

Abstract
Drug combination therapies for central nervous system (CNS) demyelination diseases including multiple sclerosis (MS) are gaining momentum over monotherapy. Over the past decade, both in vitro and in vivo studies established that statins (HMG-CoA reductase inhibitors) and rolipram (phosphodiesterase-4 inhibitor; blocks the degradation of intracellular cyclic AMP) can prevent the progression of MS in affected individuals via different mechanisms of action. In the present study, we evaluated the effectiveness of lovastatin and rolipram in combination therapy to promote neurorepair in an inflammatory CNS demyelination model of MS, experimental autoimmune encephalomyelitis (EAE). Combination treatment with suboptimal doses of these drugs in an established case of EAE (clinical disease score ≥2.0) significantly attenuated the infiltration of inflammatory cells and protected myelin sheath and axonal integrity in the CNS. It was accompanied with elevated level of cyclic AMP and activation of its associated protein kinase A. Interestingly, combination treatment with these drugs impeded neurodegeneration and promoted neurorepair in established EAE animals (clinical disease score ≥3.5) as verified by quantitative real-time polymerase chain reaction, immunohistochemistry, and electron microscopic analyses. These effects of combination therapy were minimal and/or absent with either drug alone in these settings. Together, these data suggest that combination therapy with lovastatin and rolipram has the potential to provide neuroprotection and promote neurorepair in MS, and may have uses in other related CNS demyelinating diseases.
Keywords: Combination therapy, EAE/MS, demyelination, inflammation
INTRODUCTION
Multiple sclerosis (MS) is an inflammatory central nervous system (CNS) demyelinating disease and experimental autoimmune encephalomyelitis (EAE) is an experimental model that mimics many aspects of MS (Lublin 1985). Pathophysiology of the MS disease includes breaching of the blood-brain barrier (BBB), infiltration of inflammatory cells (i.e., myelin reactive CD4+ and CD8+ T cells, and macrophages) into the CNS followed by activation of resident glial cells and secretion of inflammatory mediators leading to the formation of brain lesions (Markovic-Plese and McFarland 2001; Noseworthy et al. 2000). Demyelination in the MS lesion results from the break down of the myelin sheath and degeneration of both oligodendrocytes (OLs) and neuronal axons which may be a major determinant of progressive neurological disability in MS patients (Bjartmar et al. 2003; Wujek et al. 2002).
The concept of remyelination is a matter of interest because naturally occurring remyelination is impaired in some of the clinically silent (chronic) MS lesions due to the lack of myelinating OLs, except in the border region between the plaque and peri-plaque white matter (Barnett and Prineas 2004). Remyelination helps to preserve axons, restore conduction velocity, and repair lesions. Current therapies that essentially target CNS repair include the application of growth factors (Ransohoff et al. 2002), intravenous administration of remyelinating immunoglobulin autoantibodies (Sorensen 2003), and the transplantation of OL-progenitors (OPs) or embryonic stem cells (Brustle et al. 1999; Pluchino et al. 2003). The search for new disease modifying agents with different mechanisms of action are currently underway for use in MS because present FDA-approved therapies are only partially effective and are associated with side effects and potential toxicities (Arbizu et al. 2000; Dhib-Jalbut 2003).
Over the past decade, statins (HMG-CoA reductase inhibitors) have been exploited for their immunomodulatory characteristics i.e., immunomodulation of TH1 to TH2 immune responses for autoimmune diseases including MS (Paintlia et al. 2004; Youssef et al. 2002). Promising results were obtained in initial clinical trials of simvastatin both in MS (Vollmer et al. 2004) and rheumatoid arthritis (McCarey et al. 2004). In addition, our recent studies established that statins have the potential to promote remyelination in the MS brain (Paintlia et al. 2005). Similar to statins, inhibitors of phosphodiesterase (PDE)-4 including rolipram (which causes accumulation of intracellular cAMP) are reported to halt EAE development (Genain et al. 1995; Sommer et al. 1995) via immunomodulation of the TH1 to TH2 immune responses (Bielekova et al. 2000). In addition, elevated level of cAMP in neurons is reported to promote axonal growth and neuritis even in the presence of myelin-associated inhibitors of regeneration (Cui and So 2004; Domeniconi and Filbin 2005). Thus, an agent that augments immunomodulation of myelin-reactive T cells toward TH2 differentiation could be beneficial for combination therapy. Because cellular targets of both statins and rolipram differ with respect to their immunomodulatory activities, we hypothesized that reduced doses of these drugs in combination may be efficacious in the attenuation of inflammatory CNS demyelination and induction of neurorepair. So, in this report, we evaluated the potential of a combination of lovastatin and rolipram on neurorepair in an EAE model. We found that combination therapy with suboptimal doses of these drugs is complementary in a synergistic/additive manner to provide neuroprotection and promote neurorepair after inflammatory CNS demyelination.
MATERIALS AND METHODS
Reagents
Myelin basic protein (MBP) ~50% pure from Guinea pig brain, complete Freund’s adjuvant (CFA), HRP-tagged anti-mouse IgG antibodies, rolipram, pertussis toxin, ondansetron hydrochloride dehydrate, and other chemicals were purchased from Sigma (St. Louis, MO). Lovastatin and rolipram were purchased from Calbiochem (San Diego, CA). ‘TRIZOL’ reagent was purchased from Invitrogen (Carlsbad, CA). Anti-mouse myelin basic protein (MBP, clone 1: 129–138) antibodies were purchased from Serortec (Raleigh, NC). Anti-neurofilament heavy non-phosphorylated (SMI 32) IgG and, FITC and IgG Texas Red antibodies were purchased from Abcam Inc. (Cambridge, MA).
Animals
Female Lewis rats (Harlan Laboratory, Harlan, IN) weighing 225–250 g were housed in the animal care facility of the Medical University of South Carolina (MUSC), throughout the experiment and provided with food and water ad libitum. All experiments were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication number 80–23, revised 1985) and were approved by the MUSC Animal Care and Use Committee.
EAE induction and clinical evaluation
The procedure used for the induction of EAE has been described previously (Paintlia et al. 2004). In brief, female rats received in the hind limb a subcutaneous injection of MBP (50 μg) in 0.1 ml of phosphate-buffered saline (PBS; pH 7.4) emulsified in an equal volume of CFA supplemented with 2 mg/ml of mycobacterium tuberculosis H37Ra (Difco, Detroit., MI) on days 0 and 7. Immediately thereafter and again 24 h later, rats received pertussis toxin (200 ng, intraperitoneally, ip) in 0.1 ml of PBS. Individual animals were observed daily and clinical scores were assessed by an experimentally blinded investigator using a 0 to 5 scale: 0, no clinical score; 1, piloerection; 2, loss in tail tonicity; 3, hind leg paralysis; 4, paraplegia, and 5, moribund or dead.
Lovastatin and rolipram treatments
Lovastatin (4 mg/ml) was dissolved in 0.8% ethanol/0.6 N NaOH and adjusted to pH 7.4 and rolipram (2 mg/ml) was dissolved initially in 100 μl DMSO and then suspended in PBS (pH 7.4). For combination treatments, suboptimal doses of both drugs i.e., lovastatin (1 mg/kg) and rolipram (1 mg/kg) were administrated daily and every other day, respectively. Administration of rolipram induced salivation, vomiting, excessive grooming, and head twists, side-effects which were prevented by intramuscular injection of ondansetron hydrochloride dehydrate (0.3 mg/kg) 20 min prior to rolipram administration as reported earlier (Genain et al. 1995). Control EAE animals received an injection of vehicle (placebo, 0.8% ethanol in PBS, ip), daily. Drug treatment with suboptimal doses in combination or separately was started on the day 10th dpi after the onset of disease exhibiting clinical scores (CS) ≥2.0 and continued throughout the study. Corresponding control animals received an emulsion of CFA/PBS into their hind limb foot pads and a daily injection of vehicle (ip) or a dose of lovastatin (1 mg/kg) and rolipram (1 mg/kg) as described above as a drug control. Animals were sacrificed at disease peak at the 15th dpi to collect sera and spinal cords (SC, lumbar region). Animals developing severe EAE (CS ≥ 4.5) upon treatment with drug or placebo were euthanized immediately as per the animal protocol guidelines.
For myelin repair studies, EAE was induced in female Lewis rats as previously described but with a reduced dose of MBP (35 μg/animal). The reduced dose of MBP induced less severe EAE, resulting in spontaneous remission by the 25th dpi, enabling us to study CNS repair upon drug treatment. Drug combination treatments were started on the 13th dpi (CS ≥3.5) and continued until the end of the study as described above. Six animals in a group were used in each experimental study and repeated thrice.
Histopathology
Because the most significant histopathological changes in animals with EAE are detected in the lumbar region of the SC (Eng et al. 1989), this portion of the SC was carefully removed, fixed in 10% buffered formalin (Stephens Scientific, Riverdale, NJ) and embedded in paraffin and 4-μm sections were cut and stained with either hematoxylin/eosin (H&E), Luxol fast blue-hematoxylin (LFB), and Bielschowsky’s silver impregnation to assess inflammation, demyelination, and axonal pathology, respectively. Sections were mounted with Vectashield, an aqueous mounting media (Vector Labs, Burlingame, CA), examined under a light microscope (Olympus BX-60, Goleta, CA) and images were captured with an Olympus digital video camera (Optronics; Goleta, CA) using a dual band-pass filter using Adobe Photoshop 7 software. To quantify inflammation and demyelination in EAE, images of LFB stained SC sections (n = 6) from each group were captured (200x), coded, and processed for counting of nuclei and LFB staining intensity in a blinded manner using Image-Pro Plus 4 image software (Media Cybernetics, Silver Springs, MD).
Immunohistochemistry
For double-labeling, sections were incubated simultaneously with both types of primary antibodies after blocking with a serum-PBS solution at 4 °C overnight as described above. Then, secondary antibodies for the appropriate marker (anti-IgG conjugated with FITC or anti-IgM conjugated with Texas red) were used. Sections were also incubated with Texas red-conjugated IgM and FITC-conjugated IgG without primary antibody as negative controls and an appropriate mouse IgG and rabbit polyclonal IgG were used as isotype controls. After thorough washings, slides were mounted with aqueous mounting media and analyzed by immunofluorescence microscopy and images were captured with an Olympus digital camera as described above.
Measurement of cyclic AMP (cAMP) and Protein kinase A (PKA) activity
Supernatants of SC homogenates were used for measurement of cAMP using cyclic AMP Enzyme immunoassay kits purchased from Assay Designs (Ann Arbor, MI). Similarly, supernatants of SC homogenates were used for detection of PKA activity using a PKA radiometric (32P) assay kit from Millipore Corporation (Billerica, MA). The procedures for detection for both assays were performed according to the product protocols.
RNA extraction, cDNA synthesis, and quantitative real time-PCR (QRT-PCR) analysis
Lumbar SC tissues were carefully processed for RNA isolation using ‘TRIZOL’ reagent according to the manufacturer’s protocol described previously (Paintlia et al. 2004). Single-stranded cDNA was synthesized from SC tissue RNA from each group of animals using a superscript pre-amplification system for first-strand cDNA synthesis (BIO-RAD Laboratories, Hercules CA). QRT-PCR was performed using the BIO-RAD Laboratories iCycler iQ Real-Time PCR Detection System. Primers for target genes (Table 1) were designed using Primer quest software available free at www.idtdna.com and synthesized from integrated DNA technologies (IDT, Coralville, IA). For ORT-PCR reaction supermix (IQ™ SYBR Green) was purchased from BIO-RAD (BIO-RAD Laboratories, Hercules, CA). Thermal cycling conditions were as follows: activation of iTaq™ DNA polymerase at 95 °C for 10 min, followed by 40 cycles of amplification at 95 °C for 30 s and 58–60 °C for 1 min. The detection threshold was set above the mean baseline fluorescence determined by the first 20 cycles. Amplification reactions in which the fluorescence increased above the threshold were defined as positive. A standard curve for each template was generated using a serial dilution of the template (cDNA). Assay specificity was determined by melting-curve analysis in each experimental run of QRT-PCR. The quantities of target gene expression were normalized to the corresponding GAPDH or 18S rRNA expression in test samples and plotted.
Table 1.
Genes and DNA sequences of primers i.e., forward primer (FP) and reverse primer (RP) used for QRT-PCR amplification
| Gene Name | Primers |
|---|---|
| GAPDH | FP: 5’-cctacccccaatgtatccgttgtg-3’; RP: 5’-ggaggaatgggagttgctgttgaa-3’ |
| 18S rRNA | FP: 5’-ccagagcgaaagcatttgccaaga-3’; RP: 5’-tcggcatcgtttatggtcggaact-3’ |
| PDGF-αR | FP: 5’-cagacattgaccctgttccagagg-3’; RP: 5’-gaatctatgccaatatcatccatc-3’ |
| MBP | FP: 5’-ctctggcaaggactcacacac-3’; RP: 5’-tctgctgagggacaggcctctc-3 |
| Olig1 | FP: 5’-tggcaattaaagtgacccaagcgg-3’; RP: 5’-acttctggctctaaactggtggga-3’ |
| CNTF | FP: cttcaagagctctcacagtg-3’; RP: tgcttatctttggccccataat-3’ |
| NT3 | FP: 5’-acgtccgagcactgacttcagaaa-3’; RP: 5’-aggcacacacaggaagtgtcta-3’ |
ELISAs
Neurotrophin (NT)-3 levels in serum samples were determined using an NT-3 Emax Immunoassay kit and protocol described in the product manual (Promega, Madison WI).
Semi-thin and ultra-thin microscopy studies
For electron microscopic studies, SC of vehicle treated (n = 4) and drug treated (n = 3) EAE animals along with controls (n = 2) were selected for plastic embedding. Animals were perfused intracardially with 4% formaldehyde, and lower thoracic and lumbar SC were dissected and post-fixed in 4% formaldehyde until used for plastic embedding. Specimens were blinded until after microscopic evaluation. One- to 2-mm thick blocks were cut and rinsed with 0.1 M phosphate buffer, post-fixed in 1% osmium tetroxide, dehydrated in ethanol and propylene oxide, and embedded in Araldite. Semi-thin (1-μm) transverse sections were cut with a Reichert Ultracut E and stained with toluidine blue/pyronin B. Montages of the transverse sections were reconstructed from digitally captured images using a Spot Camera mounted on a Leitz Laborlux 12 (Spectra services, Ontario, NY). Approximately 12 images were required for each reconstruction. For quantification of demyelinated areas, the whole dorsal funiculi were outlined and then demyelinated/inflammatory regions within all untreated and treated rats were also outlined with NIH Image J. The percent area of demyelinated/inflammatory regions was then calculated. For electron microscopy, 0.5 × 0.5 mm faces of dorsal and lateral funiculi were thin sectioned and stained with uranyl acetate and lead citrate. Ultra-thin sections were imaged with a JEOL-100C microscope.
Statistical analysis
Using a student’s unpaired t-test for two set of data points and one-way ANOVA (student-Newman-Keuls to compare all pairs of columns) for multiple set of data points, p-values were determined for clinical scores, QRT-PCR analysis, ELISA, intensities, and counting data in triplicate from three independent experiments using GraphPad software (GraphPad Software Inc. San Diego, CA). The criterion for statistical significance was p <0.05.
RESULTS
Combination therapy with suboptimal doses of lovastatin and rolipram reverses cellular infiltration and provide neuroprotection
Previous studies demonstrated that the optimal dose of lovastatin and rolipram is ≥ 2 mg/kg (Paintlia et al. 2004) and ≥ 5 mg/kg (Genain et al. 1995), respectively, for blunting the progression of EAE when administered individually. In addition, combination treatment with suboptimal doses of lovastatin (1 mg/kg) and rolipram (1 mg/kg) was impressive, compared to their optimal doses when administered individually prior to the establishment of EAE (data not shown). In general, MS treatment is initiated after the patient has developed clinical signs or symptoms of CNS inflammatory demyelination. Therefore, it is imperative to test a therapeutic regimen which can prevent the development of disease and effectively reverse an established case. Thus, we first evaluated whether combination therapy with lovastatin (1 mg/kg) and rolipram (1 mg/kg) could reverse an established case of EAE and the drug treatment was initiated after the onset of disease in animals exhibiting CS ≥2.0. Interestingly, combination therapy significantly reversed the development of EAE in animals compared with vehicle, whereas no such reversal was observed in those treated with lovastatin or rolipram separately with same dose (Fig. 1A). It is noteworthy that ondansteron hydrochloride pretreatment, however, inhibited the possible side-effects of rolipram in treated EAE animals, but it had neither aggravated nor attenuated the disease associated symptoms in control EAE animals.
Figure 1. Combination treatment with lovastatin and rolipram reverses cellular infiltration into the SC of animals with established EAE.

Treatment with lovastatin (LOV; 1 mg/kg) and rolipram (RLP; 1 mg/kg) in combination or individually in established EAE animals. Plot depicts disease in EAE animals treated with LOV and RLP in combination and individually (A). Plot depicts cellular infiltration as nuclei/field in the lateral funiculi of SC of EAE animals treated with LOV and RLP in combination or individually (B). Representative photograph of the lateral funiculus of SC from each group of animals demonstrates axonal integrity with Bielschowsky’s silver impregnation staining (C). Data in plots are expressed as Mean ± SD from three independent experiments (A) and histological images of SC (n = 6)/group (B). Statistical significance * p<0.05, ** p<0.01, and NS (not significant) versus EAE (VEH). Magnifications 200× (B) and 600× (C).
Earlier, Eng et al have documented that EAE-associated pathological changes are evident in the lumbar region of the SC of animals (Eng et al. 1989). Therefore, we next evaluated the lumbar SC of animals for cellular infiltration and pathological changes. Combination treatment significantly reversed cellular infiltration into the lateral and dorsal funiculi of the SC of EAE animals compared to vehicle (Fig. 1B). Quantification of inflammation revealed that an observed increase in cellular infiltration in the various regions of transverse sections of SC of vehicle-treated EAE animals was drastically reduced by drug combination treatment (Table 2). In contrast, this reversal of cellular infiltration and pathological changes in the SC were reduced in EAE animals treated with lovastatin and rolipram alone (Fig. 1B and Table 2).
Table 2.
Degree of disease severity, cellular-inflammation and demyelination in the lumbar spinal cord of animals on peak clinical day of EAE and attenuation by suboptimal doses of lovastatin (LOV) and rolipram (RLP) when used in combination or individually and compared with vehicle (VEH).
| Groups | Clinical Score | Cellular-Infiltration | Demyelination | |||
|---|---|---|---|---|---|---|
| Subarach. Infiltrates | Perivascular Infiltrates | Cuffing | Parenchymal Infiltrates | |||
| CON | ||||||
| VEH | 0 ± 0 | ND (n = 6) | ND (n = 6) | ND (n = 6) | ND (n = 6) | ND (n = 6) |
| LOV + RLP | 0 ± 0 | ND (n = 6) | ND (n = 6) | ND (n = 6) | ND (n = 6) | ND (n = 6) |
| EAE | ||||||
| VEH | 4.6 ± 0.28 | 3+ (n= 6) | 3+(n = 6) | Y (n = 6) | Y(n = 6) | 3+(n = 6) |
| LOV | 4.33 ± 0.57NS | 2+ (n = 6) | 1+ (n = 6) | Y (n = 6) | Y (n = 6) | 2+ (n = 6) |
| RLP | 4.5 ± 0.5NS | 2+ (n = 6) | 1+ (n = 6) | Y (n = 6) | Y (n = 6) | 3+ (n = 6) |
| LOV + RLP | 2.0 ± 0.5*** | 1+ (n = 6) | 1+ (n = 6) | ND (n = 6) | ND (n= 6) | 1+ (n = 6) |
@: Clinical score data is presented as Mean ± SD from three independent experiments. Asterisks indicate *** p< 0.001 and ‘NS’ (not significant) versus VEH (EAE). Infiltration graded as 1+: one focus with few infiltrates’; 2+: two foci with moderate increase in infiltrates; 3+: three or more foci with intensive increase in infiltrates; Y: positive; ND: not detected in the posterior and lateral funiculi of SC. Demyelination graded as 1+: one or two foci with slight demyelination; 2+: two or three foci with moderate demyelination; 3+ three or more foci with severe demyelination or the complete absence of myelin and ND; no demyelination in the posterior and lateral funiculi of SC. Numbers in parenthesis indicate animals with the same pathological degree.
Because inflammatory CNS demyelination is associated with loss of both neuronal axons and OLs, which are responsible for neurological deficits in MS/EAE (Markovic-Plese and McFarland 2001; Noseworthy et al. 2000), we next evaluated neurodegeneration in the CNS by histological examination of SC transverse sections. In association with inflammation, LFB staining demonstrated an increase in demyelination with a corresponding increase in cellular infiltration in the lateral funiculi of SC of EAE animals (Table 2). Of note, demyelination and cellular infiltration were severe in the lateral funiculi of the SC of vehicle-treated EAE animals (data not shown). Conversely, drug combination therapy reduced both cellular infiltration and demyelination in the SC of EAE animals (Table 2). However, both cellular infiltration and demyelination were significantly attenuated in EAE animals upon treatment with rolipram or lovastatin individually, compared to vehicle, but these effects were not as profound as those resulting from their combination (Table 2). Likewise, silver impregnation of axons in the lateral funiculi of SC sections of EAE animals showed that drug combination therapy prevented axonal loss and preserved white matter integrity (Fig. 1C). In contrast, the loss of axons and white matter integrity of SC of EAE animals was partially protected by individually administered rolipram or lovastatin (data not shown).
Because cAMP regulates the direction of growth cones in the CNS via modulation of its associated protein kinase, PKA activity in neurons (Song et al. 1997) and PDE-4 inhibitor blocks cAMP degradation (Krause and Kuhne 1988), thus we next measured the level of cAMP and PKA activity in the SC of EAE animals. As expected, cAMP was significantly elevated in the SC of EAE animals upon treatments with the drug combination compared with vehicle (Fig. 2A). Although cAMP was also elevated significantly in the SC of EAE animals upon treatment with lovastatin or rolipram when administered individually compared with vehicle, it was significantly lower when compared with their combination treatment (Fig. 2A). Consistent with cAMP, PKA activity was also elevated in the SC of EAE animals following treatment with the drug combination compared with those treated with vehicle (Fig. 2B). Again, PKA activity was also elevated in the SC of EAE animals treated with lovastatin or rolipram alone, but this increase was relatively small compared to their combination treatment (Fig. 2B). Altogether these data provide evidence that combination therapy with lovastatin and rolipram provide neuroprotection by protecting OLs and neuronal axons against CNS inflammatory immune response.
Figure 2. Preservation of cAMP and PKA activity in the SC of EAE animals by lovastatin and rolipram combination therapy.
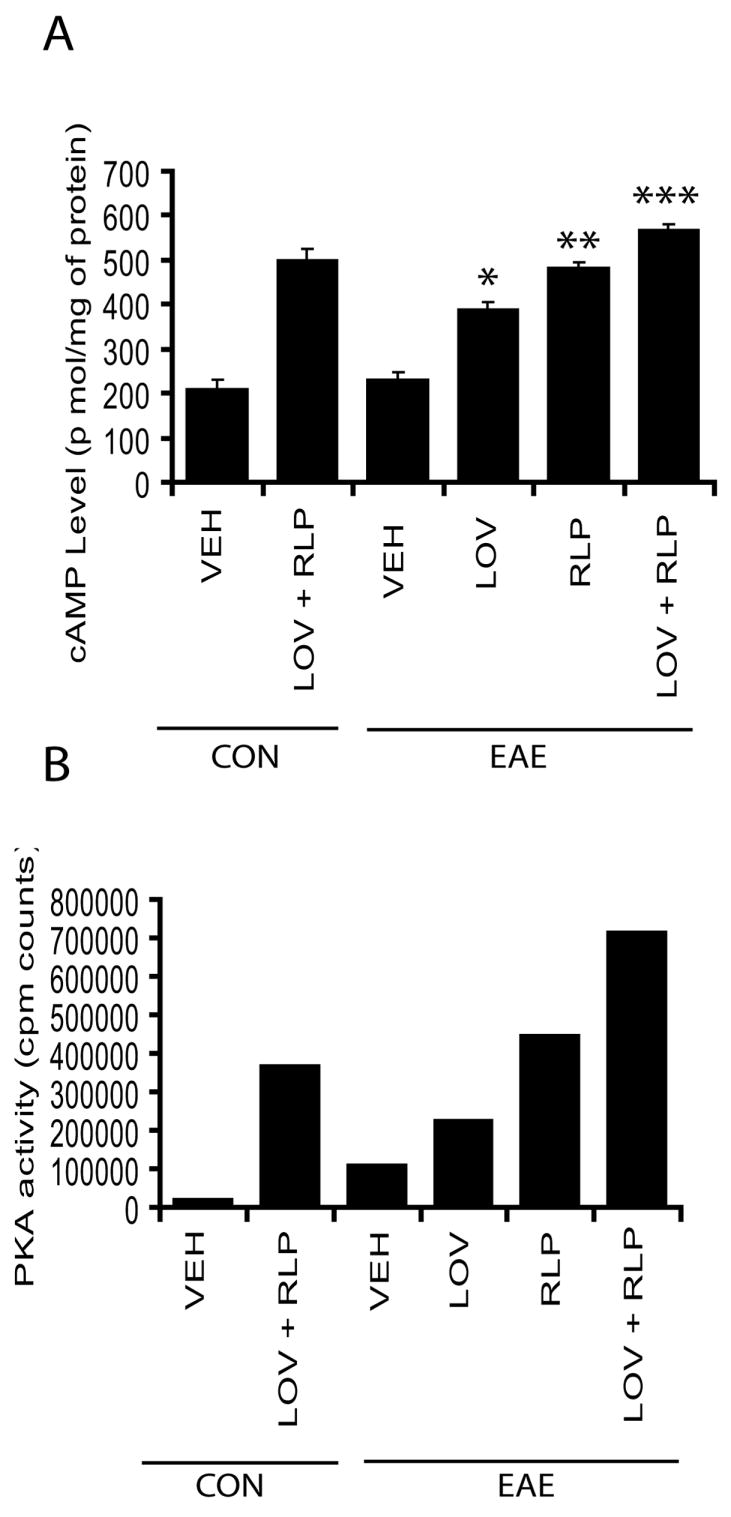
Plots depict the level of cAMP in the SC of EAE (n = 9) and control (n = 9) animals treated with drug combination or individually or vehicle (VEH) alone from three independent experiments (A). Plot depicts counts of PKA activity in the SC of EAE (n = 6) and control (n = 6) animals treated with drug combination or individually or vehicle (VEH) alone from two independent experiments (B). Data in plot is expressed as Mean ± SD (A) and average means (B). Statistical significance * p<0.05, ** p<0.01, and *** p<0.001 versus EAE (VEH).
Combination therapy with lovastatin and rolipram promotes neurorepair in the CNS
Next to address the effect of combination therapy on myelin repair, drug treatment was initiated in animals exhibiting severe EAE symptoms (CS ≥3.5) and continued till recovery (Fig. 3A). Combination therapy with lovastatin and rolipram produced rapid recovery—by the 23rd dpi—compared with vehicle (Fig. 3A). However, EAE animals treated with these drugs individually behaved similar to vehicle (data not shown). Evaluation of myelin repair induction revealed that transcripts of platelet derived growth factor-α receptor (PDGF-αR; marker for OPs, Fig. 3B) and MBP (marker of myelin-forming oligodendrocyte, Fig. 3C) in the SC of drug-combination treated ameliorating EAE animals was significantly increased over vehicle-treated ones. In line with these findings, transcripts for Olig1, an OL transcription factor associated with remyelination or myelin repair (Arnett et al. 2004) were also elevated significantly in the SC of EAE animals upon treatment with drug combination, in contrast to vehicle (Fig. 3D). In addition, transcripts for neurotrophins involved in myelin repair including ciliary neurotrophic factor (CNTF) (Stankoff et al. 2002) and NT-3 (Kirstein and Farinas 2002) in the SC of ameliorating EAE animals were significantly elevated when treated with drug combination compared with vehicle, which was suggestive of induction of promyelinating milieu in the CNS (Fig. 3E and F). In support of this data, serum NT-3 was also elevated significantly in ameliorating EAE animals treated with drug combination compared to vehicle (Fig. 3G). Moreover, vehicle-treated ameliorating EAE animals also showed induction of myelin repair, but this was relatively insignificant compared with peak clinical day of disease (Fig. 3B-G).
Figure 3. Combination treatment with lovastatin and rolipram promotes regeneration in the SC of ameliorating EAE animals.

Treatment of EAE rats with lovastatin (LOV) and rolipram (RLP) in combination was started on the 13th dpi. Plot depicts clinical score in EAE (n = 9) and control (n = 9) rats treated with drug combination or vehicle (VEH) only (A). Plots depict level of transcripts for PDGF-αR (B), MBP (C), Olig1 (D), CNTF (E), and NT-3 (F) in the SC of EAE animals as detected by QRT-PCR. Plot depicts level of NT-3 in the serum of similarly treated EAE animals (G). Data in plots are expressed as Mean ± SD from three independent experiments. Statistical significance * p<0.05, ** p<0.01 versus EAE (VEH), and # p<0.05 and non-significant (NS) versus EAE (Peak).
These results were further confirmed by toluidine blue staining and electron microscopy. SC white matter of control animals exhibited no demyelination or inflammation in all 3 funiculi (Fig. 4A). Differences in fiber diameters within and among all the funiculi were normal. The vehicle-treated EAE animal showed extensive inflammation and demyelination in the different regions of the SC (Fig. 4B). Areas of infiltration in all vehicle-treated EAE animals were mainly in the dorsal funiculus near the midline where extensive demyelination and myelin degeneration was associated with infiltration (Fig. 4B; box 1). Lateral funiculus had modest inflammation and demyelination was prominent near the pia matter (Fig. 4B; box 2). The SC section from an EAE animal treated with the drug combination was selected as a representative section to show the largest amount of demyelination observed in treated EAE animals (Fig. 4C). Demyelination was mainly present near the midline but infiltration (Fig. 4C; box 1) was considerably less than in vehicle-treated EAE animals (Fig. 4B; box 1). The lateral funiculus of an EAE animal treated with the drug combination did not show infiltration, but some fibers showed an evidence of degeneration (Fig. 4C; box 2).
Figure 4. Inflammation-mediated demyelination in the SC of EAE animals revealed by toluidine blue staining.

One-micron plastic transverse sections of SC of control (A), vehicle-treated EAE (B), and drug combination-treated ameliorating (C) EAE animals. Low magnification montages (left panel) are composed of pictures from 12 blocks of tissue. High magnification pictures of dorsal and lateral funiculi (boxed areas in low magnification pictures) make up the right panel. (A) Control SC shows tightly compacted myelinated fibers in all funiculi. Dorsal funiculus (Box 1) and lateral funiculus (Box 2) illustrates differences in fiber diameters. (B) SC section from a vehicle-treated EAE animal shows infiltration area mainly in the dorsal funiculus near the midline and lateral funiculus near the pia border. The dorsal funiculus (Box 1) shows extensive demyelination and myelin degeneration associated with infiltration. The lateral funiculus (Box 2) has very little inflammation but exhibits demyelination near the pia matter. (C) Representative SC section from EAE rats treated with drug combination was selected to show the maximum amount of demyelination observed in similarly treated rats in the group. Demyelination (Box 1) is mainly present near the midline, but infiltration is considerably less than observed in vehicle-treated EAE rats (see B). Lateral funiculus of drug combination-treated rat (Box 2) does not show infiltration. A few fibers show evidence of degeneration. Bar = 10 μm and 1 μm for low and high magnification pictures, respectively.
An induction of myelin repair (remyelination) in the SC was revealed by electron microscopy (Fig. 5). The dorsal funiculus of a control animal showed that all axons were myelinated with little intervening space between the fibers (Fig. 5A). The thickness of the myelin sheath was dependent upon axonal caliber and varies considerably from funiculi to funiculi. The lateral funiculus of a vehicle-treated EAE animal showed many myelin sheaths that were in various stages of degeneration (Fig. 5B). The intervening space was filled with gliotic processes (Fig. 5B). A picture from the dorsal funiculus of a vehicle-treated EAE animal depicted a field filled with infiltrating cells and their processes (Fig. 5C–D). In contrast, the dorsal funiculus of drug combination-treated EAE animals had fewer infiltrating cells. Importantly, while the fibers were not normal in terms of their thickness, there were many small-diameter axons surrounded by abnormally thin myelin sheaths. This pattern is suggestive of remyelination or perhaps myelin repairs (Fig. 5E). A picture from drug combination-treated EAE animals shows a myelinated zone adjacent to an area of inflammation (Fig. 5F). Abundance of small-diameter myelinated fibers is suggestive of remyelination (Fig. 5F). It is difficult to establish whether these fibers represent axons that are newly generated and remyelinating or those in the process of degeneration but protected from further degeneration by drug treatment. In either scenario, the fibers in these drug combination-treated animals appear more intact than fibers in vehicle-treated EAE animals. In the end, immunohistochemistry also demonstrated that breakdown of myelin (MBP) and axonal (SMI 32) swelling which was severe in the lateral funiculi of SC on the peak clinical day of EAE disease (Fig. 6). Drug combination treatment improved myelin repair and attenuated the cellular infiltration into the SC when compared with vehicle (Fig. 6). Hoechst dye, which stains the nucleus, demonstrated existing cellular infiltration in the SC of EAE animals.
Figure 5. Demyelination/remyelination in the SC of EAE animals revealed by electron microscopic studies.

Electron micrographs of control (A), vehicle-treated (B–D) and drug combination-treated (E–F) EAE animals. (A) Dorsal funiculus of a control rat shows all axons are myelinated. Thickness of the myelin sheath is dependent upon axonal caliber. (B) The lateral funiculus of a vehicle-treated EAE rat shows many myelin sheaths in various stages of degeneration (arrows). The intervening space is filled with gliotic processes (asterisks). (C) The dorsal funiculus of a vehicle-treated EAE rat shows an area with degenerating axons (asterisks), a myelin sheath with abnormal periodicity, and cytoplasmic accumulation (arrow) and a normal myelin sheath (crossed arrow). Much of the field is filled with infiltrating cells and their processes. (D) The dorsal funiculus of vehicle-treated EAE rat shows extensive infiltration of inflammatory cells. (E) The dorsal funiculus of drug combination-treated rat shows many, small-diameter axons surrounded by thin myelin sheaths (arrows), suggestive of remyelination. (F) The dorsal funiculus of drug combination-treated rat shows a myelinated zone adjacent to area of inflammation. Myelinated fibers are of various diameters; many small, thinly myelinated axons are conspicuous (arrows), suggestive of remyelination/repair. Bar = 1 μm (25,000×).
Figure 6. Immunohistochemistry reveals induction of myelin repair in the SC of ameliorating animals.
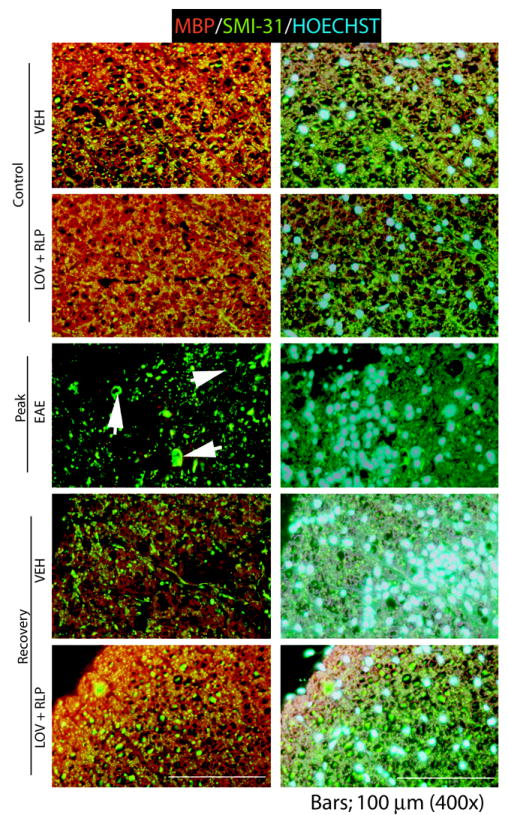
SC sections were processed for immunohistochemistry using anti-MBP and anti-SMI 32 antibodies to determine an induction of myelin repair. Examination of white matter in the lateral funiculi of SC of a control animal shows tightly compacted myelinated fibers and no degeneration (1st and 2nd panel). EAE animal on peak clinical day shows severe demyelination, axonal degeneration (arrowheads) and infiltration in the lateral funiculus of the SC (3rd panel). Vehicle-treated ameliorating EAE animal shows reduction of infiltration and induction of myelin repair (4th panel). Drug combination treatment induced reduction of infiltration and restoration of white matter integrity in the lateral funiculus of SC. Bars; 100 μm.
Collectively, these data suggest that combination therapy with lovastatin and rolipram preserves neuronal axons and promotes spontaneous myelin repair by enhancing the development of OLs and induction of a promyelinating milieu in the CNS of ameliorating EAE animals.
DISCUSSION
Combination therapies with existing or novel MS therapeutics have great clinical potential to improve MS treatment outcomes (Tullman and Lublin 2005). Combination therapies are tested in humans (Dhib-Jalbut et al. 2002; Ytterberg et al. 2007), including phase-I clinical trials (Jeffery et al. 2005; Rudick et al. 2006) with major FDA-approved immunomodulators including IFN-β and GA in the MS regimen. Moreover, other combination therapies are also being investigated in MS and EAE with FDA-approved therapies and other novel MS therapeutics including statins and minocycline (Giuliani et al. 2005; Luccarini et al. 2008; Stuve et al. 2006). Results of a recently conducted phase 1 trial of IFN-β and atorvastatin combined therapy in MS patients are quite impressive and provide a rationale for conducting phase II/III trials (Paul et al. 2008). Combination therapy is considered advantageous if both drugs (a) have different mechanisms of actions, (b) have excellent safety profiles, and (c) have no additional toxicities when used in combination for additive or synergistic effects. In this regard, lovastatin (Paintlia et al. 2004; Youssef et al. 2002) and rolipram (Bielekova et al. 2000; Sommer et al. 1995) meet these criteria and both characteristically preventing and inhibiting EAE development, thus rationale for testing these drugs in combination therapy. Also, both lovastatin (Johnson-Anuna et al. 2005) and rolipram readily cross the BBB (Krause and Kuhne 1988) to interfere in the neurodegenerative process or to induce neurorepair in the brain.
The present study established a synergistic/additive effect of combination therapy with suboptimal doses of lovastatin and rolipram, which provides neuroprotection against inflammatory CNS demyelination in EAE (Fig. 1A). Importantly, it prevented the progression of disease and promoted neurorepair in established cases of EAE and this was not achieved with either drug alone at the same dose. These observed effects of combination therapy are may be attributed to the immunomodulatory and neuroprotective effects of lovastatin and rolipram as discussed below.
Immunomodulatory effects
In organ-specific autoimmunity, the balance of cytokines is a key determinant of resistance or susceptibility. In EAE, disease susceptibility is thought to correlate with the expression of proinflammatory cytokines such as IL-23, IL-17, IFN-γ, TNF-α, IL-6, and IL-1β including iNOS. Conversely, TH2 cytokines such as IL-4, IL-10, and IL-13 have been shown to be important for preventing or ameliorating disease (Aggarwal et al. 2003; O’Garra et al. 1997). An invasion of CNS by autoreactive T cells and macrophages is associated with breaching of the BBB and expression of adhesion molecules in endothelial cells during EAE progression (Eralinna et al. 1996). Combination therapy showed significant reduction of cellular infiltration in the SC, suggestive of improved endothelial dysfunction and inhibition of CNS invasion by autoreactive T cells and macrophages (Fig. 1 and Table 2). Consistent with these findings, previously statin and rolipram are reported to attenuate EAE development by promoting TH1 to TH2-biased immune responses (Bielekova et al. 2000; Genain et al. 1995; Paintlia et al. 2004; Sommer et al. 1995; Youssef et al. 2002) in both acute and chronic EAE models.
In immune cells, statins are reported to i) inhibit IFN-γ induced MHC class II expression, in a dose dependent manner via inhibition of class II transactivator in antigen presenting cells (Kwak et al. 2001), ii) downregulate Rho-mediated migration of monocytes across the endothelium by altering isoprenoid biosynthesis (Greenwood et al. 2003), iii) inhibit the expression of adhesion molecules and matrix metalloproteinase (MMP) such as MMP-9 (Wong et al. 2001), and immunomodulate the function of T cells (Youssef et al. 2002). Likewise, rolipram inhibits the secretion of TNF-α by macrophages and monocytes via blocking the degradation of cAMP (Torphy and Undem 1991) as well as inhibits NF-κB and matrix MMP-9 activities in activated T cells (Sanchez et al. 2005).
Neuroprotective and regenerative effects
Neuroinflammation-mediated demyelination and neurological signs are well correlated during EAE pathogenesis in which OLs are considered to be essential for spontaneous myelin repair of CNS in ameliorating EAE animals (Pender et al. 1989). Importantly, immunumodulatory synergy of lovastatin and rolipram alleviated inflammation-induced neurodegeneration i.e., protection of neuronal axons and OLs (Table 2 and Fig. 1C). In addition, accumulation of cAMP and PKA activity were elevated in the SC of ameliorating EAE animals after treatment with combination of lovastatin and rolipram (Fig. 2). In vitro studies have revealed that an increase in cAMP levels by inhibition of phosphodoesterase activity increases the survival of neurons (Hanson et al. 1998) and regulates myelin formation in OLs (Bolton and Butt 2006) as well as attenuates the activation of brain glial cells (Zhang et al. 2002). In addition, cAMP induced activation of PKA is essential for the development and functioning of neurons (Song et al. 1997). Combination therapy promoted spontaneous myelin repair in the CNS after inflammatory demyelination attack in EAE animals (Fig. 3B-C). It was associated with induction of a promyelinating milieu in the CNS as showed by an increase in the expression of neurotrophins i.e., CNTF and NT-3 (Fig. 3D-F), both are involved in the induction of myelin repair following CNS injury (Kirstein and Farinas 2002; Stankoff et al. 2002). This induction of a promyelinating milieu in the CNS could be attributed to both activated immune and brain glial cells in ameliorating EAE animals. For instance, activated TH2 cells are reported to secrete neurotrophins including brain-derived neuroptophic factor and NT-3 in the CNS (Besser and Wank 1999; Moalem et al. 2000). Simvastatin has been reported to induce LIF secretion by treated T lymphocytes from MS patients (Vanderlocht et al. 2006). In addition, we documented that statins have potential to promote myelin repair in the CNS of EAE animals via increased expression of neurotrophins by activated brain glial cells exposed to proinflamamtory cytokines (Paintlia et al. 2005; Paintlia et al. 2008). In addition, statins are reported to provide neuroprotection in traumatic brain injury (Lu et al. 2007; Wu et al. 2008) and spinal cord injury (Holmberg et al. 2006; Pannu et al. 2007). Likewise, rolipram promotes brain regeneration via induction of axonal regeneration, attenuation of glial scar formation, and significant functional recovery in an SC injury model (Nikulina et al. 2004).
In addition to the induction of CNS repair by lovastatin and rolipram in vivo, these drugs are also reported to modulate the development of glial cells in vitro. For instance, statins promote the maturation of human and murine OPs into myelin-forming OLs (Miron et al. 2007; Paintlia et al. 2005) and enhance spontaneous myelin repair via lowering of isoprenoids and inhibition of small G protein functions in the brain glial cells (Paintlia et al. 2008). Statins are reported to prevent the apoptotic cell death of neurons (Johnson-Anuna et al. 2007) and promote neurite outgrowth during CNS injury (Holmberg et al. 2006; Pooler et al. 2006). Likewise, PDE-4 inhibitor-mediated preservation of cAMP has been shown to protect neurons against β-amyloid-induced neurotoxicity via induction of PKA activities (Echeverria et al. 2005).
Although other mechanisms may contribute to the observed neuroprotection and neurorepair in the CNS of EAE animals with the combination therapy of lovastatin and rolipram, we suggest that the effects of combination therapy are complementary in a synergistic/additive manner. Therefore, an observed impeded neurodegeneration and induction of neurorepair would be consistent with this hypothesis. At this time, the precise mechanism and contribution of these drugs in combination in the reversal of EAE pathogenesis and induction of neurorepair is not fully understood. An inflammatory demyelination EAE model used in the study replicates many aspects of MS pathogenesis and has advantages over other chemically (cuprizone) induced demyelination model to evaluate the neuroprotective and neuroreparative effects of an experimental drug. Therefore, in contrast to the observed outcomes of statins in our model, it could be the main cause of failed myelin-repair in simvastatin treated cuprizone demyelination model (abstract presented at the annual meeting of ‘American Academy of Neurologists, 2008, New York, USA). The observed effects of lovastatin in combination therapy could be resulting from the induction of immunomodulation (Paintlia et al. 2004; Youssef et al. 2002) and promyelinating milieu in the CNS of ameliorating EAE animals (Paintlia et al. 2005; Paintlia et al. 2008) including enhanced OL differentiation (Miron et al. 2007; Paintlia et al. 2005) and neuritis in neurons (Holmberg et al. 2006; Pooler et al. 2006). Of note, the combination of lovastatin with rolipram was equally effective to its previously reported combination with 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR), an AMP kinase activator (Paintlia et al. 2006), but with difference in their doses i.e., rolipram (1 mg/kg) and AICAR (50 mg/kg). We believe that the combination of lovastatin and rolipram is an excellent approach for MS, using their suboptimal doses to minimize any adverse toxicity or side effects of the therapeutic doses of these drugs (Miron et al. 2007; Schulz et al. 2004). Based upon these findings with combination of lovastatin and rolipram, future clinical trials are warranted to investigate the effect of these drugs in combination therapy in MS.
Acknowledgments
We thank members of Dr. Singh’s laboratory for their valuable comments and help during the course of this study and Ms. Denise Bessert in Dr. Skoff’s laboratory for the light and electron microscopic pictures. We also thank the BSB/CRI animal care at MUSC. We thank especially Dr. Jennifer G. Schnellmann for critical reading of this manuscript and Ms. Joyce Brian and Ms. Carrie Barnes for their technical assistance.
This study was supported by grants from the NIH (NS-22576, NS-34741, NS-37766, NS-40144, NS-038236, C06-RR015455, and C06-RR018823) and support from Merck & Company.
ABBREVIATIONS
- EAE
Experimental autoimmune encephalomyelitis
- MS
Multiple sclerosis
- SC
Lumbar spinal cord
- CNS
central nervous system
- OL
oligodendrocyte
- MBP
Myelin basic protein
- PDGF-αR
Platelet derived growth factor-α receptor
- dpi
Days of post-immunization
- QRT-PCR
Quantitative real-time-polymerase chain reaction
- LOV
Lovastatin
- RLP
Rolipram
- PDE-4
Phosphodiesterase-4
- CS
Clinical score
- LFB
luxol fast blue
- H & E
Hematoxylin and Eosin
- NT-3
Neurotrophin-3
- PKA
Protein kinase A
- IL
Interleukin
- IFN
Interferon
- BBB
Blood brain barrier
References
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278(3):1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Arbizu T, Alvarez-Cermeno JC, Decap G, Fernandez O, Uria DF, Garcia Merino A, Izquierdo G, Montalban X. Interferon beta-1b treatment in patients with relapsing--remitting multiple sclerosis under a standardized protocol in Spain. Acta Neurol Scand. 2000;102(4):209–17. doi: 10.1034/j.1600-0404.2000.102004209.x. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306(5704):2111–5. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55(4):458–68. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Besser M, Wank R. Cutting edge: clonally restricted production of the neurotrophins brain-derived neurotrophic factor and neurotrophin-3 mRNA by human immune cells and Th1/Th2-polarized expression of their receptors. J Immunol. 1999;162(11):6303–6. [PubMed] [Google Scholar]
- Bielekova B, Lincoln A, McFarland H, Martin R. Therapeutic potential of phosphodiesterase-4 and -3 inhibitors in Th1-mediated autoimmune diseases. J Immunol. 2000;164(2):1117–24. doi: 10.4049/jimmunol.164.2.1117. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206(2):165–71. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Bolton S, Butt AM. Cyclic AMP-mediated regulation of the resting membrane potential in myelin-forming oligodendrocytes in the isolated intact rat optic nerve. Exp Neurol. 2006;202(1):36–43. doi: 10.1016/j.expneurol.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Brustle O, Jones KN, Learish RD, Karram K, Choudhary K, Wiestler OD, Duncan ID, McKay RD. Embryonic stem cell-derived glial precursors: a source of myelinating transplants. Science. 1999;285(5428):754–6. doi: 10.1126/science.285.5428.754. [DOI] [PubMed] [Google Scholar]
- Cui Q, So KF. Involvement of cAMP in neuronal survival and axonal regeneration. Anat Sci Int. 2004;79(4):209–12. doi: 10.1111/j.1447-073x.2004.00089.x. [DOI] [PubMed] [Google Scholar]
- Dhib-Jalbut S. Glatiramer acetate (Copaxone(R)) therapy for multiple sclerosis. Pharmacol Ther. 2003;98(2):245–55. doi: 10.1016/s0163-7258(03)00036-6. [DOI] [PubMed] [Google Scholar]
- Dhib-Jalbut S, Chen M, Henschel K, Ford D, Costello K, Panitch H. Effect of combined IFNbeta-1a and glatiramer acetate therapy on GA-specific T-cell responses in multiple sclerosis. Mult Scler. 2002;8(6):485–91. doi: 10.1191/1352458502ms862oa. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Filbin MT. Overcoming inhibitors in myelin to promote axonal regeneration. J Neurol Sci. 2005;233(12):43–7. doi: 10.1016/j.jns.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Clerman A, Dore S. Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure. Eur J Neurosci. 2005;22(9):2199–206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- Eng LF, D’Amelio FE, Smith ME. Dissociation of GFAP intermediate filaments in EAE: observations in the lumbar spinal cord. Glia. 1989;2(5):308–17. doi: 10.1002/glia.440020504. [DOI] [PubMed] [Google Scholar]
- Eralinna JP, Soilu-Hanninen M, Roytta M, Hukkanen V, Salmi AA, Salonen R. Blood-brain barrier breakdown and increased intercellular adhesion molecule (ICAM-1/CD54) expression after Semliki Forest (A7) virus infection facilitates the development of experimental allergic encephalomyelitis. J Neuroimmunol. 1996;66(12):103–14. doi: 10.1016/0165-5728(96)00031-8. [DOI] [PubMed] [Google Scholar]
- Genain CP, Roberts T, Davis RL, Nguyen MH, Uccelli A, Faulds D, Li Y, Hedgpeth J, Hauser SL. Prevention of autoimmune demyelination in non-human primates by a cAMP-specific phosphodiesterase inhibitor. Proc Natl Acad Sci U S A. 1995;92(8):3601–5. doi: 10.1073/pnas.92.8.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani F, Fu SA, Metz LM, Yong VW. Effective combination of minocycline and interferon-beta in a model of multiple sclerosis. J Neuroimmunol. 2005;165(12):83–91. doi: 10.1016/j.jneuroim.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, Baker D, Adamson P. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. Faseb J. 2003;17(8):905–7. doi: 10.1096/fj.02-1014fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18(18):7361–71. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg E, Nordstrom T, Gross M, Kluge B, Zhang SX, Doolen S. Simvastatin promotes neurite outgrowth in the presence of inhibitory molecules found in central nervous system injury. J Neurotrauma. 2006;23(9):1366–78. doi: 10.1089/neu.2006.23.1366. [DOI] [PubMed] [Google Scholar]
- Jeffery DR, Chepuri N, Durden D, Burdette J. A pilot trial of combination therapy with mitoxantrone and interferon beta-1b using monthly gadolinium-enhanced magnetic resonance imaging. Mult Scler. 2005;11(3):296–301. doi: 10.1191/1352458505ms1154oa. [DOI] [PubMed] [Google Scholar]
- Johnson-Anuna LN, Eckert GP, Franke C, Igbavboa U, Muller WE, Wood WG. Simvastatin protects neurons from cytotoxicity by up-regulating Bcl-2 mRNA and protein. J Neurochem. 2007;101(1):77–86. doi: 10.1111/j.1471-4159.2006.04375.x. [DOI] [PubMed] [Google Scholar]
- Johnson-Anuna LN, Eckert GP, Keller JH, Igbavboa U, Franke C, Fechner T, Schubert-Zsilavecz M, Karas M, Muller WE, Wood WG. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Ther. 2005;312(2):786–93. doi: 10.1124/jpet.104.075028. [DOI] [PubMed] [Google Scholar]
- Kirstein M, Farinas I. Sensing life: regulation of sensory neuron survival by neurotrophins. Cell Mol Life Sci. 2002;59(11):1787–802. doi: 10.1007/PL00012506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause W, Kuhne G. Pharmacokinetics of rolipram in the rhesus and cynomolgus monkeys, the rat and the rabbit. Studies on species differences. Xenobiotica. 1988;18(5):561–71. doi: 10.3109/00498258809041693. [DOI] [PubMed] [Google Scholar]
- Kwak B, Mulhaupt F, Veillard N, Pelli G, Mach F. The HMG-CoA reductase inhibitor simvastatin inhibits IFN-gamma induced MHC class II expression in human vascular endothelial cells. Swiss Med Wkly. 2001;131(34):41–6. doi: 10.4414/smw.2001.06144. [DOI] [PubMed] [Google Scholar]
- Lu D, Qu C, Goussev A, Jiang H, Lu C, Schallert T, Mahmood A, Chen J, Li Y, Chopp M. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24(7):1132–46. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin FD. Relapsing experimental allergic encephalomyelitis. An autoimmune model of multiple sclerosis. Springer Semin Immunopathol. 1985;8(3):197–208. doi: 10.1007/BF00197296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luccarini I, Ballerini C, Biagioli T, Biamonte F, Bellucci A, Rosi MC, Grossi C, Massacesi L, Casamenti F. Combined treatment with atorvastatin and minocycline suppresses severity of EAE. Exp Neurol. 2008;211(1):214–26. doi: 10.1016/j.expneurol.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Markovic-Plese S, McFarland HF. Immunopathogenesis of the multiple sclerosis lesion. Curr Neurol Neurosci Rep. 2001;1(3):257–62. doi: 10.1007/s11910-001-0028-4. [DOI] [PubMed] [Google Scholar]
- McCarey DW, McInnes IB, Madhok R, Hampson R, Scherbakov O, Ford I, Capell HA, Sattar N. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet. 2004;363(9426):2015–21. doi: 10.1016/S0140-6736(04)16449-0. [DOI] [PubMed] [Google Scholar]
- Miron VE, Rajasekharan S, Jarjour AA, Zamvil SS, Kennedy TE, Antel JP. Simvastatin regulates oligodendroglial process dynamics and survival. Glia. 2007;55(2):130–43. doi: 10.1002/glia.20441. [DOI] [PubMed] [Google Scholar]
- Moalem G, Gdalyahu A, Shani Y, Otten U, Lazarovici P, Cohen IR, Schwartz M. Production of neurotrophins by activated T cells: implications for neuroprotective autoimmunity. J Autoimmun. 2000;15(3):331–45. doi: 10.1006/jaut.2000.0441. [DOI] [PubMed] [Google Scholar]
- Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101(23):8786–90. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- O’Garra A, Steinman L, Gijbels K. CD4+ T-cell subsets in autoimmunity. Curr Opin Immunol. 1997;9(6):872–83. doi: 10.1016/s0952-7915(97)80192-6. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Khan M, Vollmer T, Singh AK, Singh I. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. Faseb J. 2005;19(11):1407–21. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh AK, Singh I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol Pharmacol. 2008;73(5):1381–93. doi: 10.1124/mol.107.044230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh AK, Stanislaus R, Gilg AG, Barbosa E, Singh I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by Lovastatin. J Neurosci Res. 2004;77(1):63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169(3):1012–25. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu R, Christie DK, Barbosa E, Singh I, Singh AK. Post-trauma Lipitor treatment prevents endothelial dysfunction, facilitates neuroprotection, and promotes locomotor recovery following spinal cord injury. J Neurochem. 2007;101(1):182–200. doi: 10.1111/j.1471-4159.2006.04354.x. [DOI] [PubMed] [Google Scholar]
- Paul F, Waiczies S, Wuerfel J, Bellmann-Strobl J, Dorr J, Waiczies H, Haertle M, Wernecke KD, Volk HD, Aktas O, et al. Oral high-dose atorvastatin treatment in relapsing-remitting multiple sclerosis. PLoS ONE. 2008;3(4):e1928. doi: 10.1371/journal.pone.0001928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pender MP, Nguyen KB, Willenborg DO. Demyelination and early remyelination in experimental allergic encephalomyelitis passively transferred with myelin basic protein-sensitized lymphocytes in the Lewis rat. J Neuroimmunol. 1989;25(23):125–42. doi: 10.1016/0165-5728(89)90130-6. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422(6933):688–94. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- Pooler AM, Xi SC, Wurtman RJ. The 3-hydroxy-3-methylglutaryl co-enzyme A reductase inhibitor pravastatin enhances neurite outgrowth in hippocampal neurons. J Neurochem. 2006;97(3):716–23. doi: 10.1111/j.1471-4159.2006.03763.x. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Howe CL, Rodriguez M. Growth factor treatment of demyelinating disease: at last, a leap into the light. Trends Immunol. 2002;23(11):512–6. doi: 10.1016/s1471-4906(02)02321-9. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, Lublin FD, Weinstock-Guttman B, Wynn DR, Lynn F, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–23. doi: 10.1056/NEJMoa044396. [DOI] [PubMed] [Google Scholar]
- Sanchez AJ, Puerta C, Ballester S, Gonzalez P, Arriaga A, Garcia-Merino A. Rolipram impairs NF-kappaB activity and MMP-9 expression in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;168(12):13–20. doi: 10.1016/j.jneuroim.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Schulz JG, Bosel J, Stoeckel M, Megow D, Dirnagl U, Endres M. HMG-CoA reductase inhibition causes neurite loss by interfering with geranylgeranylpyrophosphate synthesis. J Neurochem. 2004;89(1):24–32. doi: 10.1046/j.1471-4159.2003.02305.x. [DOI] [PubMed] [Google Scholar]
- Sommer N, Loschmann PA, Northoff GH, Weller M, Steinbrecher A, Steinbach JP, Lichtenfels R, Meyermann R, Riethmuller A, Fontana A, et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat Med. 1995;1(3):244–8. doi: 10.1038/nm0395-244. [DOI] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388(6639):275–9. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Sorensen PS. The role of intravenous immunoglobulin in the treatment of multiple sclerosis. J Neurol Sci. 2003;206(2):123–30. doi: 10.1016/s0022-510x(02)00343-x. [DOI] [PubMed] [Google Scholar]
- Stankoff B, Aigrot MS, Noel F, Wattilliaux A, Zalc B, Lubetzki C. Ciliary neurotrophic factor (CNTF) enhances myelin formation: a novel role for CNTF and CNTF-related molecules. J Neurosci. 2002;22(21):9221–7. doi: 10.1523/JNEUROSCI.22-21-09221.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuve O, Youssef S, Weber MS, Nessler S, von Budingen HC, Hemmer B, Prod’homme T, Sobel RA, Steinman L, Zamvil SS. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006;116(4):1037–44. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torphy TJ, Undem BJ. Phosphodiesterase inhibitors: new opportunities for the treatment of asthma. Thorax. 1991;46(7):512–23. doi: 10.1136/thx.46.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullman MJ, Lublin FD. Combination therapy in multiple sclerosis. Curr Neurol Neurosci Rep. 2005;5(3):245–8. doi: 10.1007/s11910-005-0053-9. [DOI] [PubMed] [Google Scholar]
- Vanderlocht J, Hendriks JJ, Venken K, Stinissen P, Hellings N. Effects of IFN-beta, leptin and simvastatin on LIF secretion by T lymphocytes of MS patients and healthy controls. J Neuroimmunol. 2006;177(12):189–200. doi: 10.1016/j.jneuroim.2006.04.012. [DOI] [PubMed] [Google Scholar]
- Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, Preiningerova J, Rizzo M, Singh I. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363(9421):1607–8. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- Wong B, Lumma WC, Smith AM, Sisko JT, Wright SD, Cai TQ. Statins suppress THP-1 cell migration and secretion of matrix metalloproteinase 9 by inhibiting geranylgeranylation. J Leukoc Biol. 2001;69(6):959–62. [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25(2):130–9. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61(1):23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420(6911):78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- Ytterberg C, Johansson S, Andersson M, Olsson D, Link H, Holmqvist LW, von Koch L. Combination therapy with interferon-beta and glatiramer acetate in multiple sclerosis. Acta Neurol Scand. 2007;116(2):96–9. doi: 10.1111/j.1600-0404.2007.00801.x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Yang L, Konishi Y, Maeda N, Sakanaka M, Tanaka J. Suppressive effects of phosphodiesterase type IV inhibitors on rat cultured microglial cells: comparison with other types of cAMP-elevating agents. Neuropharmacology. 2002;42(2):262–9. doi: 10.1016/s0028-3908(01)00174-5. [DOI] [PubMed] [Google Scholar]