

Abstract
A critical step in the mechanism of action of inflammatory cytokines is the stimulation of sphingolipid metabolism, including activation of sphingosine kinase (SK) which produces the mitogenic and pro-inflammatory lipid sphingosine 1-phosphate (S1P). We have developed orally-bioavailable compounds that effectively inhibit SK activity in vitro, in intact cells and in cancer models in vivo. In the present study, we have assessed the effects of these SK inhibitors on cellular responses to TNFα, and evaluated their efficacies in the dextran sulfate sodium (DSS) model of ulcerative colitis in mice. Using several cell systems, it was shown that the SK inhibitors block the ability of TNFα to: activate NFκB; induce the expression of adhesion proteins; and promote the production of PGE2. In an acute model of DSS-induced ulcerative colitis, the SK inhibitors were equivalent to or more effective than Dipentum in reducing disease progression, colon shortening, and neutrophil infiltration into the colon. The effects of the SK inhibitors were associated with decreased colonic levels of the inflammatory cytokines TNFα, IL-1β, IFN-γ and IL-6, and reduction of S1P levels in the colon. A similar reduction in disease progression was provided by the SK inhibitors in a chronic model of ulcerative colitis in which the mice received three week-long cycles of DSS interspaced with week-long recovery periods. In the chronic model, immunohistochemistry for SK showed increased expression in DSS treated mice (compared to water controls) that was reduced by drug treatment. S1P levels were also elevated in the DSS group and significantly reduced by drug treatment. Together, these data indicate that SK is a critical component in inflammation, and that inhibitors of this enzyme may be useful in the treatment of inflammatory bowel diseases.
Keywords: Sphingosine kinase, Inflammatory Bowel Disease, Ulcerative colitis, TNFα
Introduction
Inflammatory bowel disease (IBD) encompasses a group of disorders characterized by pathological inflammation of the lower intestine. Crohn’s disease and ulcerative colitis are the most common forms of IBD, and while their etiologies are not fully elucidated, infectious and immunologic mediators have been clearly implicated. Studies on the development and treatment of IBD have been greatly facilitated animal models that mimic the clinical and immunopathological disorders seen in humans [1]. From studies with these models, it is clear that the manifestations of IBD are dependent on cellular immune responses and inflammatory cytokines; however, the molecular mechanisms by which this occurs are not yet clearly defined.
A common feature of immune activation in IBDs involves the influx of mast cells, monocytes, macrophages and polymorphonuclear neutrophils into the epithelium of the colon, which results in the amplification of the inflammation process, producing the clinical manifestations of the diseases [2]. This infiltration by granulocytes is accompanied by dramatic increases in levels of inflammatory cytokines in the colon [3]. Most prominently, tumor necrosis factor-α (TNFα) has been shown to play a significant role in IBD, such that antibody therapy directed against this cytokine, i.e. Remicade, is a promising new treatment [4]. TNFα activates several processes that contribute to IBD, and is necessary for both the initiation and persistence of the Th1 response. For example, TNFα has been shown act through the induction of nuclear factor kappa B (NFκB) [5] which induces the proinflammatory enzymes nitric oxide synthase (NOS) and cyclooxygenase-2 (COX-2) [6]. COX-2 plays a key role in the inflammation of IBDs through its production of prostaglandins [7,8], and oxidative stress such as that mediated by nitric oxide produced by NOS has also shown to exacerbate IBD inflammation [9].
The mechanisms and effects of the sphingolipid metabolism have been the subjects of a growing body of scientific investigation, including analyses of its roles in inflammation [10-12]. Sphingomyelin is not only a structural component of cellular membranes, but also serves as the precursor for the potent bioactive lipids ceramide and sphingosine 1-phosphate (S1P). Ceramide is produced by the hydrolysis of sphingomyelin in response to inflammatory stresses, including TNFα [13,14]. Ceramide induces apoptosis in proliferating cells by a mechanism that remains to be elucidated [15,16], and can be further hydrolyzed by ceramidase to produce sphingosine. Sphingosine is then rapidly phosphorylated by sphingosine kinase (SK) to produce S1P. Therefore, activation of ceramidase and SK by cytokines and growth factors leads to rapid increases in the intracellular levels of S1P and depletion of ceramide levels. This situation promotes cell proliferation and inhibits apoptosis. Deregulation of apoptosis in phagocytes is an important component of the chronic inflammatory state in IBDs, and S1P has been shown to protect neutrophils from apoptosis in response to Fas, TNFα and ceramide [17]. Similarly, apoptosis of macrophages is blocked by S1P [18].
In addition to its role in regulating cell proliferation and apoptosis, S1P has been shown to have several other important effects on cells that mediate immune functions. Platelets, monocytes and mast cells secrete S1P upon activation, promoting inflammatory cascades at the site of tissue damage [19,20]. Activation of SK is essential for the signaling responses to TNFα since its ability to induce adhesion molecule expression via activation of NFκB is mimicked by S1P and is blocked by the SK inhibitor dimethylsphingosine [14]. Similarly, S1P mimics the ability of TNFα to induce the expression of COX-2 and the synthesis of PGE2, and knock-down of SK by RNA interference blocks these responses to TNFα, but not S1P [21]. S1P is also a mediator of Ca2+ influx during neutrophil activation by TNFα and other stimuli, leading to the production of superoxide and other toxic radicals [22,23].
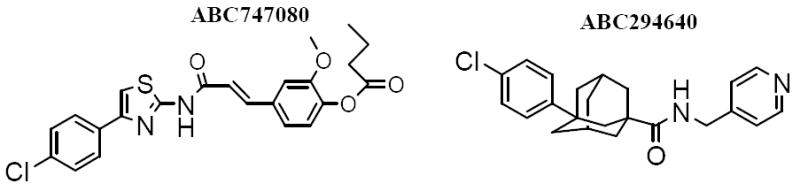
In spite of accumulating evidence for a pivotal role of SK in the regulation of inflammatory processes, pharmacological inhibition of SK is an untested means of preventing and/or treating IBDs. This is largely due to the heretofore lack of pharmacologically useful SK inhibitors. To overcome this problem, we have recently identified non-lipid compounds that inhibit recombinant human SK and are cytotoxic towards human cancer cell lines [24,25]. These compounds inhibit endogenous S1P formation in intact cells, and demonstrate a high degree of selectivity for SK versus other lipid and protein kinases. Our lead SK inhibitors, ABC747080 and ABC294640 (Figure 1), have excellent oral bioavailability and in vivo SK inhibitory activity in mice. Importantly, these SK inhibitors can be chronically administered to mice without systemic toxicity [26]. Therefore, ABC747080 and ABC294640 have been examined for their effects on TNFα-mediated cell activation, and in murine models of ulcerative colitis.
Figure 1. Structures of SK inhibitors used in these studies.
The data indicate that these compounds effectively inhibit TNFα-mediated responses including activation of NFκB and induction of adhesion proteins and PGE2 synthesis. Additionally, each compound suppresses disease progression in the in vivo models, suggesting that targeting SK is a viable new approach to the treatment of IBDs.
Materials and Methods
Reagents
Unless otherwise noted, chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO). Dipentum (Olsalazine), PEG400 and DSS were from Cellteck Pharmaceutical (Rochester, NY), J. T. Baker (Phillipsburg, NJ) and MP Biomedicals, Inc. (Solon, OH), respectively. The SK inhibitors ABC294640 and ABC747080 were synthesized as follows.
ABC294640
Adamantane-1-carboxylic acid (45 g, 0.25 mol) was added to mixture of AlCl3 (45 g, 0.34 mol) and Br2 (450 g) at 0 °C and stirred at 0 - 10 °C for 48 hr. The temperature of the mixture was then raised to 20 °C for 5 hr, before the sample was poured onto 500 g of crushed ice, diluted with 300 mL of CHCl3 and decolorized with solid Na2S2O5. The aqueous phase was extracted twice with Et2O, and the combined organic phase was washed with H2O and extracted with 10 % NaOH. The alkaline extraction was acidified with 2N H2SO4 and provided 49 g of 3-bromoadamantane-1-carboxylic acid (yield = 75.7%). Over a 30 minute period, 3-bromoadamantane-1-carboxylic acid (16.0 g, 61.7 mmol) in 50 ml of dry chlorobenzene at −10 °C was added to 100 ml dry chlorobenzene containing 9.3 g (70 mmol) of AlCl3. The mixture was warmed to room temperature for 1 hr and then heated to 90 °C for 10 hr. The mixture was then poured onto 200 g of crushed ice, and the filtered to provide 14.2 g of 3-(4-chlorophenyl)adamantane-1-carboxylic acid (yield = 79.3 %). 3-(4-chlorophenyl)adamantane-1-carboxylic acid was then reacted with 1,1’-carbonyldiimidazole to give an adamantanecarbonylimidazole intermediate, which was reacted with 4-aminomethylpyridine in toluene to produce 3-(4-chlorophenyl)-adamantane-1-carboxylic acid (pyridin-4-ylmethyl)amide (ABC294640) with a yield of 92.6% and a melting point of 128-130 °C. 1H NMR(300 MHz, CDCl3) δ 1.72-2.25(m, 12H, admant-CH), 4.44-4.46 (d, J = 6 Hz, 2H, CH2-Py), 6.18 (m, 1H, HN), 7.13-7.15 (d, J = 6Hz, 2H, H-Py), 7.15-7.30 (m, 4H, H-Ph), 8.52-8.54 (d, J = 6 Hz, 2H, H-Py); 13C NMR(300 MHz, CDCl3) δ 28.98, 35.73, 36.71, 38.77, 42.18, 42.37, 44.88, 122.38, 125.30, 126.57, 128.56, 129.26, 148.39, 150,20 177.76; MS m/z (rel intensity) 381.50 (MH+, 100), 383.41 (90), 384.35(80).
ABC747080
4-Hydroxy-3-methoxycinnamic acid (10.0 g, 51.5 mmol) was mixed with 35 mL of Bu2O to form a suspension, followed by the addition of 0.8 mL of H2SO4. After stirring for 5 min, the solution became yellow, and 200 mL of ether was added to form an emulsion. The reaction was continued for 18 hr at room temperature, and then the mixture was poured into 500 mL of ice-water and extracted with EtOAc. The EtOAc solution was dried over Na2SO4 and evaporated, producing a solid on standing overnight. After filtration, the solid was washed with hexane to provide butyric acid 4-(2-carboxy-vinyl)-2-methoxy-phenyl ester as a white solid (12.1 g, Y = 89%). Rf = 0.27 (5% MeOH in chloroform); 1H NMR (CDCl3) δ 7.75 (d, J = 15.8 Hz, 1 H), 7.00-7.20 (m, 3 H), 6.40 (d, J = 15.8 Hz, 1 H), 3.87 (s, 3 H), 2.58 (t, J = 7.2 Hz, 2 H), 1.80 (dd, J = 7.2 Hz, J = 7.2 Hz, 2 H), 1.06 (t, J = 7.2 Hz); 13C NMR (CDCl3) δ 171.2, 171.0, 151.0, 144.4, 127.7, 123.3, 122.9, 113.7, 56.1, 35.9, 18.6, 13.7.
Butyric acid 4-(2-carboxy-vinyl)-2-methoxy-phenyl ester (1.078 g, 4.08 mmol) was suspended in 12 mL of CH2Cl2, followed by addition of 2 M oxalyl chloride in 3 mL of CH2Cl2 and 0.15 mL of DMF. After 30 min of stirring, the volatile components were removed in vacuo. The white residue was suspended in 20 mL of 1,4-dioxane, followed by the addition of 4-(4-chlorophenyl)thiazol-2-ylamine (861 mg, 4.08 mmol) in 0.5 mL of pyridine. The yellow suspension was stirred at its boiling point for 30 min, cooled to room temperature, and the solvent was removed in vacuo. The residue was partitioned in 50 mL of water and 100 mL of ethyl acetate, and the resulting organic phase was further sequentially washed with 0.5 N HCl, 5% NaHCO3 and water. The organic solution was dried over Na2SO4, filtered, washed with ethyl acetate, and evaporated. The residue was washed with ethanol to provide butyric acid 4-(2-[4-(4-chlorophenyl)thiazol-2-ylcarbamoyl]-vinyl)-2-methoxy-phenyl ester (ABC747080) as a pale yellow solid (1.01 g, 2.21 mmol, Y = 54%). 1H NMR (CDCl3) δ 11.1 (br s, 1 H), 7.73 (d, J = 8.7 Hz, 2 H), 7.64 (d, J = 15.6 Hz, 1 H), 7.29 (d, J = 8.7 Hz, 2 H), 7.20 (s, 1 H), 6.97 (d, J = 7.8 Hz, 1 H), 6.85 (d, J = 1.2 Hz, 1 H), 6.76 (dd, J = 7.8 Hz, 1.2 Hz, 1 H), 6.14 (d, J = 15.3 Hz, 1 H), 3.80 (s, 3 H), 2.59 (t, J = 7.2 Hz, 2 H), 1.82 (dd, J = 7.2 Hz, 7.2 Hz, 2 H), 1.07 (t, J = 7.2 Hz, 3 H); 13C NMR (DMSO-d6) δ 170.7, 163.2, 157.9, 150.9, 147.7, 141.7, 140.7, 133.1, 133.0, 132.1, 128.6 (2C), 127.2 (2C), 123.3, 120.2, 119.6, 112.2, 109.1, 55.8, 35.0, 18.0, 13.3; MS (MALDI) m/z calcd for C23H22ClN2O4S (M + H+) 457, found 457.
Cellular S1P formation assay
Cells were grown to confluency in 24-well tissue culture plates and then treated with 1% DMSO (as the drug vehicle), or the indicated concentration of ABC294640 or ABC747080 for 5 hours. The cells were then incubated with [3H]sphingosine for 15 min, and the formation of [3H]S1P was measured as previously described [24].
Assay of NFκB activation
Human 293T embryonic kidney cells expressing the large T antigen, and a chromosomally integrated luciferase reporter construct regulated by six copies of the NFκB response element were purchased from Panomics, Inc. (Redwood City, CA), and grown as recommended by the vendor. For experiments on TNFα-stimulation of NFκB activity, the cells were plated into 24-well culture plates and grown to confluence. The medium was then removed and replaced with serum-free medium for 16 hr before 100 ng of TNFα per mL was added. After 6 hr, the cells were washed and lysed and the amount of luciferase was quantified using an assay kit from Promega Corporation (Madison, WI) and a Perkin Elmer HTS 7000+ Bioassay plate reader in the luminescence mode.
Assay of adhesion protein expression
Human retinal endothelial cells were purchased from Cell Technologies (ACBRI181, Kirkland, WA), and maintained in growth medium consisting of Minimum Essential Medium with D-valine supplemented with 20% fetal calf serum (Gibco, Rockville, MD), 50 μg/mL of endothelial cell growth supplement (Vec Technologies, Rensslar, NY), 16 U/mL heparin (Fisher Scientific, Pittsburg, PA), MEM vitamins and glutamine, and antibiotic/antimycotic (Gibco, Rockville, MD). The cells were plated on a 25 cm2 tissue culture flask coated with fibronectin at 2 μg/cm2, and were grown in a humidified incubator at 37 °C. For experiments on TNFα signaling, the culture medium was replaced with fresh medium that lacked fetal calf serum for 24 hr before the addition of 100 μg of TNFα per mL, and whole cell lysates were prepared 6 hr later. The protein concentrations in the samples were determined using the fluorescamine assay [27] with bovine serum albumin as the standard. Samples were normalized for equal amounts of protein per lane (100 μg), separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis and electrotransferred to nitrocellulose membranes. The blots were then blocked and probed with antibodies to VCAM-1 (CBL206, Chemicon International, Temecula, CA) or ICAM (sc-7891, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and developed with horseradish peroxidase-conjugated secondary antibodies.
Assay of PGE2 production
Human retinal endothelial cells were cultured as described above, and rat IEC6 intestinal epithelial cells were grown in DMEM medium containing 10% fetal bovine serum. In each case, the cells were plated into 24-well culture plates, allowed to grown to confluence, and then serum-starved for 8 hr before the addition of buffer or 100 ng of TNFα per mL, and 10 μg/mL of ABC294640 or ABC747080. After 18 hr, samples of the medium were removed and levels of PGE2 were quantified using an ELISA kit from Caymen Chemical (Ann Arbor, MI).
Ulcerative colitis models
The acute model of DSS-induced ulcerative colitis utilized male C57BL/6 mice, which were provided standard rodent diet and water ad libitum. After their acclimation, the animals were randomly divided into groups of 5 or 6 for DSS- (40,000 MW) and drug-treatments. The SK inhibitors were dissolved in polyethylene glycol (PEG, 400 average MW), and given once daily by oral gavage in a volume of 0.1 mL per dose. Dipentum (Olsalazine Sodium) was used as a positive control. Dipentum is an FDA-approved anti-colitis drug whose active ingredient is metabolized to 5-aminosalicylic acid in vivo. The mice were treated as follows:
| Group | DSS | Oral Treatment (Once daily on Days 0-6) |
| 1 | None | PEG alone |
| 2 | 2% in drinking water | PEG alone |
| 3 | 2% in drinking water | 50 mg/kg ABC294640 in PEG |
| 4 | 2% in drinking water | 50 mg/kg ABC747080 in PEG |
| 5 | 2% in drinking water | 50 mg/kg Dipentum in PEG |
The Disease Activity Index (detailed below) which monitors weight loss, stool consistency and blood in the stool as a measure of disease severity, was scored for each animal on Days 4-6. On Day 7, the mice were sacrificed by CO2 asphyxiation followed by cervical dislocation and the entire colon was removed and measured to the nearest 0.1 cm. The colon was then divided into thirds and used for histologic and biochemical studies described below.
A chronic 35 day model of IBD was used to evaluate the effectiveness of the SK inhibitors in mice that have experienced multiple cycles of DSS-induced inflammation. The chronic model is similar to the acute model, except that the DSS concentration in the drinking water is lower and animals receive periodic exposure to DSS (DSS on days 1-7, water on Days 8-13, DSS on day 14-21, water on Days 22-27 and then DSS until the completion of the study on Day 35). In these experiments, treatment of the mice with an SK inhibitor or Dipentum began on Day 28 and continued daily until the completion of the study. The DAI index was monitored every other day until Day 28 and then daily until Day 35.
Disease Activity Index (DAI)
The DAI was calculated for each animal on each as described by Fitzpatrick, et al. [28] according to the following scoring system:
| Score | Weight Loss | Stool consistency | Blood in stool |
| 0 | None | Normal | Negative |
| 1 | 1-5% | ||
| 2 | 6-10% | Loose stool | Positive |
| 3 | 11-15% | ||
| 4 | >15% | Diarrhea | Gross rectal bleeding |
With this scoring system, the DAI is calculated by scoring each animal for weight loss, stool consistency and blood in the stool and then dividing the total score by three. For example, an animal that lost 11% of its body weight (score of 3) with evidence of loose stool (score of 2) plus gross rectal bleeding (score of 4) would have a calculated DAI of 3.
Histology Score
The distal one-third (~2.5 cm) of the colon was transected, pinned open, rinsed with PBS and the incidence of gross inflammation (continuous areas of redness) were recorded. The tissue was then fixed with formalin overnight, followed by embedding in paraffin, sectioning and staining with hematoxylin-eosin. The sections were microscopically examined for histopathologic changes using the following scoring system. The Histology Score is determined by multiplying the percent involvement for each of the 3 following histological features by the percent area of involvement [29-32]. Therefore, the minimal score is 0 and the maximal score is 40.
| FEATURE SCORED | SCORE | DESCRIPTION |
| Inflammation Severity | 0 | None |
| 1 | Mild | |
| 2 | Moderate | |
| 3 | Severe | |
| Inflammation Extent | 0 | None |
| 1 | Mucosa | |
| 2 | Mucosa and Submucosa | |
| 3 | Transmural | |
| Crypt Damage | 0 | None |
| 1 | 1/3 of crypt damaged | |
| 2 | 2/3 of crypt damaged | |
| 3 | Crypts lost, surface epithelium intact | |
| 4 | Crypts lost, surface epithelium lost | |
| Percent Involvement | 0 | 0% |
| 1 | 1-25% | |
| 2 | 26-50% | |
| 3 | 51-75% | |
| 4 | 76-100% |
Myeloperoxidase assay
Mucosal myeloperoxidase (MPO) activity was determined as a measure of granulocyte and monocytes infiltration into the colon. The middle one-third of the colon was removed and snap frozen in a methanol/dry ice bath and stored at -80°C until time of assay. The colon was then thawed and homogenized in 10 volumes (w/v) of cold PBS. The homogenate was then centrifuged at 22,000 × g for 15 min, and the resulting pellet was resuspended and assayed for MPO by quantifying the metabolism of tetramethylbenzidine as described by Fitzpatrick et al. [28].
Sphingosine 1-phosphate assay
Samples of each colon were homogenized as indicated above and plasma samples, taken at the time of sacrifice, were extracted with ethyl acetate / isopropanol / water (60/30/10, by volume). The organic phase, containing the sphingolipids, was dried under nitrogen and then dissolved in methanol. Internal standards were used as follows: C17 base D-erythro-sphingosine for sphingosine and C17 SIP for S1P. Fractionation of the lipids by HPLC utilized a C8-reverse phase column eluted with 1 mM methanolic ammonium formate / 2 mM aqueous ammonium formate as described by Pettus et al. [21]. The method provides baseline resolution of the internal standards and analytes. Continuous reaction monitoring ESI positive ionization mode was used to acquire ions at the appropriate retention times with m/z of 286 (precursor) → 268 (intermediate) → 250 (product ion) for C17 base D-erythro-sphingosine, 300 → 282 → 264 for D-erythro-sphingosine, 366 → 348 → 250 for C17 SIP, and 380 → 362 → 264 for D-erythro-S1P. Calibration curves were generated by plotting the area ratios of the synthetic standards for each sphingolipid, and were used to determine the absolute amounts of sphingosine and S1P in the serum and colon samples from control and SK inhibitor-treated mice.
Cytokine assays
Supernatants for the homogenates of each colon were used to determine the levels of TNFα, IL-1β, IFN-γ, IL-6 and IL-10 using Luminex assays performed by the Cytokine Core Laboratory at the University of Maryland, Baltimore.
Immunohistochemical analysis of SK expression in colon
The expression of SK in the colons from the chronic DSS-model was assessed. Briefly, sections were deparaffinized in xylene and rehydrated in a series of ethanol dilutions. Sections were permeabilized in 0.5% Triton X-100 in PBS and boiled for 10 minutes in 10mM sodium citrate buffer for antigen retrieval. Slides were then incubated 10 min in 3% hydrogen peroxide to quench endogenous peroxidase. After washing in PBS, sections were blocked for 1 hour with the appropriate reagent from the VECTASTAIN ABC Kit (Vector Laboratories Inc.) used for the remaining steps. Sections were then incubated for 30 min at room temperature with primary SK antibody (rabbit polyclonal AB675; Biosynthesis Inc, Lewisville, TX; raised against a seven amino acid peptide evaluated to be exclusive to only SK) followed by PBS washes and 30 min incubation with diluted biotinylated secondary antibody and then 30 min incubation with DAB reagent. Sections were lightly counterstained with hematoxylin, rehydrated, and mounted for analysis by bright field microscopy.
Data analyses
Differences among treatment groups were analyzed by two-tailed unpaired t-test analyses, with a difference of P < 0.05 is considered to be statistically significant.
Results
Sphingosine kinase activity in intestinal epithelial and endothelial cells
To confirm that SK is present in endothelial cells, we previously demonstrated its expression in bovine, rat and human endothelial cells by western analyses using polyclonal antibodies that cross-react with SK from multiple species [33]. Each of several preparations of human endothelial cells contained high amounts of SK protein, demonstrating that these cells consistently express SK.
To evaluate the effects of compounds on the activity of SK in intact cells, we developed a cell-based assay in which the phosphorylation of exogenously added [3H]sphingosine to [3H]S1P by endogenous SK can be quantified [24]. The assay is patterned after our in vitro SK assay in which [3H]sphingosine and [3H]S1P are separated by extraction and levels of both species are determined by scintillation counting. We have used a number of cell lines in this assay to confirm that the SK inhibitors are active in multiple intact cell systems. Most relevant to IBD, we have demonstrated that the lead SK inhibitors reduce cellular levels of S1P synthesis human endothelial cells and rat IEC6 cells (Figure 2). ACB294640 and ABC747080 each caused dose-dependent suppression of SK activity in each of the cell types, with the endothelial cells being somewhat more sensitive than the epithelial cells.
Figure 2. Inhibition of cellular SK by ABC294640 and ABC747080.
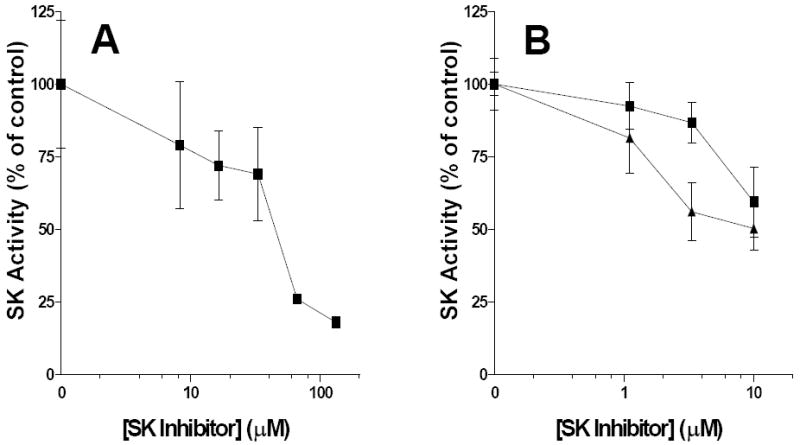
Rat IEC6 cells (Panel A) or human endothelial cells (Panel B) were incubated with the indicated concentration of ABC294640 (■) or ABC747080 (▲) before the addition of 0.4 μCi of [3H]sphingosine. After 15 min, cells were lysed and extracted with chloroform : methanol, and the amounts of [3H]sphingosine in the organic phase and [3H]S1P in the aqueous phase were then determined. Values represent the mean ± sd SK activity in triplicate samples in a typical experiment.
In vitro effects of SK inhibitors on signaling by TNFα
The effects of the two lead SK inhibitors on responses of human endothelial cells to TNFα were characterized. The excellent aqueous solubility of ABC294640 allowed it to be evaluated in an NFκB reporter cell line (Figure 3). Fibroblasts transfected with an NFκB response element linked to luciferase produce high levels of the enzyme upon exposure to TNFα. Activation of NFκB by TNFα was dose-dependently suppressed by the SK inhibitor, ABC294640, with an IC50 of approximately 20 μM, which corresponds well to its potency for inhibiting S1P production in cells. The sensitivity of these cells to organic solvents precluded similar studies with ABC747080, which is typically solubilized with DMSO or EtOH for in vitro studies.
Figure 3. Inhibition of TNFα-induced activation of NFκB by ABC294640.
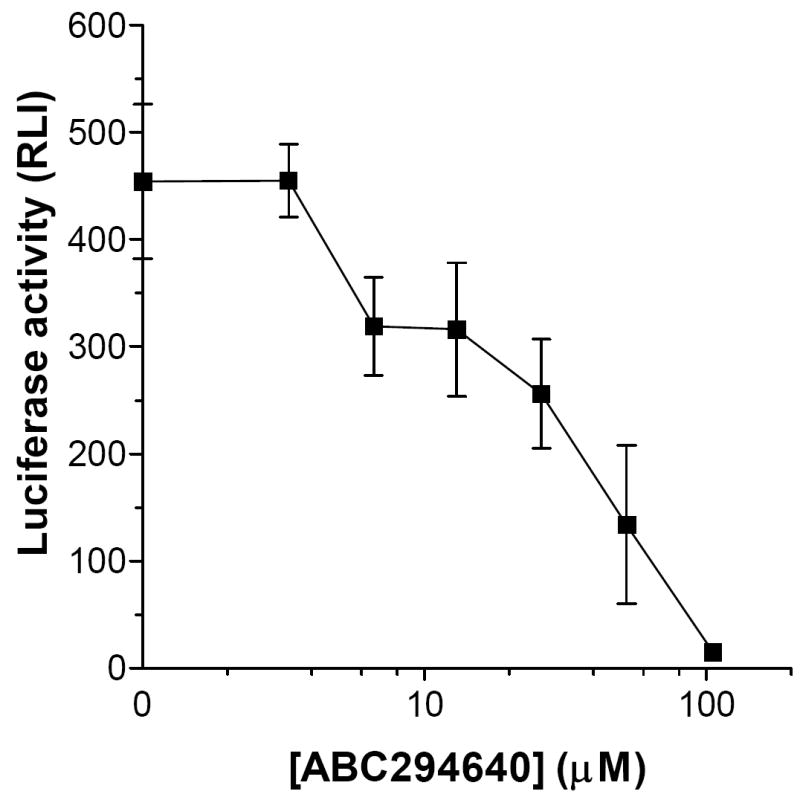
Fibroblasts transfected with a TNFα-responsive promoter linked to luciferase were treated with the indicated concentrations of ABC294640 and then treated with TNFα for 6 hr. The amount of luciferase expressed by the cells was then measured by luminescence. Values represent the mean ± sd luciferase activity in triplicate samples in a typical experiment, each experiment was performed twice.
Western analyses were conducted with human endothelial cells to evaluate the effects of the SK inhibitors on signaling proteins known to be regulated by TNFα. In these experiments, the cells were serum-starved for 24 hours and then exposed to TNFα (100 ng/mL) for 6 hours. Cell lysates from treated cells were assayed for the adhesion molecules ICAM and VCAM. As indicated in Figure 4, TNFα caused marked increases in the expression levels of adhesion proteins involved in leukocyte recruitment, including ICAM-1 and VCAM-1. These effects of TNFα were attenuated by treating the cells with either ABC294640 or ABC747080, such that the induction of both proteins was completely abrogated by 25 μM ABC294640.
Figure 4. Inhibition of TNFα-induced adhesion molecule expression by SK inhibitors.
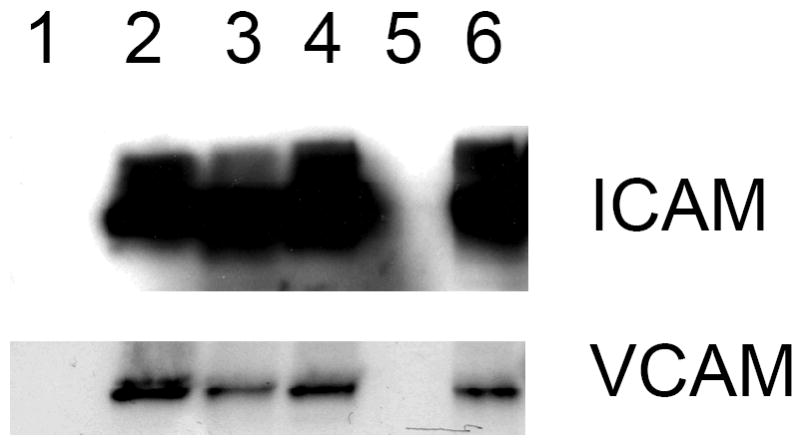
Human endothelial cells were left untreated (Lane 1) or treated with TNFα (Lanes 2-6) in the presence of DMSO (lane 2), 25 μM or 6 μM ABC747080 (lanes 3 and 4), or 25 μM or 6 μM ABC294640 (lanes 5 and 6). Adhesion molecule expression was then measured by western blotting. Actin was measured and showed equal loading of all lanes (data not shown).
To determine the effect of the SK inhibitors on COX-2 activity, an ELISA assay was used to measure PGE2 production by IEC6 rat intestinal epithelial cells and human endothelial cells treated with TNFα. Exposure of either type of cell to TNFα resulted in marked increases in Cox-2 activity, measured as the production of PGE2 (Figure 5). This induction of Cox-2 activity by TNFα was strongly suppressed by ABC294640 or ABC747080.
Figure 5. Inhibition of TNFα-induced Cox-2 activity by SK inhibitors.
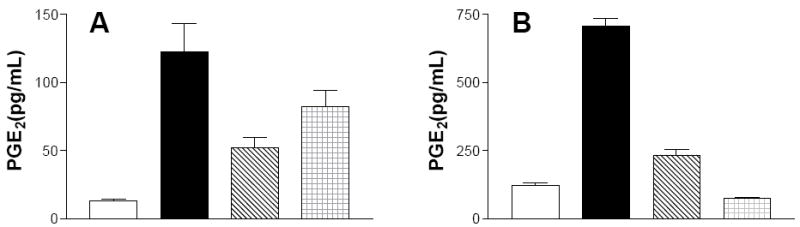
Rat IEC6 cells (Panel A) or human endothelial cells (Panel B) were incubated for 18 h with DMSO as a solvent control (open bars), or 100 ng TNFα / mL in the presence of DMSO (solid bars) or 10 μg/mL ABC294640 (cross-hatched bars) or ABC747080 (checkered bars). Levels of PGE2 secreted into the medium were quantified by ELISA. Values represent the mean ± sd for triplicate samples in a typical experiment, each experiment was performed twice.
In vivo effects of SK inhibitors in DSS-models of ulcerative colitis
The acute model of DSS-induced ulcerative colitis utilized male C57BL/6 mice, which were randomly divided into groups of 5 or 6 for DSS- and drug-treatments. The SK inhibitors were dissolved in PEG400, and given once daily by oral gavage in a volume of 0.1 mL per dose. Dipentum, an FDA-approved anti-colitis drug whose active ingredient, olsalazine, is converted to 5-aminosalicylic acid in vivo, was used as a positive control. The mice were given normal drinking water or 2% DSS and treated orally with an SK inhibitor or Dipentum at a dose of 50 mk/kg daily, and the DAI, which monitors weight loss, stool consistency and blood in the stool as a measure of disease severity, was scored for each animal on Days 4-6.
Animals receiving normal drinking water and PEG as a solvent control had very low DAIs throughout the experiment (Figure 6). Exposure of the mice to DSS in their drinking water markedly induced IBD symptoms, including weight loss and the production of loose, bloody stools. The intensity of the disease progressively increased from Day 4 to the time the mice were sacrificed on Day 6. Treatment of the animals receiving DSS with ABC294640, ABC747080 or Dipentum reduced the intensity of the IBD manifestations in the mice, most dramatically on Day 6. The SK inhibitors and Dipentum were essentially equivalent in their abilities to reduce the DAI of mice receiving DSS. It should be noted that this acute model produces rapid and dramatic symptoms of IBD, making it a very stringent assay for drug testing.
Figure 6. Effects of SK inhibitors and Dipentum on the DAI in the acute DSS-colitis model.
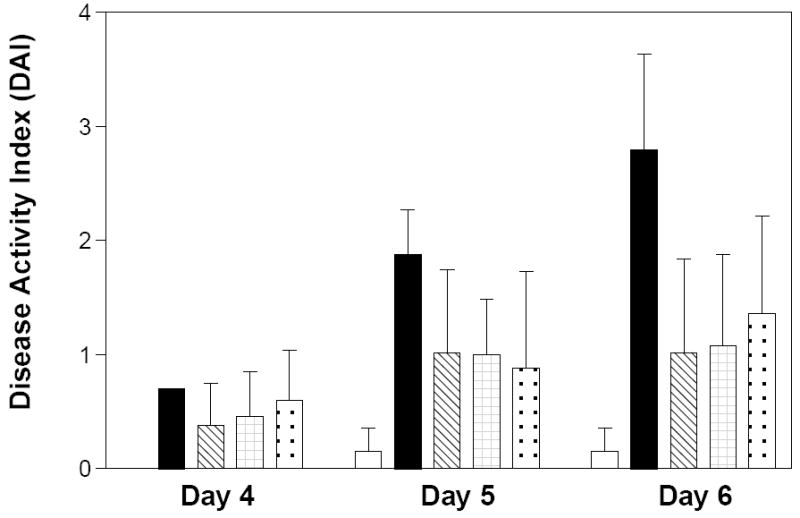
C57BL/6 mice were treated for 6 days as follows: normal drinking water and daily oral administration of PEG (open bars), 2% DSS in the drinking water and daily oral administration of PEG (solid bars); 2% DSS in the drinking water and daily oral administration of 50 mg/kg ABC294640 in PEG (cross-hatched bars), 2% DSS in the drinking water and daily oral administration of 50 mg/kg ABC747080 in PEG (checkered bars), 2% DSS in the drinking water and daily oral administration of 50 mg/kg Dipentum in PEG (dotted bars). On the indicated day, the Disease Activity Index was calculated for each group. Values represent the mean ± sd for 5 -6 mice per group.
On Day 6, the animals were sacrificed by cervical dislocation and the entire colon was measured to assess shortening due to scarring and damage, and then fixed, sectioned and examined histologically on a blinded basis. Compared with the water control group, the colons of mice treated with DSS and PEG were significantly shortened (Figure 7). DSS-treated mice that were also treated with ABC294640, ABC747080 or Dipentum had colons of intermediate length, indicating substantial protection by the drugs. Again, the response to either of the SK inhibitors was at least as good as that of mice treated with Dipentum.
Figure 7. Effects of SK inhibitors and Dipentum on colon length in the acute DSS-colitis model.
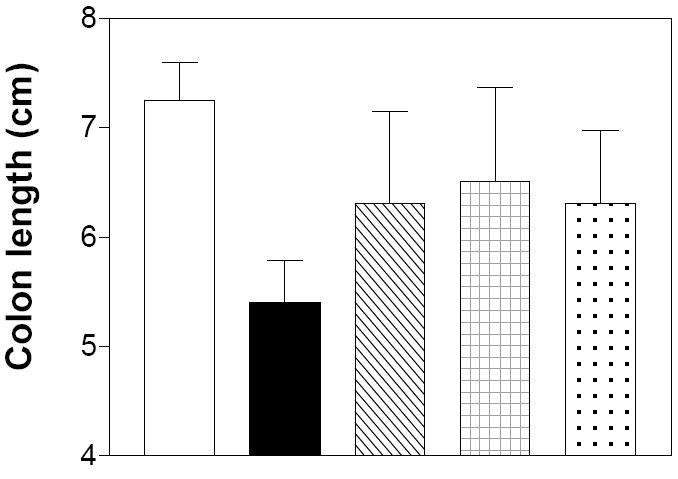
Mice from the experiment described in Figure 6 were sacrificed on Day 6, and the colon was harvested from each animal and measured. Data represent the mean ± sd colon length.
Histological examination of colon sections from the various treatment groups were consistent with the DAI endpoint, revealing marked damage in the DSS-alone group that was reduced or negated in the SK inhibitor-treated animals (Figure 8). Panel A shows a normal colon from a water-treated control animal, while Panel B shows a severely inflamed and damaged colon from a DSS-treated animal. Numerous neutrophils were present throughout the section (arrow a), along with severely damaged crypts (arrow b), and moderate inflammatory infiltration with submucosal edema (arrow c). A section from a partial responder to ABC294640 is shown in Panel C and indicates only mild crypt damage, a low level of inflammatory cell infiltration and no edema in the submucosa. A section from a complete responder to ABC294640 is shown in Panel D, which demonstrated normal crypt morphology and few infiltrating inflammatory cells, similar to the control specimens.
Figure 8. Histology of colons from mice in the acute DSS-colitis model.
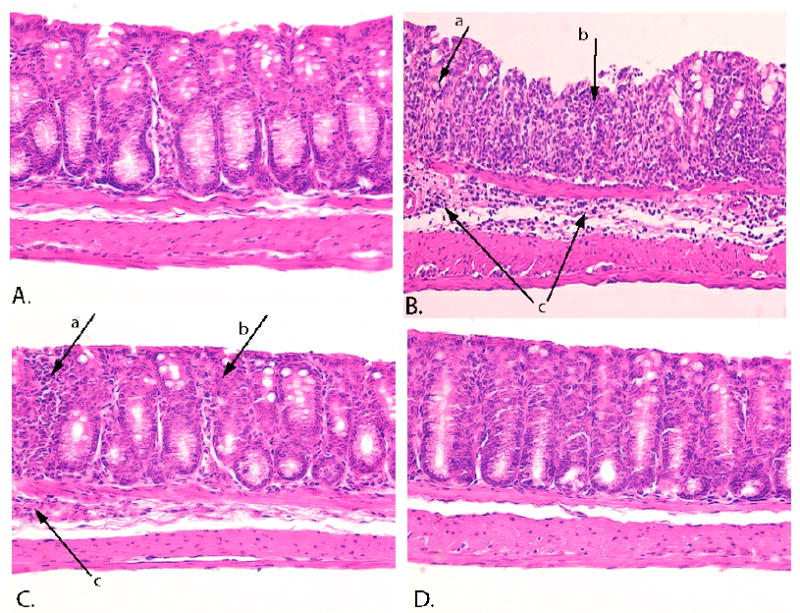
Thin sections of colons from the animals described in Figure 6 were stained with H&E and examined for pathologic changes. Panel A - water-treated control animal. Panel B - DSS-treated animal. Panel C - a moderate responder to ABC294640. Panel D - a complete responder to ABC294640.
As a quantifiable measure of damage, the colons were graded for their Histology Score, which is based on inflammation severity, inflammation extent, crypt damage and the percentage of surface area demonstrating the characteristic. These morphologies were scored on a blinded basis. As indicated in Figure 9, animals receiving DSS in their drinking water had substantially higher Histology Scores (representing moderate-to-severe IBD) than animals receiving normal drinking water (which had some mild inflammation, possibly due to the PEG vehicle). As with the other assays, the Histology Scores of mice given an SK inhibitor or Dipentum were consistently lower than the DSS-alone animals, although not all animals were fully protected. DAI scores and histology scores correlated well for the individual animals (data not shown), confirming that the DAI score as an excellent indicator of colon inflammation and damage.
Figure 9. Effects of SK inhibitors and Dipentum on the colon Histology Score in the acute DSS-colitis model.
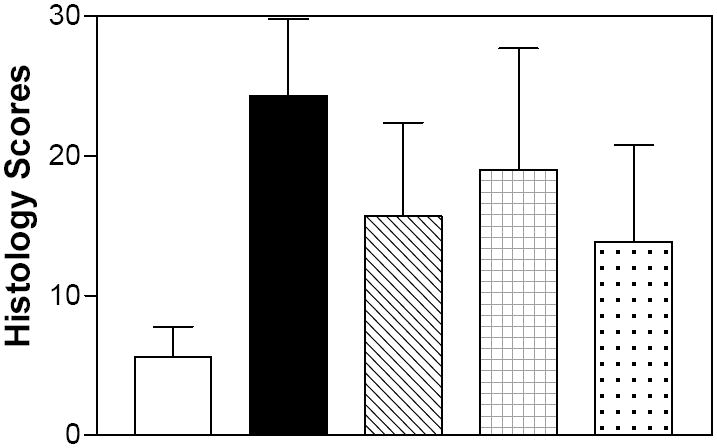
Mice from the experiment described in Figure 6 were sacrificed on Day 6, and the colon was harvested from each animal and the Histology Score was determined. Values represent the mean ± sd for 5 – 6 mice per group.
Myeloperoxidase (MPO) activity, which is reflective of neutrophil influx into the colon, is often used as measure of inflammation, and was assayed in the colons of the mice from the DSS-colitis studies. As indicated in Figure 10, MPO activity was highly elevated in the DSS-alone animals compared to water controls. The increase in MPO activity was markedly attenuated in mice receiving daily doses of ABC294640, ABC747080 or Dipentum. This reduction in the activity of the neutrophil marker is consistent with the decreased occurrence of granulocytes observed in the H&E-stained colon sections (Figure 8). Therefore, the level of colonic MPO appears to be an excellent biomarker for the extent of tissue infiltration by inflammatory leukocytes.
Figure 10. Effects of SK inhibitors and Dipentum on neutrophil infiltration into the colon in the acute DSS-colitis model.
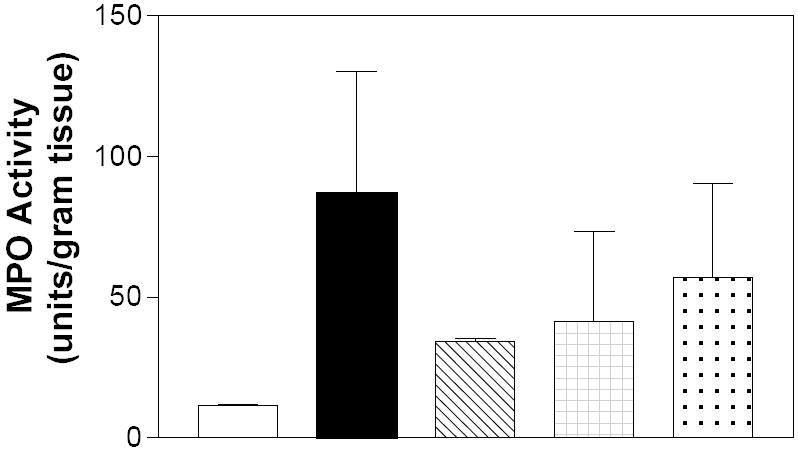
Myeloperoxidase activity from the colons of the animals described in Figure 6 was measured as described in the Materials and Methods section. Values the mean ± sd MPO activity in units per gram of tissue.
Several cytokines involved in inflammation were measured by the Cytokine Core Laboratory at the University of Maryland, Baltimore, using the Luminex 100 System that allows the quantification of multiple cytokines and growth factors in a small sample volume. We examined the Th1 cytokine IFN-γ, the regulatory IL-10 cytokine, as well as the macrophage-derived pro-inflammatory cytokines, TNFα, IL-1β, IL-6 in colon samples from mice in the DSS model of colitis. Figure 11 depicts the results of these assays, and indicates that DSS-treatment promoted the accumulation of all of the cytokines in the colon. Importantly, the elevations of all of the pro-inflammatory proteins, i.e. IFN-γ, IL-1β, IL-6 and TNFα, were attenuated in mice treated with either an SK inhibitor or Dipentum. Conversely, levels of the anti-inflammatory cytokine IL-10 were not suppressed by the SK inhibitors.
Figure 11. Effects of SK inhibitors and Dipentum on colonic cytokine levels in the acute DSS-colitis model.

Colon samples from mice described in Figure 6 were extracted and assayed for the levels of the indicated cytokines as described in the Material and Methods section. Values represent the mean ± sd amount of each cytokine in 4-5 samples per group.
As a final measure of the effects of the SK inhibitors in this acute model, S1P levels were assayed in the colons of the DSS-treated animals using an LC-MS/MS method. This technique allows us to examine correlations between biologic activity and changes in S1P levels in animals treated with the SK inhibitors. Samples of colons from animals from the DSS-colitis experiments were homogenized in cold PBS, spiked with internal standards (C17 analogs of sphingosine and S1P) and processed by liquid-liquid extraction. Ratios of analyte to internal standard for each sphingolipid were determined. S1P levels were markedly higher in the colons from DSS-treated mice as compared to the water controls (Figure 12). Importantly, animals that were treated with either ABC294640 or ABC747080 had markedly lower levels of colonic S1P than the DSS-alone samples.
Figure 12. Effects of SK inhibitors on S1P levels in the colons of the animals in the DSS-colitis model.
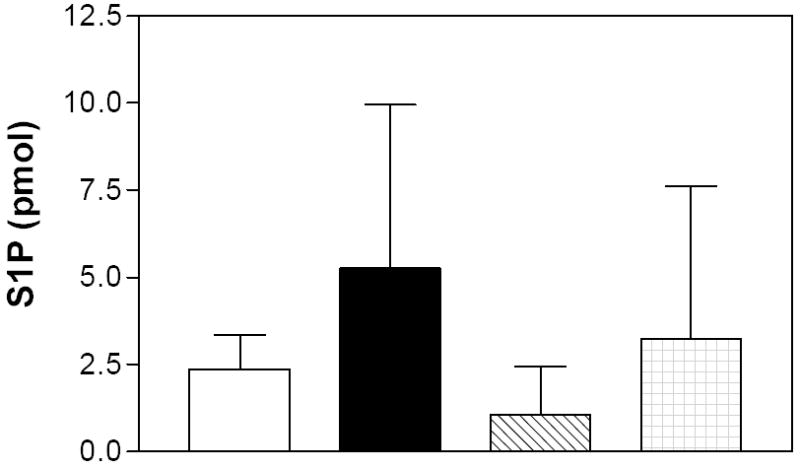
Colon samples from mice described in Figure 6 were extracted and assayed for the levels of S1P by LC/MS/MS as described in the Material and Methods section. Values represent the mean ± sd for 4-5 samples per group.
CHRONIC MODEL
A 35-day model of IBD was used to evaluate the effectiveness of the SK inhibitors in mice that experience multiple cycles of DSS-induced inflammation. This chronic model is similar to the acute model, except that the DSS concentration in the drinking water is lower and animals receive periodic exposure to DSS (DSS on days 1-7, water on Days 8-13, DSS on day 14-21, water on Days 22-27 and then DSS until the completion of the study on Day 35). In these experiments, treatment of the mice with an SK inhibitor or Dipentum began on Day 28 and continued daily until the completion of the study. The DAI index was monitored every other day until Day 28 and then daily until Day 35. Animals were sacrificed on Day 35, and changes in the colon length and cytokine profiles were measured.
As previously described by others, cyclic exposure of mice to DSS in their drinking water caused reversible increases in the DAI (Figure 13). Treatment of the mice with ABC294640, ABC747080 or Dipentum during the third exposure to DSS significantly suppressed the increase in DAI experienced by the control mice (P < 0.001 for all three compounds on Day 35).
Figure 13. Effects of SK inhibitors on the DAI in the chronic DSS-colitis model.
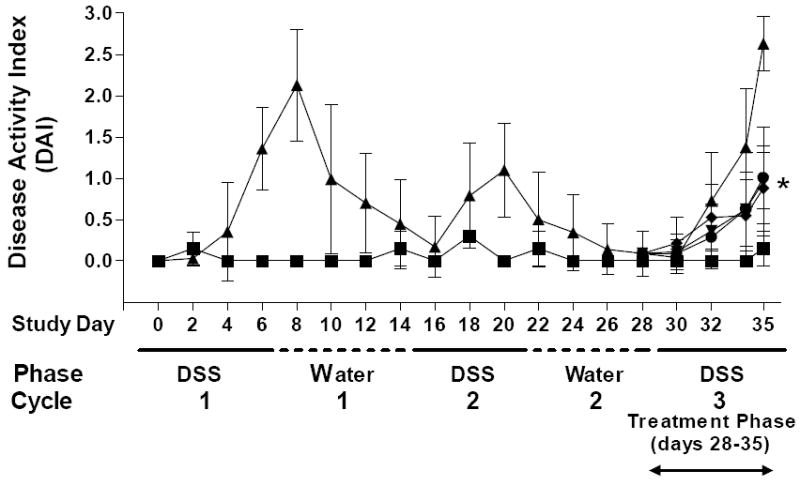
Mice received 2 cycles (7 days per cycle) of DSS (1.5% cycle 1 and 1% cycle 2), 2 cycles of normal drinking water and were randomized by DAI on Day 28 into groups of 8 mice. The mice were then treated as follows: Group 1 (■) - normal drinking water and orally dosed with PEG400 every day for 7 days (water control); Group 2 (▲) - drinking water containing 1.5% DSS and orally dosed with PEG daily for 7 days; Group 3 (▼) - drinking water containing 1.5% DSS and orally dosed with ABC294640 (50 mg/kg) every day for 7 days; Group 4 (●) - drinking water containing 1.5% DSS and orally dosed with ABC747080 (50 mg/kg) daily for 7 days; Group 5 (◆) - drinking water containing 1.5% DSS and orally dosed with Dipentum (50 mg/kg). *p < 0.001.
The colon lengths of DSS-treated mice were significantly shorter than the water-treated control animals (4.9 ± 0.2 cm vs. 7.8 ± 0.3 cm) reflecting inflammation-induced scarring. As in the acute model, the colons of animals treated with ABC294640, ABC747080 or Dipentum were of intermediate length (6.2 ± 0.2, 5.8 ± 0.1 and 6.1 ± 0.2 cm, respectively). This is a significant finding since the animals were untreated for the first and second DSS cycles. Therefore, suppression of inflammation-induced colon contraction can be reversed by effective anti-IBD drugs.
Immunohistochemistry revealed that SK expression was present in low levels in the colons of control, non-DSS treated mice. SK expression was elevated in the colons of DSS treated mice compared to water controls with this expression clearly reduced in DSS mice also receiving compound ABC292640 (Figure 14).
Figure 14. Immunohistochemistry of SK expression in mice undergoing a 35-day chronic DSS model.
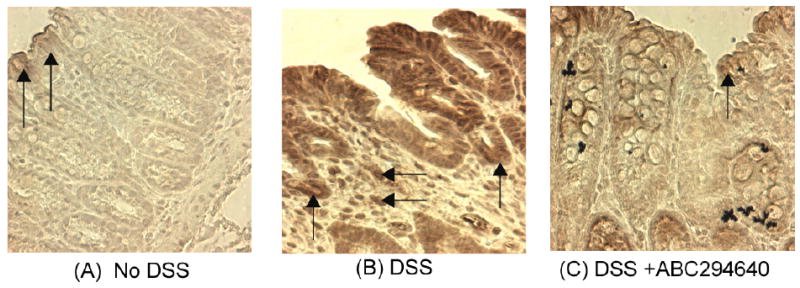
Panel A shows minimal expression of SK in water control mice with light staining on epithelial cells (vertical arrows). Panel B shows a DSS only treated mouse with clear evidence of enhanced staining on the colonic crypts (vertical arrows) as well as in cells in the lamina propria (horizontal arrows). Panel C shows a DSS treated mouse receiving 50 mg/kg ABC292640 for the 7 days prior to sacrifice. There is reduced SK staining as compared to the DSS alone group with light to moderate staining throughout including on epithelial cells (vertical arrows).
S1P levels in the colons of the chronic colitis model mice were assessed in an identical manner as described for the acute model and revealed results similar to those in the acute model with elevated S1P levels in DSS alone treated mice as compared to water controls (Figure 15). Treatment with compounds ABC29460 and ABC747080 (oral 50mg/kg daily; 7 days prior to sacrifice) resulted in significant reductions of S1P levels (Figure 15).
Figure 15. Effects of SK inhibitors on S1P levels in the colons of the animals in the chronic DSS-colitis model.
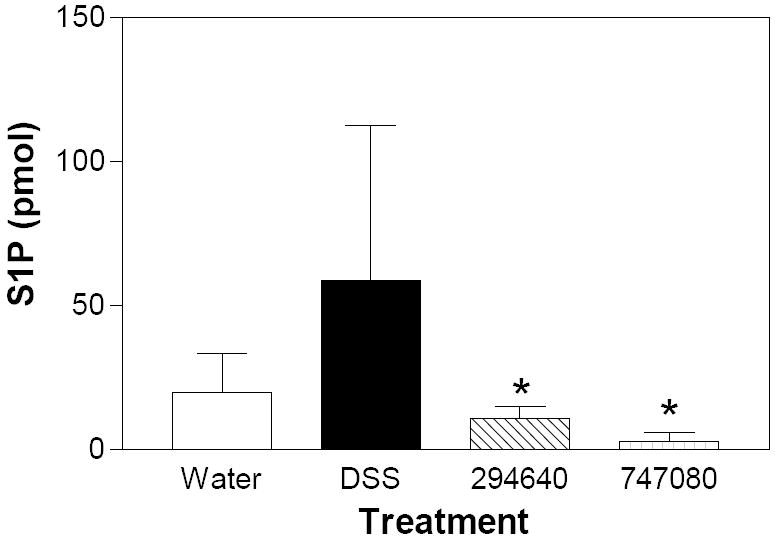
Colon samples from mice described in Figure 13 were extracted and assayed for the levels of S1P by LC/MS/MS as described in the Material and Methods section. Values represent the mean ± sd for 8 samples per group; * p<0.05.
The levels of the pro-inflammatory cytokines TNFα, IL-1β, IFN-γ and IL-6 were substantially increased in the colons of mice treated chronically with DSS; whereas, the level of IL-10 was unchanged (Figure 16). Mice treated with ABC294640 during the final DSS cycle had reduced levels of the pro-inflammatory cytokines, while animals treated with ABC747080 or Dipentum expressed cytokine profiles equivalent to the DSS-alone group. This may reflect the presence of high numbers of resident immune cells in the colons of mice exposed chronically to DSS. However, the elevation in cytokine levels in the SK inhibitor-treated mice does not result in increased DAI or colon shortening, indicating that signaling induced by the inflammatory cytokines had been blocked.
Figure 16. Effects of SK inhibitors and Dipentum on colonic cytokine levels in the chronic DSS-colitis model.

Colon samples from mice described in Figure 13 were extracted, and assayed for the levels of the indicated cytokines as described in the Material and Methods section. Control (clear bars), DSS alone (filled bars), DSS plus ABC294640 (cross-hatched bars), DSS plus ABC747080 (checkered bars), and DSS plus Dipentum (dotted bars). Values represent the mean ± SD amount in 8 samples per group.
For comparison, the levels of the same cytokines in the serum of the mice at the time of sacrifice were also determined. As indicated in Figure 17, the circulating levels of these cytokines are markedly lower than the colonic levels reflecting the local inflammation in this model. DSS increased the circulating levels of IL-1β, IFN-γ, IL-6 and IL-10, while TNFα remained below the detection limit of the assay. None of the test compounds affected the circulating levels of IL-1β or INF-γ; however, both ABC294640 and Dipentum reduced the serum level of IL-6. Therefore, serum levels of IL-6 may be a useful pharmacodynamic marker for the anti-inflammatory effects of the SK inhibitors during clinical testing.
Figure 17. Effects of SK inhibitors and Dipentum on serum cytokine levels in the chronic DSS-colitis model.

Serum from mice described in Figure 13 was assayed for the levels of the indicated cytokines as described in the Materials and Methods section. Control (clear bars), DSS alone (filled bars), DSS plus ABC294640 (cross-hatched bars), DSS plus ABC747080 (checkered bars), and DSS plus Dipentum (dotted bars). Values represent the mean ± SD amount in 8 samples per group.
Discussion
The SK inhibitors described in this report were originally envisioned to provide new anti-proliferative and pro-apoptotic agents for the treatment of cancer. However, recent recognition of the importance of sphingolipid metabolism in mediating the effects of inflammatory cytokines promoted our exploration of the effects of these compounds using in vitro and in vivo models of IBD. Overall, these results indicate that the SK inhibitors under development are promising new agents for evaluation in the clinical treatment of IBDs. In vitro experiments showed that the SK inhibitors attenuate many of the effects of TNFα, including expression of VCAM and ICAM. This is interesting as all three of these proteins are current targets for IBD treatment, as Remicade (a TNFα scavenger) is on the market, Tysabri (which blocks VCAM-1) is in phase III trials for Crohn’s Disease and Alicaforsen (an antisense inhibitor of ICAM-1) recently completed a successful phase II trial. Therefore, the combined mechanistic data support the hypothesis that SK inhibitors are likely to be effective for clinically preventing and/or treating IBD mediated by inflammatory cytokines.
Evidence to support the advancement of the SK inhibitors toward the clinic was provided in both acute and chronic mouse models of ulcerative colitis. In both models, the Disease Activity Index was reduced by the SK inhibitors to a similar, or lower level, than the positive control drug, Dipentum, which is currently on the market for mild-to-moderate IBDs. Treatment of the DSS-exposed mice with an SK inhibitor resulted in colons that were longer and less damaged as determined by histological evaluation. Reflecting this protection, a broad spectrum of inflammatory mediators, including S1P, NFκB, TNFα, VCAM-1, ICAM-1, COX-2, myeloperoxidase activity, IL-1β, IFN-γ, and IL-6, were favorably modulated by treatment of the animals with an SK inhibitor. The efficacy of these findings provide further evidence that the anti-IBD activity of the compounds under study is linked to SK inhibition and decreased S1P formation in the colons of the mice.
The findings of elevated SK and S1P levels in the colons of mice experiencing ulcerative colitis are novel, and lend further evidence that these agents play important roles in the etiology of IBD. Interestingly, colon samples from patients with Crohn’s Disease demonstrate similar elevations of SK expression (unpublished observations), indicating that this pathway is also aberrantly activated in human IBD. Studies are currently underway to determine the relationships between SK overexpression, IBD severity and response to chemotherapy.
A model for the roles of sphingolipid metabolites in the pathology of IBDs is summarized in Figure 18. A combination of events occurs in the intestinal endothelial and epithelial cells and recruited mast cells, macrophages and neutrophils. Early in the disease, immunologic reactions or other activating signals promote the release of inflammatory cytokines, particularly TNFα from macrophages and mast cells. Several studies have shown that the actions of TNFα are mediated through its activation of S1P production. For example, TNFα induces S1P production in endothelial cells [14,34], hepatocytes [35], neutrophils [36], monocytes [37], fibroblasts [21] and lung adenocarcinoma cells [21] by activation of sphingomyelinase, ceramidase and SK. S1P is a central player in the pathway since it has pleiotropic actions on the mucosal epithelial cells, macrophages, mast cells and neutrophils. Within the intestinal endothelial cells, S1P activates NFκB thereby inducing the expression of adhesion molecules, COX-2 resulting in PGE2 synthesis, and NOS producing nitric oxide. Together, these chemoattractants and the adhesion molecules promote neutrophil infiltration into the mucosa. Sphingosine kinase has been directly demonstrated to play a significant role in the recruitment and function of these cells [22,38]. At the same time, S1P activates the neutrophils resulting in the release of oxygen free radicals that further inflame and destroy epithelial tissue. Similarly, S1P promotes the activation and degranulation of mast cells.
Figure 18. A model for the role of sphingolipid metabolism in IBD.
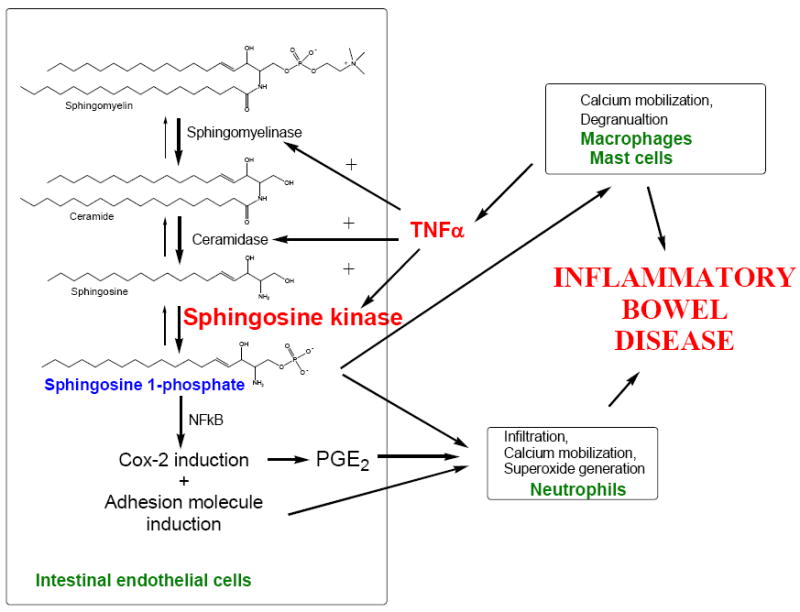
According to this model, two major targets for new anti-IBD therapies can be defined: TNFα and S1P. A great deal of effort has focused on developing anti-TNFα agents leading to the introduction of Remicade and related agents to the clinic. The use of inhibitors of SK as anti-IBD agents has not been previously addressed, largely because of the heretofore lack of potent, specific SK inhibitors that are pharmacologically active. We have now identified novel small molecule inhibitors of SK that have in vivo anti-IBD activity. We believe that these compounds will be useful for the treatment and/or prevention of IBDs by blocking the production of S1P in response to TNFα. This will prevent the deleterious activation of neutrophils, macrophages and mast cells, as well as prevent neutrophil infiltration into the mucosa. Overall, it is clear that S1P is a critical component in inflammation, making SK an excellent target for the development of new anti-inflammatory drugs for the treatment of ulcerative colitis and/or Crohn’s Disease.
Acknowledgments
This work was supported by grant 1 R43 DK071395 from the National Institutes of Health.
Abbreviations
- IBD
inflammatory bowel disease
- SK
sphingosine kinase
- S1P
sphingosine-1-phosphate
- TNFα
tumor necrosis factor alpha
- PGE2
prostaglandin E2
- NFkB
nuclear factor kB
- DSS
dextran sulfate sodium
- IL
interleukin
- IFNγ
interferon gamma
- COX-2
cyclooxygenase-2
- ICAM-1
intracellular adhesion molecule-1
- VCAM-1
vascular cell adhesion molecule-1
- PEG
polyethylene glycol
- DAI
disease activity index
- MPO
myeloperoxidase
- PBS
phosphate buffered saline
- LC-MS/MS
liquid chromatography mass spectroscopy
References
- 1.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rask-Madsen J. Soluble mediators and the interaction of drugs in IBD. Drugs today (Barc) 1998;34:45–63. doi: 10.1358/dot.1998.34.1.485200. [DOI] [PubMed] [Google Scholar]
- 3.He S. Key role of mast cells and their major secretory products in inflammotory bowel disease. World J Gastroenterology. 2004;10(3):309–318. doi: 10.3748/wjg.v10.i3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandborn WJ. Strategies for targeting tumour necrosis factor in IBD. Best Pract Res Clin Gastroenterol. 2003;17:105–117. doi: 10.1053/bega.2002.0345. [DOI] [PubMed] [Google Scholar]
- 5.Loncar M, Al-azzeh ED, Sommer PS, Marinovic M, Schmehl K, Kruschewski M, Blin N, Stohwasser R, Gott P, Kayademir T. Tumour necrosis factor alpha and nuclear factor kappa B inhibits transcription of human TFF3 encoding a gastrointestinal healing peptide. Gut. 2003;52:1297–1303. doi: 10.1136/gut.52.9.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van der Woude C, Kleibeuker JH, Jansen PL, Moshage H. Chronic inflammation, apoptosis and (pre-)malignant lesions in the gastro-intestinal tract. Apoptosis. 2004;9:123–130. doi: 10.1023/B:APPT.0000018794.26438.22. [DOI] [PubMed] [Google Scholar]
- 7.Singh V, Patil CS, Jain NK, Singh A, Kulkarni SK. Effecto of nimesulide on acetic acid- and leukotriene-induced inflammatory bowel disease in rats. Prostaglandins Other Lipid Mediat. 2003;71:163–175. doi: 10.1016/s1098-8823(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 8.Mahadevan U, Loftus EV, Tremaine WJ, Sandborn WJ. Safety of selective cyclooxygenase-2 inhibitors in inflammatory bowel disease. American Journal of Gastroenterology. 2002;97:910–914. doi: 10.1111/j.1572-0241.2002.05608.x. [DOI] [PubMed] [Google Scholar]
- 9.Kruidenier L, K I, Lamers CB, Verspaget HW. Intestinal oxicative damage in inflammatory bowel disease: semi-quantification, localization and association with mucosal antioxidants. Journal of Pathology. 2003;201:28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 10.Olivera A, Spiegel S. Sphingosine kinase: a mediator of vital cellular functions. Prostaglandins. 2001;64:123–134. doi: 10.1016/s0090-6980(01)00108-3. [DOI] [PubMed] [Google Scholar]
- 11.Spiegel S. Sphingosine 1-phosphate: a prototype of a new class of second messengers. J Leukoc Biol. 1999;65:341–344. doi: 10.1002/jlb.65.3.341. [DOI] [PubMed] [Google Scholar]
- 12.Alessenko AV. The role of sphingomyelin cycle metabolites in transduction of signals of cell proliferation, differentiation and death. Membr Cell Biol. 2000;13:303–320. [PubMed] [Google Scholar]
- 13.Mathias S, Dressler KA, Kolesnick RN. Characterization of a ceramide-activated protein kinase: stimulation by tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 1991;88:10009–10013. doi: 10.1073/pnas.88.22.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia P, Gamble JR, Rye KA, Wang L, Hii CS, Cockerill P, Khew-Goodall Y, Bert AG, Barter PJ, Vadas MA. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci U S A. 1998;95:14196–14201. doi: 10.1073/pnas.95.24.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathias S, Pena LA, Kolesnick RN. Signal transduction of stress via ceramide. Biochem J. 1998;335(Pt 3):465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry DK, Hannun YA. The role of ceramide in cell signaling. Biochim Biophys Acta. 1998;1436:233–243. doi: 10.1016/s0005-2760(98)00145-3. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa M, Jayadev S, Tsujimoto M, Hannun YA, Ito F. Role of ceramide in stimulation of the transcription of cytosolic phospholipase A2 and cyclooxygenase 2. Biochem Biophys Res Commun. 1996;220:681–686. doi: 10.1006/bbrc.1996.0464. [DOI] [PubMed] [Google Scholar]
- 18.Rabano M, Pena A, Brizuela L, Marino A, Macarulla JM, Trueba M, Gomez-Munoz A. Sphingosine-1-phosphate stimulates cortisol secretion. FEBS Lett. 2003;535:101–105. doi: 10.1016/s0014-5793(02)03882-6. [DOI] [PubMed] [Google Scholar]
- 19.Yatomi Y, Ruan F, Hakomori S, Igarashi Y. Sphingosine-1-phosphate: a platelet-activating sphingolipid released from agonist-stimulated human platelets. Blood. 1995;86:193–202. [PubMed] [Google Scholar]
- 20.Prieschl EE, Csonga R, Novotny V, Kikuchi GE, Baumruker T. The balance between sphingosine and sphingosine-1-phosphate is decisive for mast cell activation after Fc epsilon receptor I triggering. J Exp Med. 1999;190:1–8. doi: 10.1084/jem.190.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. Faseb J. 2003;17:1411–1421. doi: 10.1096/fj.02-1038com. [DOI] [PubMed] [Google Scholar]
- 22.MacKinnon AC, Buckley A, Chilvers ER, Rossi AG, Haslett C, Sethi T. Sphingosine kinase: a point of convergence in the action of diverse neutrophil priming agents. J Immunol. 2002;169:6394–6400. doi: 10.4049/jimmunol.169.11.6394. [DOI] [PubMed] [Google Scholar]
- 23.Itagaki K, Kannan KB, Hauser CJ. Lysophosphatidic acid triggers calcium entry through a non-store-operated pathway in human neutrophils. J Leukoc Biol. 2005;77:181–189. doi: 10.1189/jlb.0704390. [DOI] [PubMed] [Google Scholar]
- 24.French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 25.French KJ, Upson JJ, Smith SN, Woll M, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. doi: 10.1124/jpet.106.101345. submitted. [DOI] [PubMed] [Google Scholar]
- 26.French KJ, Zhuang Y, Xia Z, Upson JJ, Smith CD. Pharmacokinetics of orally-available inhibitors of sphingosine kinase. doi: 10.1007/s10620-007-0133-6. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bohlen P, Stein S, Dairman W, Udenfriend S. Fluormetric assay of proteins in the nanogram range. Arch Biochem Biophys. 1973;155:213–220. doi: 10.1016/s0003-9861(73)80023-2. [DOI] [PubMed] [Google Scholar]
- 28.Fitzpatrick LR, Wang J, Le M. In vitro and in vivo effects of gliotoxin, a fungal metabolite: Efficacy against dextran sodium sulfate-induced colitis in rats. Dig Dis Sci. 2000;45:2327–2336. doi: 10.1023/a:1005630723111. [DOI] [PubMed] [Google Scholar]
- 29.McCafferty DM, Miampamba M, Sihota E, Sharkey KA, Kubes P. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut. 1999;45:864–873. doi: 10.1136/gut.45.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roviezzo F, Bucci M, Delisle C, Brancaleone V, Di Lorenzo A, Mayo IP, Fiorucci S, Fontana A, Gratton JP, Cirino G. Essential requirement for sphingosine kinase activity in eNOS-dependent NO release and vasorelaxation. Faseb J. 2006;20:340–342. doi: 10.1096/fj.05-4647fje. [DOI] [PubMed] [Google Scholar]
- 31.Williams KL, Fuller CR, Dieleman LA, DaCosta CM, Haldeman KM, Sartor RB, Lund PK. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 2001;120:925–937. doi: 10.1053/gast.2001.22470. [DOI] [PubMed] [Google Scholar]
- 32.Culmsee C, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JH, Mattson MP, Krieglstein J. Nerve growth factor survival signaling in cultured hippocampal neurons is mediated through TrkA and requires the common neurotrophin receptor P75. Neuroscience. 2002;115:1089–1108. doi: 10.1016/s0306-4522(02)00539-0. [DOI] [PubMed] [Google Scholar]
- 33.Maines LW, Antonetti DA, Wolpert EB, Smith CD. Evaluation of the role of P-glycoprotein in the uptake of paroxetine, clozapine, phenytoin and carbamazapine by bovine retinal endothelial cells. Neuropharmacology. 2005;49:610–617. doi: 10.1016/j.neuropharm.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 34.Xia P, Wang L, Gamble JR, Vadas MA. Activation of sphingosine kinase by tumor necrosis factor-alpha inhibits apoptosis in human endothelial cells. J Biol Chem. 1999;274:34499–34505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 35.Osawa Y, Banno Y, Nagaki M, Brenner DA, Naiki T, Nozawa Y, Nakashima S, Moriwaki H. TNF-alpha-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J Immunol. 2001;167:173–180. doi: 10.4049/jimmunol.167.1.173. [DOI] [PubMed] [Google Scholar]
- 36.Niwa M, Kozawa O, Matsuno H, Kanamori Y, Hara A, Uematsu T. Tumor necrosis factor-alpha-mediated signal transduction in human neutrophils: involvement of sphingomyelin metabolites in the priming effect of TNF-alpha on the fMLP-stimulated superoxide production. Life Sci. 2000;66:245–256. doi: 10.1016/s0024-3205(99)00587-1. [DOI] [PubMed] [Google Scholar]
- 37.Fueller M, Wang de A, Tigyi G, Siess W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell Signal. 2003;15:367–375. doi: 10.1016/s0898-6568(02)00117-1. [DOI] [PubMed] [Google Scholar]
- 38.Jolly P, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. Transactivation of sphingosine-1-phospate receptors by Fc{varepsilon}RI triggering is required for normal mast cell degranulation and chemotaxis. Journal of Experimental Medicine. 2004;199:959–970. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]