A, normal control; B and C, tafazzin deletion mutants (ΔTAZ). Cristae (inner membranes plus intercristae space) are shown in blue, the peripheral compartment (inner boundary membrane plus outer membrane plus intermembrane space) is shown in orange, and aggregated inner membranes are shown in purple. In the upper row, a single slice of the tomogram is presented as background. In panel C, white lines are drawn through some of the cylindrical cavities that penetrate the block of aggregated inner membranes in order to enhance the 3-D impression. ΔTAZ mitochondria show regional cristae remodeling (panel B) and hyperdense malformations consisting of stacks of aggregated inner membranes (panel C). Bars: 500 nm.
4. Discussion
Deletion of tafazzin caused profound changes in the lipid composition of the inner mitochondrial membrane in differentiated and undifferentiated mouse cells and in Drosophila, including loss of cardiolipin, changes in cardiolipin composition, and increases in monolysocardiolipin (Fig. 2 and Fig. 5). These metabolic changes were associated with functional defects in flight muscle but not in cardiac muscle of Drosophila, and the presence of abnormal mitochondria correlated with the functional defects (Table 1, Fig. 6). To characterize the structural consequences of the altered cardiolipin metabolism, we created models of mitochondria by electron microscopic tomography, a technique that allows three-dimensional visualization (Frey et al. 2000). Although our sections had only a thickness of 100 nm, we have previously demonstrated that useful information can be extracted even from small tomographic volumes, in particular with respect to near-range spatial relations between neighboring membranes (Acehan et al., 2007). We found significant alterations of the mitochondrial ultrastructure in particular types of differentiated tissues, but not in embryonic stem cells (Figs. 3, 4, 7). The selective nature of these defects suggests that normal concentration and composition of cardiolipin is essential only in certain types of mitochondria. In particular, our data support and further expand the idea that tafazzin plays a role in tissue development and differentiation (Khuchua et al. 2006).
Thus, the question arises as to which differentiation pathway requires the presence of tafazzin? Although tafazzin was found to be ubiquitously expressed in zebrafish tissues, at least during the early stages of development (Khuchua et al. 2006), the clinical effects of tafazzin deficiency in humans are rather tissue-specific. In particular, heart and skeletal muscles are affected (Kelley et al. 1991; Barth et al. 1999; Spencer et al. 2006), which are tissues where mitochondria contain highly organized and densely packed cristae. Likewise, in Drosophila, the key “symptom” of tafazzin deficiency is motor weakness of the indirect wing muscles, an organ in which mitochondria are densely populated with cristae. Initially, we expected to find the same effect in Drosophila heart and were surprised that the mutants did not demonstrate any signs of cardiac dysfunction, even when advanced techniques for the measurement of chamber size and contractility were used (Table 1). However, electron microscopy revealed that fly heart mitochondria do not share the same kind of cristae organization that is characteristic of mitochondria from mammalian heart and Drosophila wing muscles (Fig. 6). The data suggest that mitochondria are more susceptible to tafazzin deletion if their cristae form an organized network of densely packed membranes. High cristae density is, however, not an absolute requirement for morphologic alterations, as demonstrated earlier by our observations of defective mitochondria in Barth patient lymphoblasts (Acehan et al. 2007).
Why are certain types of mitochondria more sensitive to tafazzin deficiency? Tafazzin is required to maintain normal cardiolipin levels and to form cardiolipin with a normal molecular composition; in particular it is required for the synthesis of tetralinoleoyl-cardiolipin and other uniformly acylated species (for a review see (Schlame 2008)). Although those species have been identified in diverse cell types and organisms (Schlame et al. 2002; Valianpour et al. 2002; Schlame et al. 2003; Gu et al. 2004; Kuijpers et al. 2004; Xu et al. 2006b), the abundance of tetralinoleoyl-cardiolipin tends to be higher in mitochondria with high cristae density (Schlame et al. 1999), suggesting that tetralinoleoyl-cardiolipin and equivalent molecular species have a specific function in such mitochondria. We have analyzed the 3-D ultrastructure of abnormal cristae that form in the absence of tafazzin and observed extensive remodeling of inner membranes and the formation of inner-membrane aggregates (Fig. 3 and Fig. 7). Since membrane remodeling in cardiomyocyte mitochondria bears resemblance to the mitochondrial fragmentation pattern of apoptotic cells (Sun et al. 2007), future studies must clarify whether tafazzin deficiency predisposes to apoptosis or whether it stimulates fission activity in general. Any of the structural changes may result from the loss of uniformly substituted cardiolipin or from the accumulation of monolysocardiolipin because the latter is also present in the inner membrane (Fig. 5). These changes may lead to altered bilayer properties and/or to altered organization of protein complexes, which may lead to protein unfolding and aggregation, which in turn may induce non-specific membrane adhesion. While we found some membrane adhesion zones in tafazzin-deficient lymphoblasts (Acehan et al. 2007), they are more dominant in flight muscle (Fig. 7), where high cristae density and a high concentration of membrane proteins are likely to make mitochondria more susceptible to membrane aggregation. Therefore, membrane aggregation may accelerate in times of stress, increased energy demand, or active mitochondrial biogenesis, i.e. whenever the membrane protein concentration increases. This may perhaps explain why Barth patients often deteriorate during infection, growth spurt, and other episodes of physiologic strain.
Footnotes
This work was supported in part by grants from the National Institutes of Health (HL078788, HL083065, and GM071044), the Barth Syndrome Foundation, the United Mitochondrial Disease Foundation, and the Prinses Beatrix Fonds (WAR05-0126). Electron tomography relied on the facilities at the New York Structural Biology Center, which is a STAR center supported by the New York State Office of Science, Technology and Academic Research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87:40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, Van’t Veer-Korthof ET, Van der Harten JJ, Sobotka-Plojhar MA. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. 1983;62:327–355. doi: 10.1016/0022-510x(83)90209-5. [DOI] [PubMed] [Google Scholar]
- Barth PG, Wanders RJ, Vreken P, Janssen EA, Lam J, Baas F. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) J Inherit Metab Dis. 1999;22:555–567. doi: 10.1023/a:1005568609936. [DOI] [PubMed] [Google Scholar]
- Benzer S. Behavioral mutants of Drosophila isolated by countercurrent distribution. Proc Natl Acad Sci U S A. 1967;58:1112–1119. doi: 10.1073/pnas.58.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell. 2005;16:5202–5214. doi: 10.1091/mbc.E05-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou J, McInnes RR, Jay V, Wilson G, Becker LE, Lehotay DC, Platt BA, Bridge PJ, Robinson BH, Clarke JT. Barth syndrome: clinical observations and genetic linkage studies. Am J Med Genet. 1994;50:255–264. doi: 10.1002/ajmg.1320500309. [DOI] [PubMed] [Google Scholar]
- Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci. 2000;25:319–324. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- Greenawalt JW. The isolation of outer and inner mitochondrial membranes. Methods Enzymol. 1974;31:310–323. doi: 10.1016/0076-6879(74)31033-6. [DOI] [PubMed] [Google Scholar]
- Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol. 2004;51:149–158. doi: 10.1046/j.1365-2958.2003.03802.x. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Akbari H, van Lenthe H, Kulik W, Wanders RJ, Frentzen M, Vaz FM. Identification and characterization of human cardiolipin synthase. FEBS Lett. 2006;580:3059–3064. doi: 10.1016/j.febslet.2006.04.054. [DOI] [PubMed] [Google Scholar]
- Hug G, Schwartz DC, Tuuri D, Dillon T. Sex-linked dilated cardiomyopathy, characteristic mitochondrial ultrastructure in liver, heart and muscle, effect of carnitine treatment. Pediatr Res. 1990;27:20–28. [Google Scholar]
- Ino T, Sherwood WG, Cutz E, Benson LN, Rose V, Freedom RM. Dilated cardiomyopathy with neutropenia, short stature, and abnormal carnitine metabolism. J Pediatr. 1988;113:511–514. doi: 10.1016/s0022-3476(88)80642-5. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Cheatham JP, Clark BJ, Nigro MA, Powell BR, Sherwood GW, Sladky JT, Swisher WP. X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr. 1991;119:738–747. doi: 10.1016/s0022-3476(05)80289-6. [DOI] [PubMed] [Google Scholar]
- Khuchua Z, Yue Z, Batts L, Strauss AW. A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function. Circ Res. 2006;99:201–208. doi: 10.1161/01.RES.0000233378.95325.ce. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kuijpers TW, Maianski NA, Tool AT, Becker K, Plecko B, Valianpour F, Wanders RJ, Pereira R, Van Hove J, Verhoeven AJ, Roos D, Baas F, Barth PG. Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood. 2004;103:3915–3923. doi: 10.1182/blood-2003-11-3940. [DOI] [PubMed] [Google Scholar]
- Kulik W, van Lenthe H, Stet FS, Houtkooper RH, Kemp H, Stone JE, Steward CG, Wanders RJ, Vaz FM. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem. 2008;54:371–378. doi: 10.1373/clinchem.2007.095711. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Vaz FM, Gu Z, Wanders RJ, Greenberg ML. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J Biol Chem. 2004;279:44394–44399. doi: 10.1074/jbc.M405479200. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Matise MP, Auerbac W, Joyner AL. Gene Targeting. In: Joyner AL, editor. A Practical Approach. 2003. pp. 101–132. [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Schlame M. Assays of cardiolipin levels. Methods Cell Biol. 2007;80:223–240. doi: 10.1016/S0091-679X(06)80011-7. [DOI] [PubMed] [Google Scholar]
- Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res. 2008;49:1607–1620. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, Wanders RJ, DiMauro S, Blanck TJ. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- Schlame M, Shanske S, Doty S, Konig T, Sculco T, DiMauro S, Blanck TJ. Microanalysis of cardiolipin in small biopsies including skeletal muscle from patients with mitochondrial disease. J Lipid Res. 1999;40:1585–1592. [PubMed] [Google Scholar]
- Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJ. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51:634–637. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, Berthy J, Redfearn SP, Byrne BJ. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118:e337–e346. doi: 10.1542/peds.2005-2667. [DOI] [PubMed] [Google Scholar]
- Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, Frey TG. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–1065. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- Valianpour F, Mitsakos V, Schlemmer D, Towbin JA, Taylor JM, Ekert PG, Thorburn DR, Munnich A, Wanders RJ, Barth PG, Vaz FM. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J Lipid Res. 2005;46:1182–1195. doi: 10.1194/jlr.M500056-JLR200. [DOI] [PubMed] [Google Scholar]
- Valianpour F, Wanders RJ, Overmars H, Vreken P, Van Gennip AH, Baas F, Plecko B, Santer R, Becker K, Barth PG. Cardiolipin deficiency in X-linked cardioskeletal myopathy and neutropenia (Barth syndrome, MIM 302060): a study in cultured skin fibroblasts. J Pediatr. 2002;141:729–733. doi: 10.1067/mpd.2002.129174. [DOI] [PubMed] [Google Scholar]
- van Werkhoven MA, Thorburn DR, Gedeon AK, Pitt JJ. Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth Syndrome. J Lipid Res. 2006;47:2346–2351. doi: 10.1194/jlr.D600024-JLR200. [DOI] [PubMed] [Google Scholar]
- Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279:378–382. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- Wasserthal LT. Drosophila flies combine periodic heartbeat reversal with a circulation in the anterior body mediated by a newly discovered anterior pair of ostial valves and ‘venous’ channels. J Exp Biol. 2007;210:3707–3719. doi: 10.1242/jeb.007864. [DOI] [PubMed] [Google Scholar]
- Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci U S A. 2006;103:1394–1399. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Sutachan JJ, Plesken H, Kelley RI, Schlame M. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab Invest. 2005;85:823–830. doi: 10.1038/labinvest.3700274. [DOI] [PubMed] [Google Scholar]
- Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006a;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, Schlame M. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A. 2006b;103:11584–11588. doi: 10.1073/pnas.0603242103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa S, Itabashi Y, Koshimizu U, Tanaka T, Sugimura K, Kinoshita M, Hattori F, Fukami S, Shimazaki T, Ogawa S, Okano H, Fukuda K. Transient inhibition of BMP signaling by Noggin induces cardiomyocyte differentiation of mouse embryonic stem cells. Nat Biotechnol. 2005;23:607–611. doi: 10.1038/nbt1093. [DOI] [PubMed] [Google Scholar]