

Abstract
The small all-β protein tendamistat folds and unfolds with two-state kinetics. We determined the volume changes associated with the folding process by performing kinetic and equilibrium measurements at variable pressure between 0.1 and 100 MPa (1 to 1,000 bar). GdmCl-induced equilibrium unfolding transitions reveal that the volume of the native state is increased by 41.4 ± 2.0 cm3/mol relative to the unfolded state. This value is virtually independent of denaturant concentration. The use of a high-pressure stopped-flow instrument enabled us to measure the activation volumes for the refolding (ΔVf0‡) and unfolding reaction (ΔVu0‡) over a broad range of GdmCl concentrations. The volume of the transition state is 60% native-like (ΔVf0‡ = 25.0 ± 1.2 cm3/mol) in the absence of denaturant, indicating partial solvent accessibility of the core residues. The volume of the transition state increases linearly with denaturant concentration and exceeds the volume of the native state above 6 M GdmCl. This result argues for a largely desolvated transition state with packing deficiencies at high denaturant concentrations and shows that the structure of the transition state depends strongly on the experimental conditions.
The characterization of the energy barriers between unfolded and native proteins is one of the major goals of protein folding studies and has been targeted by several experimental approaches. Kinetic analysis of mutant proteins allowed to probe the presence of individual structural elements in the transition state for folding of several proteins (1, 2). These studies showed that many native interactions are already weakly formed in the transition state. Conclusions on the solvent accessibility of the transition state are commonly drawn from the denaturant dependence of the rate constants for unfolding and refolding (3). The results on a large number of proteins suggest that protein folding reactions have rather native-like transition states in terms of solvent exposure. Information on the energetics of the transition state was obtained from measurements of the temperature dependence of the folding kinetics and revealed both enthalpic and entropic contributions to the free energy barriers at room temperature (4–7).
Despite the wealth of experimental data on protein folding, little is known about the effect of pressure on folding kinetics. The pressure dependence of a chemical reaction rate reflects the volume change between the initial state and the transition state of the reaction. In protein folding formation of the native state usually is accompanied by an increase in volume, which is believed to be caused by (i) the volume changes of released water molecules (8–10) and in the polypeptide chain (11, 12) upon formation of a compact state with a solvent-inaccessible core and (ii) inefficient packing of the native state (13). Therefore, the pressure dependence of folding and unfolding rates can give valuable information on the structure and the hydration properties of the transition state. Up to now, kinetic studies on the effect of pressure on folding kinetics were restricted to pressure-jump experiments (14, 15). Volume changes in protein folding are commonly in the region of 20–100 cm3/mol for single-chain proteins (8). Thus, pressure jumps of 100–250 bar, which have been applied in protein folding studies, cause only minor changes in the equilibrium constant and consequently result in small amplitudes of the relaxation kinetics. In addition, the experiments are limited to conditions in the transition region between native and unfolded protein.
We used the small all-β-sheet protein tendamistat (16) as a model to investigate the effect of pressure on protein folding reactions. The fast folding reaction of tendamistat with disulfide bonds intact is a two-state process under all applied conditions (17), which allows straightforward determination of the microscopic rate constants for folding and unfolding. The use of a pressurized stopped-flow instrument enabled us to perform kinetic measurements over a broad range of GdmCl concentrations at pressures between 0.1 and 100 MPa (1 MPa = 10 bar). Global analysis of the effects of pressure and GdmCl on the folding kinetics in combination with equilibrium unfolding transitions yielded the complete volume profile for tendamistat folding. The volume of the native state is increased by 41.4 ± 2.0 cm3/mol compared with the unfolded state. This value is virtually independent of denaturant concentration. The activation volumes for refolding and unfolding both depend strongly on the GdmCl concentration and show that the transition state becomes significantly more native-like with increasing denaturant concentration.
Materials and Methods
Materials.
Tendamistat was a gift from Klaus Koller (Hoechst, Frankfurt, Germany). Purity was checked by MS and reversed-phase HPLC and exceeded 99%. Ultra-pure GdmCl (AA grade) was from Nigu Chemie (Waldkraiburg, Germany).
Kinetics of Unfolding and Refolding at Various Pressures and GdmCl Concentrations.
Kinetic measurements were performed by 1:1 mixing of either native tendamistat (in 10 mM NaOAc/HOAc, pH 5.35, various concentrations of GdmCl below 6.7 M) or unfolded protein (in 20 mM NaCl/HCl, pH 1.5, at various concentrations of GdmCl above 2.0 M) with the appropriate buffer to yield the desired concentration of GdmCl at final conditions of 50 mM glycine/HCl, pH 2.0, 35°C. The use of a high-pressure stopped-flow instrument (18) allowed kinetic measurements between 1 and 100 MPa monitored by the change in fluorescence above 305 nm after excitation at 286 nm. The final protein concentration was 6 μM. At each pressure and GdmCl concentration equilibration of the system was allowed for 15 min before 4–7 traces were averaged. The rate constants were independent of the sequence of applied pressures. Reactions with τ > 3.5 s could not be measured accurately because of drifts in the fluorescence signal after 10–20 s, presumably caused by gravity-induced flow of the solutions of different densities in the stopped-flow apparatus. Additional to the fast folding reaction a small fraction (15%) of unfolded tendamistat folds on a slow parallel pathway, limited by proline isomerization. This reaction occurs on a slower timescale and thus not influence the analysis of the fast folding process (17, 19).
The ionization of glycine at low pH involves a volume change of ΔV0 = −6.8 cm3/mol (20), leading to a decrease of the pKA of 0.1 units between 0.1 and 100 MPa. The resulting change in pH would lead to about 4% decrease of the refolding rate and 6% increase of the unfolding rate. The pKA values of the protein carboxylic groups, however, decrease similarly, such that the overall effect on the rates will be even less significant; this effect thus was not taken into account in data analysis.
GdmCl-Induced Equilibrium Unfolding Transitions at Various Pressures.
Tendamistat was incubated at different concentrations of GdmCl between 0 M and 6.5 M (40 μM in 50 mM Gly/HCl, pH 2.0) in a “Le Noble” piston-type cell (21) with an optical path length of 2 cm. The cells were pressurized between 2 and 100 MPa at 35°C in a double-beam, temperature-controlled pressure bomb (22). For every pressure and GdmCl concentration, equilibration of the system was allowed for 15 min before absorbance spectra were recorded between 240 and 350 nm on a Perkin–Elmer Lambda 19 double-beam spectrometer. The transitions were monitored by the absorbance change at 280.5 nm, and the resulting values were corrected for variations in the baseline absorption. These values are independent of the order of pressures applied. The slopes of the baselines of native and unfolded protein were independent of pressure and thus were fit globally for all transitions. The data were normalized to the fraction of native protein by setting the baseline of native protein to 1 and the baseline of the unfolded protein to 0.
Effect of Pressure and GdmCl on Protein Stability.
The Gibbs fundamental equation of chemical thermodynamics:
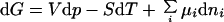 |
1 |
can be adapted for protein folding transitions including the effect of a chemical denaturant like GdmCl on protein stability:
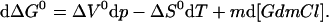 |
2 |
ΔG0 is the difference in Gibbs free energy between the native state and the unfolded state under standard conditions. ΔV0 and ΔS0 are defined accordingly. The m value represents the change in ΔG0 with the GdmCl concentration (m = (∂ΔG0/∂[GdmCl])p, T), which is empirically found to be constant over a wide range of GdmCl concentrations for protein folding transitions. At constant temperature Eq. 2 simplifies to:
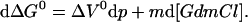 |
3 |
To determine ΔG0 at a certain pressure (p) and GdmCl concentration ([GdmCl]) the effects of pressure and GdmCl on the m value and on ΔV0 have to be taken into account. The experimental data show that the m value can be approximated to be independent of the GdmCl concentration and ΔV0 to be independent of pressure (see Results), indicating similar compressibilities of the native and the unfolded state. Because Eq. 3 represents an exact differential, we know that:
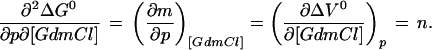 |
4 |
Thus, integration of Eq. 3 gives:
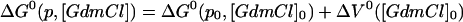 |
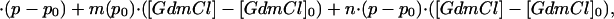 |
5 |
where p0 and [GdmCl]0 indicate reference conditions of 0.1 MPa and 0 M GdmCl, respectively. Eq. 5 assumes that n is independent of pressure and GdmCl, which is in agreement with the experimental data (see Results).
Effect of Pressure and GdmCl on Folding Kinetics.
The rate constants of folding (kf) and unfolding (ku) can be related to Gibbs free energies of activation, ΔGf0‡ and ΔGu0‡, respectively, using transition state theory (23):
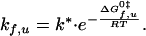 |
6 |
The value of the pre-exponential factor k* represents the maximum rate for the folding or unfolding reaction that is assumed to be independent of pressure and GdmCl. The value of k* is currently under investigation and thus the values of ΔGf,u0‡ must be regarded as apparent Gibbs free energies of activation. By treating ΔGf,u0‡ the same way as ΔG0 (Eqs. 1-5), we obtain in analogy to Eq. 5:
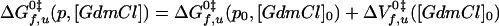 |
7 |
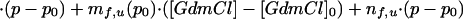 |
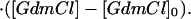 |
Again, it is assumed that mu,f are independent of GdmCl concentration, that ΔVf,u0‡ are independent of pressure and that nf,u are independent of both pressure and denaturant concentration, which is in agreement with the experimental data (see Results).
Analysis of Experimental Data.
Equilibrium unfolding curves at each pressure were analyzed according to the method of Santoro and Bolen (24), assuming a two-state unfolding transition with linear baselines for the GdmCl and pressure dependence of the absorbance of native and unfolded protein and a linear GdmCl dependence of ΔG0 [(∂ΔG0/∂[GdmCl]]p = m = constant). This yields ΔG0 at each pressure in the absence of denaturant [ΔG0(H2O)] and the associated m values. For the analysis of the pressure dependence of the equilibrium constant (K) at constant GdmCl concentration Eq. 3 was combined with:
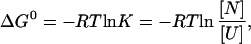 |
8 |
which gives the well-known equation (25):
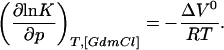 |
9 |
In tendamistat folding about 15% of the unfolded molecules fold slowly on a parallel pathway limited by a prolyl isomerization reaction (Us molecules). To take the two different populations of unfolded molecules into account the actual equilibrium constant for the folding transitions (K = [N]/[UF]) was determined from the measured equilibrium constant (Kobs) using (19, 26):
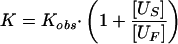 |
10 |
with [US] = 0.15 and [UF] = 1 − [US] = 0.85 and assuming that both unfolded species have the same volume. This correction had only minor effects on the results.
The GdmCl dependence of the apparent rate constant (λ) at each pressure was analyzed by using the two-state model with
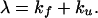 |
11 |
The microscopic rate constants for refolding and unfolding in water, kf(H2O) and ku(H2O), respectively, were obtained from the measurements of the apparent rate constant (λ) at various concentrations of denaturant by fitting the complete GdmCl dependence of ln λ according to
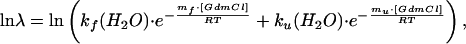 |
12 |
where mf,u represent the GdmCl dependencies of ln kf,u. The pressure dependence of kf and ku at constant denaturant concentration was analyzed by using
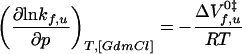 |
13 |
in analogy to Eq. 9.
Global Analysis of Equilibrium and Kinetic Data.
For two-state folding the equilibrium constant (K) represents the ratio of the folding (kf) and unfolding (ku) rate constants (K = kf/ku). In combination with Eqs. 5 and 7, this relationship provides links between all kinetic and thermodynamic parameters:
 |
14 |
These relationships together with Eqs. 5, 7, 8, 10, and 11 were used for a global fitting of the pressure and GdmCl dependence of equilibrium and kinetic data.
Data Fitting.
The program profit (Quantum Soft, Zurich, Switzerland) was used for data fitting. Errors and confidence intervals were evaluated by using the implemented “error analysis” routine.
Results
We studied the effect of pressure on tendamistat folding and stability at pH 2.0, 35°C. Fig. 1 shows time traces for refolding at 2.0 M GdmCl (Fig. 1A) and unfolding at 6.5 M GdmCl (Fig. 1B) at varying pressure. Increasing the pressure from 0.1 to 100 MPa (1 to 1,000 bar) decreases the folding rate at 2.0 M GdmCl 3-fold whereas the unfolding rate at 6.5 M GdmCl decreases only 1.15-fold. To determine the effect of pressure on the microscopic rate constants for folding (kf) and unfolding (ku) we measured the pressure dependence of the apparent rate constant (λ = kf + ku) at a variety of different GdmCl concentrations between 1 and 7.5 M (Fig. 2A). Under all conditions single exponential kinetics were observed both for unfolding and refolding. Rate constants for GdmCl concentrations between 2.5 and 5.5 M could not be obtained with sufficient accuracy because of instability of the kinetic traces at times longer than 10 s. Fig. 2A shows that refolding becomes progressively slower with increasing pressure at GdmCl concentrations between 1 and 2.33 M GdmCl. Unfolding becomes slower with increasing pressure above 6 M GdmCl and faster with increasing pressure below 6 M GdmCl.
Figure 1.

Pressure-dependent kinetics of tendamistat folding at 2.0 M GdmCl (A) and unfolding at 6.5 M GdmCl (B) monitored by the change in fluorescence above 305 nm. Measurements were carried out at 5 MPa (black), 20 MPa (dark blue), 40 MPa (green), 60 MPa (red), 80 MPa (light blue), and 100 MPa (magenta). Experimental conditions were 50 mM Gly/HCl, pH 2.0, 35°C.
Figure 2.

Effect of pressure on the GdmCl dependence of the apparent rate constant (A) and the equilibrium transition (B) for tendamistat folding at pH 2.0, 35°C. Experiments were carried out at 5 MPa (black), 20 MPa (dark blue), 40 MPa (green), 60 MPa (red), 80 MPa (light blue), and 100 MPa (magenta). The lines represent the result of the global fit of the kinetic and the equilibrium data according to Eqs. 5, 7, 8, 10 and 11. The residuals of the global fits are shown below each panel.
The GdmCl dependence of ln λ was fit according to Eq. 12 to yield kf and ku in the absence of denaturant [ku(H2O) and kf(H2O), respectively] and their respective changes with the GdmCl concentration, mf/RT and mu/RT. The results show that both mf and mu change with pressure, i.e., that (∂mf,u/∂p)[GdmCl] = nf,u ≠ 0 (Eq. 4). These parameters allow the calculation of ku and kf at any given pressure and GdmCl concentration and the determination of the activation volumes for folding (ΔVf0‡) and unfolding (ΔVu0‡) at each GdmCl concentration by using Eq. 13. Both ln kf (Fig. 3A) and ln ku (Fig. 3B) change linearly with pressure at all GdmCl concentrations. However, the slope of the pressure dependence of ln ku and ln kf changes with the denaturant concentrations, indicating a GdmCl dependence of the activation volumes [(∂ΔVf,u0‡/∂[GdmCl])p = nf,u; Eq. 4]. ΔVf0‡ increases from 27 ± 1.4 cm3/mol at 1.0 M GdmCl to 35 ± 1.4 cm3/mol at 2.3 M GdmCl, and ΔVu0‡ increases from about −1.0 ± 0.9 cm3/mol at 5.5 M GdmCl to 9.0 ± 0.9 cm3/mol at 7.5 M GdmCl (Fig. 4A). Comparison of the change in m-values with pressure (Fig. 2A) with the change in the activation volumes with the GdmCl concentration (Fig. 3 A and B) gives values of 6.1 ± 2.5 and 5.8 ± 1.5 (cm3/mol)/M for nf and values of 4.4 ± 1.0 and 4.8 ± 1.0 (cm3/mol)/M for nu, respectively. This good agreement demonstrates the validity of Eq. 4.
Figure 3.

Pressure dependence of the microscopic rate constants, kf (A) and ku (B), and the equilibrium constant, K (C) at various GdmCl concentrations indicated on the right. Microscopic rate constants were determined by fitting the data in Fig. 2A to Eq. 12. The equilibrium constants were taken from Fig. 2B by using Eq. 8. The lines represent fits according to Eq. 13 for the microscopic rate constants and according to Eq. 9 for the equilibrium constant. The results of the fits are shown in Fig. 4A.
Figure 4.

(A) GdmCl dependence of the activation volumes and the reaction volume for tendamistat folding at 50 mM Gly/HCl, pH 2.0, 35°C. The data points show the activation volumes for refolding (red) and unfolding (blue) and the reaction volume (black) determined in individual fits of the kinetic and equilibrium data displayed in Fig. 3. The lines represent the results from the global fit (solid line) of kinetic and equilibrium data shown in Fig. 2 together with the 2σ confidence intervals (dashed lines). The parameters of the global fit are: ΔG0 (H2O, 0.1 MPa) = −16.38 ± 0.14 kJ/mol; m(0.1 MPa) = 4.62 ± 0.04 (kJ/mol)/M; ΔV0(H2O) = 41.4 ± 2.0 cm3/mol; n = −0.6 ± 0.7 (cm3/mol)/M; kf(H2O, 0.1 MPa) = 10.2 ± 0.3 s−1; mf(0.1 MPa) = 3.04 ± 0.03 (kJ/mol)/M; ku(H2O) = 1.36 ± 0.06 × 10−2 s−1; mu(0.1 MPa) = −1.58 ± 0.02 (kJ/mol)/M; ΔVf0‡(H2O) = 25.0 ± 1.2 cm3/mol; nf = 2.5 ± 0.6 (cm3/mol)/M; ΔVu0‡(H2O) = −16.4 ± 1.4 cm3/mol; nu = 3.1 ± 0.2 (cm3/mol)/M. The results change only slightly if a pressure dependence of the GdmCl concentration in the range observed for solutions of other monovalent salts is assumed (36). (B) Volume changes along the reaction coordinate of tendamistat folding. The volume of the unfolded state was set to zero. The solid line represents the experimentally determined volume changes. The dashed line represents the hypothetical continuation of the profile to the native state. The symbols on the line indicate the location of the transition state at 0 M GdmCl (●) and 8 M GdmCl (⧫). The transition state moves to higher volumes with increasing GdmCl concentrations as indicated by the arrow. The yellow circle represents the protein and the blue circles the water molecules.
To compare the kinetic data for tendamistat folding with the thermodynamic parameters we measured the effect of pressure on the GdmCl-induced equilibrium unfolding transition under the same conditions as in the kinetic experiments (Fig. 2B). Tendamistat is destabilized by increasing pressure, resulting in a shift of the unfolding transition to lower GdmCl concentrations. Fitting the transitions according to the two-state model and analyzing the effect of pressure on the equilibrium constant (Eq. 9) shows that the volume of the native state is increased by 41.6 ± 2.7 cm3/mol compared with the unfolded state (Fig. 3C). This value shows no systematic variation with denaturant concentration (Fig. 4A). Comparison of the kinetic and thermodynamic parameters at each pressure according to Eq. 14 shows that the two-state character of tendamistat folding is conserved under all conditions. This enables us to simultaneously fit the pressure and the GdmCl dependence of the equilibrium constant (Fig. 2B) and the apparent rate constant (Fig. 2A) in a global fitting procedure by using Eqs. 5, 7, 8, 10, and 11. The fit is able to describe both the kinetic data (Fig. 2A) and the equilibrium data (Fig. 2B) very well and without systematic deviations. As expected from the individual fits of the kinetic data the nf and nu values are nonzero, indicating a GdmCl-dependent change in the activation volume and a pressure-dependent change in the m values (Eq. 4). Fig. 4 compares the results from the global fit with the reaction volume and the activation volumes from the individual fits of the kinetic and equilibrium (Fig. 3). Qualitatively both methods give the same results, but the global analysis has significantly smaller errors. In the absence of denaturant the refolding and unfolding process have activation volumes of 25.0 ± 1.2 cm3/mol and −16.4 ± 1.4 cm3/mol, resulting in a reaction volume of 41.4 ± 2.0 cm3/mol. Both activation volumes increase by virtually the same amount with increasing denaturant concentration resulting in a negligible GdmCl dependence of the reaction volume of −0.6 ± 0.7 cm3/mol per M of GdmCl. As a result of the increase in activation volumes the volume of the transition state exceeds the volume of the native state at GdmCl concentrations above 6 M. Under these conditions both unfolding and refolding become slower with increasing pressure.
Discussion
Effect of Pressure on Tendamistat Folding and Stability.
Global analysis of the effect of pressure and GdmCl on tendamistat folding and stability allowed the determination of the reaction volume (ΔV0) and the activation volumes for the refolding (ΔVf0‡) and the unfolding reaction (ΔVu0‡) between 0 and 7.5 M GdmCl. The volume of native tendamistat is increased by 41.4 ± 2.0 cm3/mol compared with the denatured form at pH 2.0 and 35°C (Figs. 3C and 4). This value is virtually independent of the denaturant concentrations (Fig. 4) and is similar to reaction volumes for folding of many other small single domain proteins (8). Contributions to ΔV0 may arise from packing deficiencies in the native state (13) and differences in solvent interactions between native and unfolded protein (8–10). The structure of native tendamistat shows no indication for cavities (16, 27). However, the observed volume change corresponds to only about 1% of the total protein volume. Thus, even minor packing deficiencies in the native protein might contribute to the reaction volume. Other contributions to ΔV0 are known to arise from changes in interactions between the solvent and the protein chain. This effect is especially pronounced around charged groups (electrostriction). Our experiments were performed at pH 2, which minimizes changes in electrostriction because all acid side chains are protonated both in the native and in the unfolded state. Contributions from electrostriction around positive charges also are not expected because no basic groups are buried in the native state (16). The origin of the usually observed small decrease in volume associated with protein unfolding has been subject of controversial discussions (10–12, 28). In the picture that emerges the interior of a folded protein resembles an almost crystalline state. Consequently, parts of the polypeptide chain that are shielded from solvent have a smaller volume than solvent exposed parts (12) and the volume of these groups increases upon unfolding. This effect is opposed by a decrease in volume of water molecules that become bound to the polypeptide chain upon exposure of hydrophobic and polar groups (12). For most proteins the result of these compensating effects in combination with small packing deficiencies in the native state leads to the small decrease in volume commonly observed in protein unfolding.
Structure of the Transition State.
Because volume changes during tendamistat folding arise mainly from the changes in solvent interactions of the polypeptide chain and packing deficiencies in the hydrophobic core, measurement of the activation volumes for the refolding and unfolding reaction gives structural information on the transition state. The activation volumes for the refolding and unfolding reaction of 25.0 ± 1.2 and −16.4 ± 1.4 cm3/mol in the absence of denaturant show that the volume of the transition state is 60% native-like and suggests that it is still partially solvated. It is unlikely that cavities contribute significantly to the volume of the transition state. The presence of cavities would indicate minor contributions from desolvation, and a highly solvated transition state is not able to form solvent inaccessible cavities. The m value, i.e., the change in ΔG0 with denaturant concentration, also is believed to reflect differences in solvent accessibility between the unfolded state and the native state. Accordingly, the changes in the free energy of activation with denaturant are commonly used as a measure for the solvent exposure of the transition state. By this criterion the transition state for tendamistat folding is 67% native-like (Fig. 2A), which is slightly less solvent accessible than expected from the activation volumes.
Effect of GdmCl on the Structure of the Transition State.
The activation volumes for refolding and unfolding both strongly increase with the GdmCl concentration, although the reaction volume is virtually independent of denaturant concentration (Fig. 4). This effect cannot be attributed to an influence of GdmCl on the solvent, because water associated with the unfolded polypeptide chain is released in the folding step and bound in the unfolding step. Thus, denaturants should have adverse effects on the activation volumes of the folding and unfolding reaction. In addition, the maximum difference in bound water occurs between the native state and the unfolded state and thus contributions from effects of the denaturant on water structure would most prominently affect the reaction volume. Consequently, the large and compensating changes in the activation volumes for the refolding and unfolding reaction indicate GdmCl-induced changes in the structure of the transition state.
As a result of the GdmCl-induced structural movement of the transition state its volume becomes larger than the volume of the native state above 6 M GdmCl. This finding suggests contributions from both dehydration and packing deficiencies to the volume of the transition state at high denaturant concentrations, because the volume increase caused by dehydration should not exceed the reaction volume. As packing deficiencies can become important only when solvent is excluded from major parts of the hydrophobic core these results point at a largely desolvated, but not yet tightly packed, transition state under strongly destabilizing conditions. Obviously, the transition state becomes more native-like with increasing denaturant concentration. This observation is in agreement with the Hammond postulate, which states that the structure of the transition state of a chemical reaction becomes more product like, when the product is destabilized (29). For protein folding reactions a denaturant-induced structural movement of the transition state might be explained by the weakening of hydrophobic interactions and hydrogen bonding with increasing concentrations of denaturant. As a result, a larger number of interactions are required to compensate for the loss in conformational entropy during the folding process. This effect leads to a more native-like transition state. The Hammond postulate also was proposed to explain movements of the transition state for barnase and CI2 in response to mutations affecting protein stability (30). Our results show that changes in protein stability induced by altered solvent conditions also can give rise to Hammond behavior.
Because the kinetic m values are believed to reflect changes in solvent accessibility between the unfolded state (mf) or the native state (mu) and the transition state (3), they also should change when the structure of the transition state changes (31). However, mf and mu are constant between 1 and 7.5 M GdmCl although the activation volumes indicate significant GdmCl-induced structural changes of the transition state. This finding implies that the kinetic m values contain contributions from both the changes in solvent accessibility and a structural movement of the transition state along the reaction coordinate. The difference in the structural organization in the transition state calculated from activation volumes (60% native-like) and the m value analysis (67% native-like) might reflect this effect. These results show that the interpretation of kinetic m values in terms of the structure of the transition state is not straightforward, because the m values may contain information both on structure and denaturant-induced structural changes of the transition state.
If the structure of the transition state is sensitive to changes in protein stability, the activation volume also should change with increasing pressure, which is not observed. Similar to the kinetic m values, the activation volumes might contain contributions from the actual transition state volume and a pressure-induced structural movement of the transition state. This model is supported by the compensating changes in the pressure dependencies of mf and mu, which indicate a more native-like transition state at higher pressure. However, tendamistat stability only decreases by 4 kJ/mol between 0.1 and 100 MPa. Thus, the effect of pressure in the applied range on the movement of the transition state should be less pronounced than the effect observed for GdmCl, which destabilizes native tendamistat by 35 kJ/mol between 0 and 7.5 M.
Measurement of the activation volumes for staphylococcal nuclease folding by using pressure-jump experiments showed that the volume of the transition state is larger than the volume of the native protein in the region of the pressure-induced unfolding transition (14). The volume of the transition state for staphylococcal nuclease was suggested to be independent of denaturant concentration between 0.5 and 0.75 M GdmCl (32). Our results show that the volume of the transition state for tendamistat folding is also similar to the volume of the native protein in the transition region. However, the much wider stability range explored in our experiments reveal major denaturant-dependent changes in the activation volumes. Under strongly native solvent conditions the transition state is much less native-like and its volume is between the volumes of unfolded and native tendamistat.
Assuming that the observed denaturant-induced changes in the activation volume reflect a movement of the transition state on the reaction coordinate allows us to map the volume changes along the reaction coordinate for tendamistat folding (Fig. 4B). Our data show that the folding polypeptide chain goes through a point of maximum volume before it reaches the native state. In a certain region on the reaction coordinate, the solvent seems to be excluded from the core but the native set of tertiary interactions is not yet formed. This observation is in agreement with the concept of a dry molten globule, which was predicted theoretically (33) and observed experimentally during unfolding of ribonuclease A (34) and dihydrofolate reductase (35). Our results suggest that exclusion of water precedes formation of the correct side-chain contacts. The strong denaturant dependence of the activation volumes argues for a strong structural plasticity of the transition state for protein folding and shows that the activated state can be either solvent accessible or shielded from solvent, depending on the experimental conditions.
Acknowledgments
We thank Dr. Klaus Koller (Hoechst, Frankfurt) for the gift of tendamistat, Ansgar Philippsen for the calculation of the volume of native tendamistat, and Buzz Baldwin for discussions and comments on the manuscript. This work was supported by grants from the Swiss National Science Foundation (to A.E.M. and T.K.).
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Otzen D E, Itzhaki L S, elMasry N F, Jackson S E, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villegas V, Martinez J C, Avilés F X, Serrano L. J Mol Biol. 1998;283:1027–1036. doi: 10.1006/jmbi.1998.2158. [DOI] [PubMed] [Google Scholar]
- 3.Tanford C. Adv Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 4.Pohl F M. FEBS Lett. 1976;65:293–296. doi: 10.1016/0014-5793(76)80132-9. [DOI] [PubMed] [Google Scholar]
- 5.Oliveberg M, Tan Y-J, Fersht A R. Proc Natl Acad Sci USA. 1995;92:8926–8929. doi: 10.1073/pnas.92.19.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindler T, Schmid F X. Biochemistry. 1996;51:16833–16842. doi: 10.1021/bi962090j. [DOI] [PubMed] [Google Scholar]
- 7.Schönbrunner N, Pappenberger G, Scharf M, Engels J, Kiefhaber T. Biochemistry. 1997;36:9057–9065. doi: 10.1021/bi970594r. [DOI] [PubMed] [Google Scholar]
- 8.Heremans K. Annu Rev Biophys Bioeng. 1982;11:1–21. doi: 10.1146/annurev.bb.11.060182.000245. [DOI] [PubMed] [Google Scholar]
- 9.Dill K A. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 10.Hummer G, Garde S, Garcia A E, Paulaitis M E, Pratt L R. Proc Natl Acad Sci USA. 1998;95:1552–1555. doi: 10.1073/pnas.95.4.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klapper M H. Biochem Biophys Acta. 1971;229:557–566. doi: 10.1016/0005-2795(71)90271-6. [DOI] [PubMed] [Google Scholar]
- 12.Gerstein M, Chothia C. Proc Natl Acad Sci USA. 1996;93:10167–10172. doi: 10.1073/pnas.93.19.10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frye K J, Royer C A. Protein Sci. 1998;7:2217–2222. doi: 10.1002/pro.5560071020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vidugiris G J A, Markley J L, Royer C A. Biochemistry. 1995;34:4909–4912. doi: 10.1021/bi00015a001. [DOI] [PubMed] [Google Scholar]
- 15.Jacob M, Holtermann G, Perl D, Reinstein J, Schindler T, Geeves M, Schmid F X. Biochemistry. 1999;38:2882–2891. doi: 10.1021/bi982487i. [DOI] [PubMed] [Google Scholar]
- 16.Pflugrath J, Wiegand I, Huber R, Vértesy L. J Mol Biol. 1986;189:383–386. doi: 10.1016/0022-2836(86)90520-6. [DOI] [PubMed] [Google Scholar]
- 17.Schönbrunner N, Koller K-P, Kiefhaber T. J Mol Biol. 1997;268:526–538. doi: 10.1006/jmbi.1997.0960. [DOI] [PubMed] [Google Scholar]
- 18.Bugnon P, Laurenczy G, Ducommun Y, Sauvageat P-Y, Merbach A E, Ith R, Tschanz R, Doludda M, Bergbauer R, Grell E. Anal Chem. 1996;68:3045–3049. doi: 10.1021/ac960382k. [DOI] [PubMed] [Google Scholar]
- 19.Kiefhaber T, Kohler H H, Schmid F X. J Mol Biol. 1992;224:217–229. doi: 10.1016/0022-2836(92)90585-8. [DOI] [PubMed] [Google Scholar]
- 20.Morild E. Adv Protein Chem. 1981;34:93–166. doi: 10.1016/s0065-3233(08)60519-7. [DOI] [PubMed] [Google Scholar]
- 21.le Noble W J, Schlott R. Rev Sci Instrum. 1976;47:770–771. [Google Scholar]
- 22.Richens D T, Ducommun Y, Merbach A E. J Am Chem Soc. 1987;109:603–604. [Google Scholar]
- 23.Eyring H. J Chem Phys. 1935;3:107–115. [Google Scholar]
- 24.Santoro M M, Bolen D W. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 25.Planck M. Ann Phys Chem. 1893;32:462–503. [Google Scholar]
- 26.Kuwajima K, Mitani M, Sugai S. J Mol Biol. 1989;206:547–561. doi: 10.1016/0022-2836(89)90500-7. [DOI] [PubMed] [Google Scholar]
- 27.Kline A D, Braun W, Wüthrich K. J Mol Biol. 1988;204:675–724. doi: 10.1016/0022-2836(88)90364-6. [DOI] [PubMed] [Google Scholar]
- 28.Kauzmann W. Nature (London) 1987;325:763–764. [Google Scholar]
- 29.Hammond G S. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 30.Matouschek A, Otzen D E, Itzhaki L, Jackson S E, Fersht A R. Biochemistry. 1995;34:13656–13662. doi: 10.1021/bi00041a047. [DOI] [PubMed] [Google Scholar]
- 31.Silow M, Oliveberg M. Biochemistry. 1997;36:7633–7637. doi: 10.1021/bi970210x. [DOI] [PubMed] [Google Scholar]
- 32.Vidugiris G J A, Truckses D M, Markley J L, Royer C A. Biochemistry. 1996;35:3857–3864. doi: 10.1021/bi952012g. [DOI] [PubMed] [Google Scholar]
- 33.Shaknovich E I, Finkelstein A V. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]
- 34.Kiefhaber T, Labhardt A M, Baldwin R L. Nature (London) 1995;375:513–515. doi: 10.1038/375513a0. [DOI] [PubMed] [Google Scholar]
- 35.Hoeltzli S D, Frieden C. Proc Natl Acad Sci USA. 1995;92:9318–9322. doi: 10.1073/pnas.92.20.9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibson R E. J Am Chem Soc. 1935;57:284–293. [Google Scholar]