

Abstract
The child’s brain is more malleable or plastic than that of adults and this accounts for the ability of children to learn new skills quickly or recovery from brain injuries. Several mechanisms contribute to this ability including overproduction and deletion of neurons and synapses, and activity-dependent stabilization of synapses. The molecular mechanisms for activity dependent synaptic plasticity are being discovered and this is leading to a better understanding of the pathogenesis of several disorders including neurofibromatosis, tuberous sclerosis, Fragile X syndrome and Rett syndrome. Many of the same pathways involved in synaptic plasticity, such as glutamate-mediated excitation, can also mediate brain injury when the brain is exposed to stress or energy failure such as hypoxia-ischemia. Recent evidence indicates that cell death pathways activated by injury differ between males and females. This new information about the molecular pathways involved in brain plasticity and injury are leading to insights that will provide better therapies for pediatric neurological disorders.
Keywords: Plasticity, Injury, Fragile X Syndrome, Rett Syndrome, Hypoxia-Ischemia, NMDA, AMPA, Periventricular Leukomalacia
Many disorders and injuries of the developing brain affect the basic mechanisms that allow the nervous system to be shaped by experience during childhood. These mechanisms provide the substrate for brain plasticity (kasosei in Japanese), which is much more active in children than in adults. Plasticity in the child’s brain is enhanced because the organization of networks of neuronal synapses as well as white matter pathways remain “under construction” well into adolescence and even later(1). Accordingly, the effects of intensive learning in school, exposure to a second language or practice in athletics has a much greater impact on children than adults. Several neurobiological mechanism contribute to brain plasticity, including an over-production of neurons in early development, apoptosis or programmed cell death of excessive neurons, overproduction and elimination of immature synapses in childhood, and continuous stabilization and strengthening of synaptic connections later in life(2). In this review we focus on some mechanisms for synaptic plasticity, and emerging evidence that these processes are disrupted in several pediatric neurological disorders.
Synaptic Plasticity
Synaptic plasticity is the most important mechanism that allows the developing brain to adapt to environmental influences and store information throughout life(3). This term includes changes that increase or decrease the strength or efficacy of synapses as well as the addition or pruning of synapses. Changes in the number of synapses are especially dynamic in the cerebral cortex in infancy and childhood in the human brain(4). Synapses are produced at a rapid rate in the postnatal period and reach a density that is twice the adult level by age two years, and then fall to the adult level by early adolescence. This process of synapse proliferation and pruning appears to be under the control of both intrinsic programs and environmental influences. The balance of activity between excitatory synapses that use glutamate as their neurotransmitter and inhibitory synapses that use γ-aminobutyric acid (GABA) as their neurotransmitter influence the stabilization of synapses and neuronal circuits (Figure 1) (2;5). Neurons that form synapses with the same neuron and which fire together repeatedly are more likely to form lasting circuits than those whose firing is not coordinated(6;7). Other neurotransmitters including acetylcholine and serotonin projections to the cerebral cortex influence the proliferation and pruning of synapses as well as the ability of neuronal circuits to rearrange in response to changes in sensory information. For example, the organization of cortical maps for somatosensory and auditory information in rodents is strongly influenced by release of acetylcholine from axons projected from the cholinergic nucleus basalis(8;9).
Figure 1.
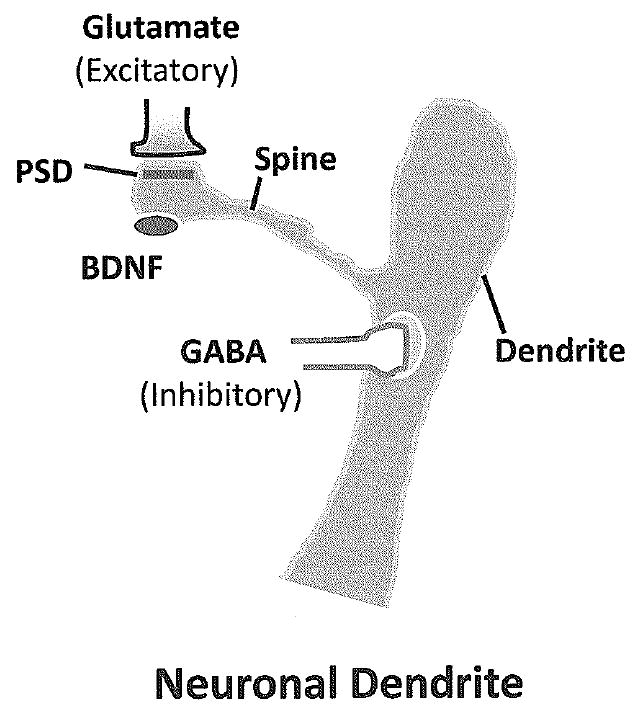
Diagram of a neuronal dendrite and spine. Excitatory synapses that use glutamate form synapses on dendritic spins but inhibitory neurons form synapses on the body of the dendrite. Spines change shape in response to excitatory activity, mature spines are shorter than immature spines, which are receiving less excitatory input. PSD = postsynaptic density; BDNF= brain derived neurotrophic factor.
Changes in the strength of excitatory synapses are responsible for encoding memories in the brain as well as for other forms of plasticity of neural circuits(10). As shown in Figure 2, three major types of glutamate receptors respond to the neurotransmitter glutamate, including N-methyl-D-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and metabotropic receptors linked to second messengers, such as the mGluR5 receptors(11;12). These glutamate receptors are anchored in the postsynaptic density (PSD) that is characteristic of excitatory but not inhibitory synapses(13). The PSD is a scaffold-like structure made up of hundreds of proteins including cytoskeletal elements and signaling molecules that change in response to development and synaptic activity(14). AMPA receptors linked to channels that carry sodium and/or calcium are responsible for most of the fast excitatory activity in the brain, and their number in the postsynaptic membrane determines the strength of the excitatory synaptic activity. NMDA channels also carry sodium and calcium, and they are opened by activation of specific receptors for glutamate and glycine. NMDA receptors are voltage-dependent and open only when there is enough AMPA receptor activity to depolarize the synaptic membrane(15).
Figure 2.
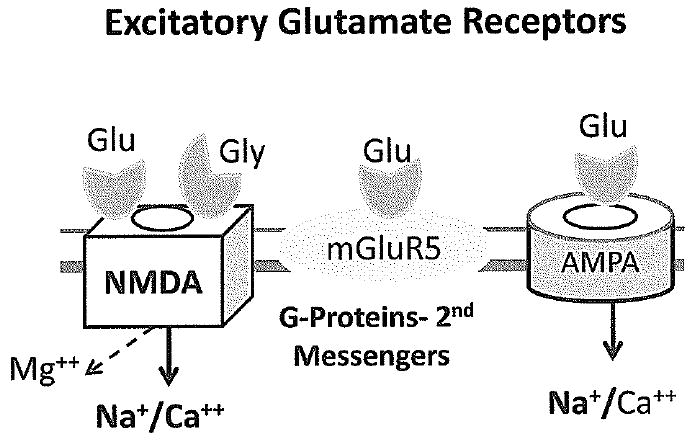
Diagram of the three major types of excitatory neurotransmitter receptors for glutamate. NMDA receptors are activated when glutamate (glu) and glycine (gly) both occupy receptor sites and the membrane depolarizes, allowing magnesium (Mg++) to leave the channel. Relief of the magnesium block allows calcium and sodium to pass through the channel. Most fast excitatory activity in the brain is mediated by AMPA receptor channels, which flux mostly sodium, but channels lacking the GluR2 subunit also pass calcium. In contrast to NMDA and AMPA receptors, metabotropic glutamate receptors are not linked to ion channels but to G proteins and second messenger systems such as phosphoinositide turnover that regulate intracellular calcium levels and protein translation.
Activation of NMDA receptors induces long term potentiation (LTP) which is associated with an increase in synaptic strength and is thought to be a physiologic correlate of memory formation in the hippocampus (Figure 3)(16). LTP induced by activation of NMDA receptors has been reported to be increased in the immature rodent brain compared to the adult brain, due in part to developmental changes in NMDA receptor subunits(16;17). The overall balance of excitation versus inhibition in the neonatal brain also appears to be shifted towards excitation in the neonatal period because receptors for GABA mediate excitatory activity at this stage due to developmental changes in chloride transporters(18). In contrast to activation of NMDA receptors, stimulation of mGluR5 receptors is associated with long term depression (LTD) of synaptic strength(19). AMPA receptors move back and forth between the postsynaptic membrane and the cytoplasm in a process called trafficking that is controlled by activity at NMDA and mGluR5 receptors(20;21). In LTP, high levels of NMDA receptor activity lead to insertion of more AMPA receptors into the postsynaptic membrane resulting in greater synaptic strength. Activation of NMDA receptors associated with physiologic induction of LTP also enhances production of brain derived neurotrophic factor (BDNF) by neurons (Figure 1)(22). BDNF binds to specific receptors on neurons and induces morphologic changes associated with LTP, including insertion of AMPA receptors into the postsynaptic membrane and changes in spine morphology(23). In contrast, activation of mGluR5 receptors leads to internalization of AMPA receptors, LTD and reduced synaptic strength(19).
Figure 3.
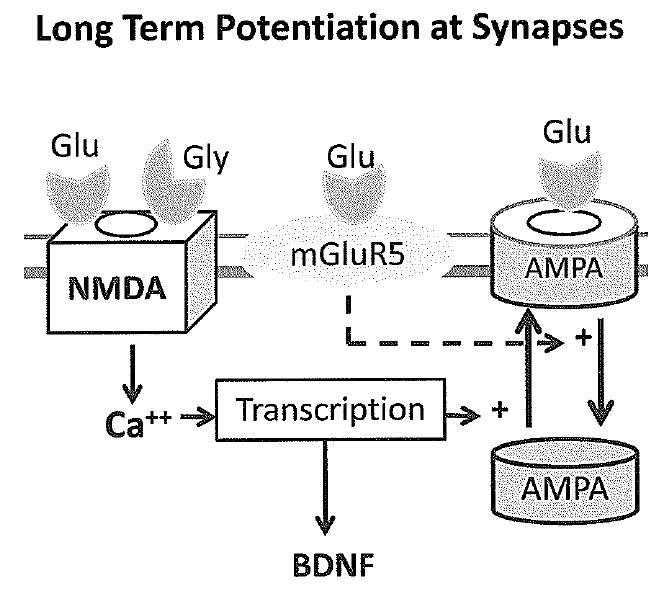
Long term potentiation (LTP) is a form of synaptic plasticity that increases the strength of synapses, and it is a physiological correlate of memory. NMDA receptor activation is necessary for LTP, and it stimulates insertion of more AMPA receptors into the synaptic membrane. Activation of NMDA receptors also stimulates the production of brain derived neurotrophic factor (BDNF) which also enhances insertion of NMDA receptors. Stimulation of mGluR5 receptors regulates protein translation and antagonizes LTP by stimulating trafficking of AMPA receptors away from the synapse into the cytoplasm. This increases long term depression (LTD).
Abnormal Plasticity in Pediatric Brain Disorders
Several acquired and genetic pediatric disorders disrupt brain function primarily by targeting synaptic mechanisms involved in neuronal plasticity of the developing brain(1;2). The common theme for these disorders is that they disrupt specific steps in the pathways that lead to changes in numbers of synapses or in the strength of synapses based on synaptic activity(24). For example, neurofibromatosis type 1 (NF-1) is caused by a mutation in the tumor suppressor gene for the protein neurofibromin, which is a GTPase activating protein (GAP)(25). Mutations in neurofibromin result in over-activity of Ras, a small GTPase switch that controls signaling from growth factor receptors in the neuronal membrane to intracellular signaling pathways in the nucleus(26). Neurofibromin controls the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI-3) pathways that are involved in cell growth as well as learning and memory. Work in a mouse model of NF indicates that learning problems are associated with excessive inhibition by GABA neurons in the hippocampus, and learning can be improved by a drug that antagonizes GABA(26). The cholesterol lowering drug lovastatin has also been shown to improve learning in the NF-1 animal model by reducing Ras activity(27). Mice that lack neurofibromin in the majority of cortical neurons and astrocytes fail to form cortical barrels in the somatosensory cortex(28). Excessive activity of the similar small GTPase Ras-like protein Rheb is also involved in the pathogenesis of tuberous sclerosis (TS)(29). TS is caused by mutations in tuberin (TSC2) or hamartin (TSC1), which binds to TSC2, and normally they function together to inhibit Rheb. Excessive activity of Rheb caused by mutations in TSC1 or TSC2 results in excessive activity of a protein kinase known as murine target of rapamycin (mTOR) which regulates protein synthesis and cell growth(30). Mice with haploinsufficiency in TSC1 showed social and cognitive deficits without cerebral pathology or seizures, suggesting that cognitive problems in the disorder result primarily from disrupted neuronal function rather than effects of tubers or other lesions(31).
Neuronal Plasticity in Fragile X Syndrome
Fragile X syndrome is well known to pediatric neurologists as the most common inherited form of mental retardation with characteristic dysmorphic features and neurobehavioral abnormalities including social avoidance, anxiety and autistic-like behaviors(32). Neuropathological studies of human brain as well as studies in a mouse model of fragile-X syndrome showed that neuronal architecture is abnormal in fragile X syndrome with long, thin and tortuous dendritic spines which appear immature(33). Fragile X syndrome is caused by a loss of function mutation in the fragile-X mental retardation protein (FMRP), an mRNA binding protein that regulates translation(34). In 2002 Huber et al reported that LTD is increased in the hippocampus from mice without FMRP(35). LTD is normally triggered by protein synthesis in response to with activation of metabotropic glutamate receptors, and these results suggest that FMRP normally antagonizes protein synthesis induced by these receptors (Figure 4)(36). Additional experiments in cell culture showed that internalization of AMPA receptors is increased in neurons lacking FMRP compared to controls, which is consistent with the finding that LTD is enhanced(36). Experiments in slices of visual cortex from FMRP deficient mice also showed that LTP is very impaired, and it could be restored by a general antagonist of metabotropic glutamate receptors(37). These data suggest that the deficit in LTP is mediated by excessive activity of metabotropic receptors. They are consistent with the observation that a metabotropic antagonist can also reverse behavioral and anatomic abnormalities in fruit flies in which a homologue of Fmr1 has knocked out(38). These results are also consistent with another experiment in which mice were bred to produce both knockout of Fmr1 and a 50% reduction in the expression of mGlur5 receptors(39). The reduction in mGluR5 receptors in this model corrected the defect in ocular dominance plasticity caused by Fmr1 knockout. A study of plasticity in the neocortex of Fmr1 knockout mice in the early postnatal period showed that spike timing-dependent plasticity (STDP), which depends on NMDA receptors, was also absent while LTD was robust(40). Another study in young adult Fmr1 knockout mice showed that impaired induction of LTP induced by theta burst afferent activity in the hippocampus could be restored by infusion of BDNF(41). Another recent study in the Drosophila model of fragile X syndrome found that dFMRP is positively regulated by sensory input during late brain development and is required to limit axon growth and activity-dependent pruning of axons branches(42). These studies indicate that there is morphological, biochemical and electrophysiological evidence of impaired synaptic plasticity in fragile X syndrome, and suggest that pharmacological intervention might be possible in the future.
Figure 4.
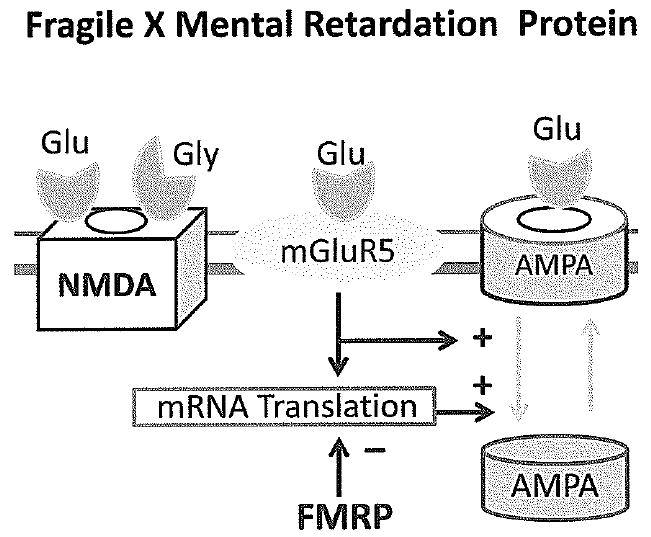
The gene for Fragile X mental retardation protein (FMRP) is mutated in Fragile X syndrome. The normal function of FMRP is to antagonize the effects of stimulation of mGluR5 receptors which causes internalization of AMPA receptors and long term depression (LTD). A mutation in the gene for FMRP therefore leaves mGluR5 action unopposed, causing AMPA receptors to become internalized and depressing synaptic function.
Synaptic Abnormalities in Rett Syndrome
Like Fragile X syndrome, Rett syndrome (RTT) is an X-linked developmental disorder of cognition and behavior that has a major impact on the development and plasticity of synapses(43). Most males with RTT do not survive, but girls develop characteristic stereotyped hand-wringing movements, severe cognitive impairment, acquired microcephaly, seizures, and disorders of breathing and autonomic dysfunction after a period of relative normality in the first year of life. Early clinical descriptions as well as neuropathology and imaging studies suggested a disorder of neuronal development, and studies of neurotransmitter receptors in postmortem brain found abnormalities in the expression of glutamate and GABA in cerebral cortex(44;45). Microarray studies of postmortem brain also found abnormalities in expression of genes associated with developing synapses(46). Mutations in the transcriptional repressor MeCP2 are responsible for most cases of RTT, and this protein is expressed primarily in neurons(47). The timing of its expression during development is delayed until just before the formation of synapses(48-50). Biopsies of nasal epithelium from girls with RS found that neurogeneis and early development of neurons are normal but establishment of mature synapses is blocked(51). Mecp2 deficient mice have morphologic abnormalities in cerebral cortex with a reduced number of thin dendritic spines and immature postsynaptic densities at excitatory synapses(52). These mice have impaired learning and cognition along with deficits in hippocampal plasticity including both LTP and LTD(53;54). Altered basal inhibitory rhythms and enhanced hyperexcitability have also been recorded in the hippocampus of Mecp2 deficient mice(55). This is consistent with the finding that neurons from Mecp2 mice are more sensitive to excitotoxic cell death and hypoxia than controls(56). The maturation and gene expression in hippocampal neurons has also been found to be abnormal(57;58). Phosphorylation of Mecp2 has been shown to regulate activity dependent transcription of the brain derived neurotrophic factor (BDNF) as well as enhancing the growth of dendrites and dendritic spines(59). In contrast, over-expression of Mecp2 in neurons in vitro has been shown to increase axonal length and dendritic complexity(60). The level of BDNF expression has been shown to affect the progression of neurologic impairment in Mecp2 mutant mice, with loss of BDNF worsening impairment and BDNF overexpression prolonging life(59;61). Administration of an ampakine drug that activates AMPA glutamate receptors has been shown to increase expression of BDNF and improve breathing abnormalities in Mecp2 null mice(62). RTT is an important example of the broad impact that disruption in synaptic developmental and activity dependent neuronal plasticity have on the developing brain.
Synaptic Plasticity and Vulnerability to Hypoxia-Ischemia
Some of the same mechanisms responsible for synaptic plasticity can also become mechanisms for injury if the developing brain is subjected to stresses such as hypoxia-ischemia, infection or certain metabolic disorders(1;11). For example the voltage dependent NMDA glutamate receptors can be opened in response to hypoxia-ischemia due to a combination of membrane depolarization from energy deficiency and accumulation of glutamate in the synaptic cleft due to inadequate removal by energy dependent transporters (Figure 5)(63). In this situation, the enhanced function of immature NMDA receptors that enables heightened plasticity in the immature brain can become a liability, making developing excitatory neuronal circuits vulnerable to injury. Enhanced function of NMDA receptors contributes to neuronal injury that occurs in response to the asphyxia in term infants who can develop preferential injury to circuits in the cortex and/or basal ganglia in response to severe asphyxia(15). Nearly complete asphyxia from cord compression often results in preferential injury to the peri-rolandic cortex, putamen and thalamus, which are connected by circuits that use glutamate as their neurotransmitter(64). In contrast, less severe but more prolonged asphyxia is more likely to produce multi-cystic encephalomalacia involving the cerebral cortex but sparing the basal ganglia. These special patterns of injury are probably related to the selective vulnerability of developing neuronal circuits that reflect their normal adaptive role in brain plasticity(63;65).
Figure 5.
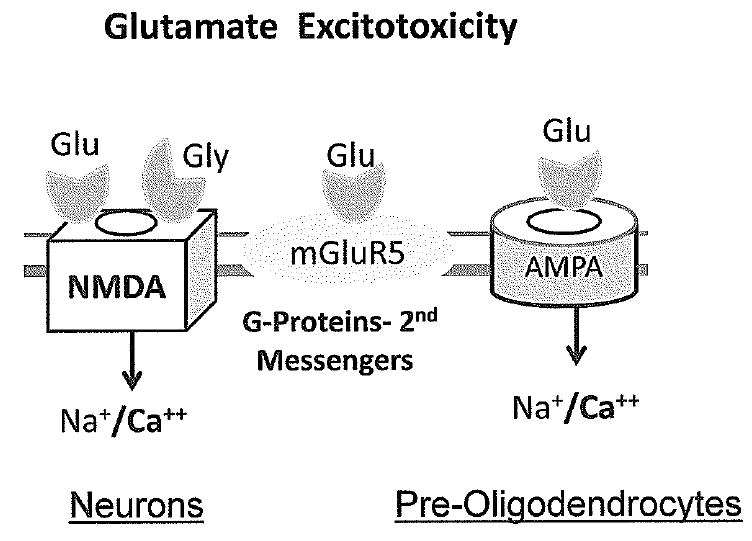
Glutamate excitotoxicity results from over-activity of glutamate ionotropic receptors. Injury to neurons is caused by over-stimulation of NMDA receptors which flood the cytoplasm with calcium. AMPA receptor stimulation is also necessary to depolarize the neuronal membrane, allowing NMDA channels to open. Damage to pre-oligodendrocytes in the premature brain is caused predominantly by activation of AMPA receptors which lack GluR2 subunits and are permeable to calcium at that stage in development.
In contrast to brain injuries that occur in term infants, periventricular leukomalaciawhite (PVL) is a prominent feature of injuries that occur prior to 32 weeks gestation(66). This pattern of selective vulnerability has been linked to the sensitivity of late oligodendrocyte progenitor cells to damage from glutamate mediated excitotoxicity and oxidative stress(67). The lower incidence of PVL seen in infants older than 32 week gestation is correlated with a large decline in populations of oligodendrocyte precursors and onset of myelination at this time. The vulnerability of the late oligodendrocyte progenitor cells to injury depends on their expression of AMPA receptors that lack GluR2 subunits which makes them able to flux high amounts of calcium that is toxic to cells(68;69). The inadequacy of enzymes that detoxify nitric oxide and other oxygen free radicals in the oligodendrocyte progenitors also contributes to their vulnerability in the preterm brain(70). Studies in human postmortem brain from babies of different gestational ages indicates that the period of vulnerability of the white matter in the fetal and preterm brain coincides with expression of GluR2 lacking AMPA receptors(71). Oligodendrocytes in the term and neonatal brain express AMPA receptors with GluR2 subunits, making them less vulnerable, while neurons in the cerebral cortex are expressing receptors without GluR2 receptors, making them more vulnerable(71). These data indicate that the age dependent selective vulnerability of white matter and neuronal structures in the developing brain is related to developmental programs for expression of glutamate receptors. Although little is known at this point about the normal function of glutamate receptors in oligodendrocyte development, it is possible that they mediate communication between activity in axons and oligodendrocytes.
Excitatory Receptors and the Cascade of Injury
Excessive activation of glutamate receptors in neurons and oligodendroglia initiates a cascade of events that result in injury to neurons or pre-oligodendrocytes as shown in Figure 6. Calcium that enters cells though NMDA and AMPA glutamate receptors as well as additional calcium entering through voltage sensitive calcium channels can flood the cytoplasm and mitochondria(72). This enhances the production of the toxic free radical gas nitric oxide produced by activation of nitric oxide synthase (nNOS) and oxygen free radicals(73;73-76). Nitric oxide alone or combined with superoxide ions to form peroxynitrite is toxic for mitochondria, and mitochondria increase production of their own reactive oxygen species (ROS) in the face of hypoxia(77). Calcium flooding and enhanced production of ROS in mitochondria combined with impaired adenosine triphosphase (ATP) production secondary to hypoxia increases levels of lactic acid and can cause cerebral edema. Very severe hypoxia-ischemia and mitochondrial injury can lead to critical energy failure with implosion of cell membranes and the pathological process of necrosis (Figure 6). However, less severe insults are more likely to lead either survival and recovery or apoptosis or programmed cell death, which is triggered by events within the nucleus that cause chromatin condensation and DNA fragmentation(78). Apoptosis is especially prominent in the developing brain compared with the adult, probably because it is naturally activated in the developing brain to eliminate excess cells that will not be needed in the mature brain(78;79). After a hypoxic-ischemic injury to the brain in neonatal rodents, apoptosis is observed for a week or more after the insult, suggesting that it continues to be triggered long after the insult.
Figure 6.
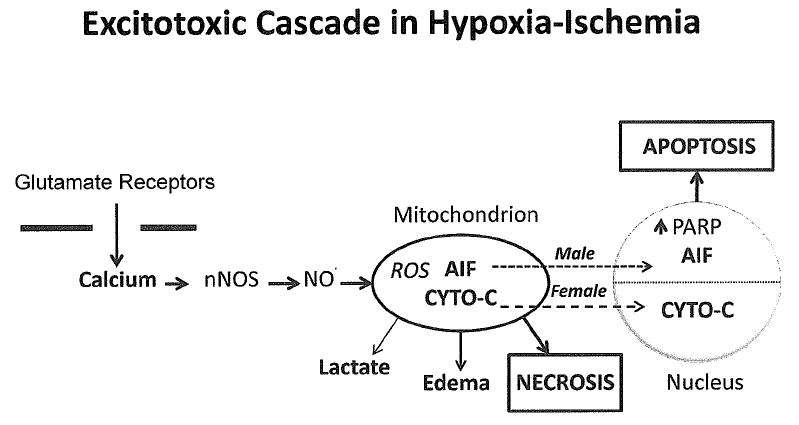
Cascade of events that follows a hypoxic-ischemic insult and over-stimulation of glutamate receptors in the brain. This cascade evolves over days to weeks and involves signaling to the mitochondria and nucleus. Very severe injury to the mitochondria leads to complete energy failure and destruction of cell membranes associated with necrosis. Milder insults activate apoptosis, which is much more prominent in infants than in adults. In addition to an extrinsic FAS death pathway (not shown), caspase-dependent and non-caspase dependent pathways activate apoptosis in the nucleus. Work in rodents indicates that these pathways are sexually dimorphic with males preferring the PARP-AIF non-caspase pathway more than females. This pathway is also more easily activated by glutamate receptor stimulation. In contrast, the cytochrome C- caspase 3 pathway is more active in females. ROS = reactive oxygen species; AIF = apoptosis inducing factor; CYTO-C = cytochrome C; PARP = Poly (ADP-ribose) polymerase.
Apoptosis can be triggered either by signals from the mitochondria to the nucleus or by signaling from the extrinsic cell surface Fas death receptor to the nucleus, and both these pathways are enhanced by oxidative stress(80;81). As shown in Figure 6, energy failure in mitochondria can trigger two types of cell death signals to the nucleus, one mediated by activation of caspase 3 and a second non-caspase pathway triggered by apoptosis inducing factor (AIF). AIF is a flavoprotein that is released from stressed mitochondria and travels into the nucleus where it activates apoptosis(82). The major signal for release of AIF from mitochondria is a reduction in levels of the high energy substrate nicotinamide adenine dinucleotide (NAD+) in mitochondria in response to activation of the DNA repair enzyme (ADP-ribose) polymerase (PARP) in the nucleus(83). PARP is activated during hypoxia-ischemia by oxidative damage to DNA. The second major apoptotic cell death pathway depends on activation of caspase 3, and is triggered by movement of cytochrome C from mitochondria into the nucleus(84). Recent data indicate that these two cell death pathways are sexually dimorphic, with the caspase-dependent pathway more prominent in females and the PARP-AIF pathway more prominent in males(85;86). The non-caspase PARP-AIF pathway appears to be more easily triggered by activation of excitatory glutamate receptors and activation of nNOS than the caspase-dependent pathway. Neurons from males have been shown to preferentially release more AIF from mitochondria into the nucleus in response to glutamate and NO. than females, and females preferentially release more cytochrome C(87). In neonatal mice, genetic knockout of the Parp gene is protective in males but not females while caspase 3 inhibiting drugs protect females but not males(88). Inhibitors of PARP and nNOS also protect adult male rodents from injury, but they paradoxically increase injury in females(89;90). This information indicates the cell death pathways in the brain may differ according to genetic sex and this could be relevant to the excess of males with cerebral palsy and other forms of brain injury.
Neuroprotective Therapies for Brain Injury
The perspective that many cell signaling pathways that are involved in brain plasticity are also involved in the pathogenesis of brain injury in the developing brain is an important one for thinking about how to design neuroprotective therapies (Table 1)(91). The corollary of this concept is that some therapies that protect the brain could also impair plasticity or kill neurons if applied in excessive amounts. For example it is clear that NMDA glutamate channel blockers and drugs that activate GABA receptors can cause apoptosis in the developing brain(92). There is clearly a benefit to risk assessment with all agents that are likely to have protective effects, and careful study in animal models as well as follow-up in human trials is warranted. The antibiotic minocycline, which is protective in some models of brain injury, can also enhance injury in neonatal hypoxia-ischemia(93). The observation that signaling pathways involved in cell death are sexually dimorphic is also important both for pre-clinical studies and clinical trials. Recent data indicate that drugs that block glutamate receptors, nNOS, caspase 3 and PARP are likely to act differently in males and females(89;94;95). The cytokine erythropoietin, which has neurotrophic and neuroprotective effects and is protective against hypoxia-ischemia in neonatal mice, also appears to be more effect in females than in males(96;97). Although at least one study found that hypothermia was more protective in female than in male 7 day old rats, the two reported successful clinical trials of hypothermia for term infants with asphyxia did not show any differences according to sex(98;99).
Table 1.
Potential Interventions for Neuroprotection Against Brain Injury
| Hypothermia |
| NMDA, AMPA glutamate receptor antagonists |
| Nitric Oxide Synthase Inhibitors |
| Caspase Inhibitors |
| Acetylcysteine |
| PARP Inhibitors |
| Erythropoietin |
| Growth Factors (NGF, BDNF) |
| Mitochondrial ATP-sensitive K+ Activators |
Conclusion
A major difference between the nervous system in infants and children compared to adults is the capacity for greater plasticity in the developing brain. The molecular signaling pathways involved in brain plasticity are being discovered at an increasing rate, and it is clear that they are disrupted in some common pediatric disorders. These discoveries may lead to better treatments for currently untreatable disorders such as fragile X syndrome, neurofibromatosis and tuberous sclerosis. Brain damage in response to hypoxia-ischemia and other insults often involves overstimulation of these same plasticity mechanisms. Recent evidence indicates that cell death pathways are strongly influenced by genetic sex. A better understanding of molecular pathways involved in plasticity and injury will lead to progress in pediatric neurology.
Acknowledgments
Supported by NIH grants R01 NS28208 and P01 HD24448
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael V. JOHNSTON, Departments of Neurology and Pediatrics, Kennedy Krieger Institute and Johns Hopkins University School of Medicine, Baltimore, Maryland USA.
Akira ISHIDA, Department of Pediatrics, Akita University School of Medicine, Akita, Japan
Wako Nakajima ISHIDA, Department of Pediatrics, Akita University School of Medicine, Akita, Japan
Hiroko Baber MATSUSHITA, Department of Pediatrics, Kyoto City Hospital Kyoto, Japan
Akira NISHIMURA, Department of Pediatrics, Kyoto Prefectural University of Medicine Kyoto, Japan
Masahiro TSUJI, Department of Pediatrics, Kobe Medical Center General Hospital Kobe, Japan
Reference List
- 1.Johnston MV. Clinical disorders of brain plasticity. Brain Dev. 2004 Mar;26(2):73–80. doi: 10.1016/S0387-7604(03)00102-5. [DOI] [PubMed] [Google Scholar]
- 2.Johnston MV, Nishimura A, Harum K, Pekar J, Blue ME. Sculpting the developing brain. Adv Pediatr. 2001;48:1–38. [PubMed] [Google Scholar]
- 3.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007 Jun;8(6):413–26. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 4.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997 Oct 20;387(2):167–78. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 5.Segal M, Andersen P. Dendritic spines shaped by synaptic activity. Curr Opin Neurobiol. 2000 Oct;10(5):582–6. doi: 10.1016/s0959-4388(00)00123-9. [DOI] [PubMed] [Google Scholar]
- 6.Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 2002 Oct 25;298(5594):770–6. doi: 10.1126/science.1075510. [DOI] [PubMed] [Google Scholar]
- 7.Cohen-Cory S, Lom B. Neurotrophic regulation of retinal ganglion cell synaptic connectivity: from axons and dendrites to synapses. Int J Dev Biol. 2004;48(89):947–56. doi: 10.1387/ijdb.041883sc. [DOI] [PubMed] [Google Scholar]
- 8.Nishimura A, Hohmann CF, Johnston MV, Blue ME. Neonatal electrolytic lesions of the basal forebrain stunt plasticity in mouse barrel field cortex. Int J Dev Neurosci. 2002 Oct;20(6):481–9. doi: 10.1016/s0736-5748(02)00078-3. [DOI] [PubMed] [Google Scholar]
- 9.Froemke RC, Merzenich MM, Schreiner CE. A synaptic memory trace for cortical receptive field plasticity. Nature. 2007 Nov 15;450(7168):425–9. doi: 10.1038/nature06289. [DOI] [PubMed] [Google Scholar]
- 10.Tsumoto T. Long-term potentiation and depression in the cerebral neocortex. Jpn J Physiol. 1990;40(5):573–93. doi: 10.2170/jjphysiol.40.573. [DOI] [PubMed] [Google Scholar]
- 11.McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev. 1990 Jan;15(1):41–70. doi: 10.1016/0165-0173(90)90011-c. [DOI] [PubMed] [Google Scholar]
- 12.Benarroch EE. Metabotropic glutamate receptors: synaptic modulators and therapeutic targets for neurologic disease. Neurology. 2008 Mar 18;70(12):964–8. doi: 10.1212/01.wnl.0000306315.03021.2a. [DOI] [PubMed] [Google Scholar]
- 13.Beique JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003 Feb 1;546(Pt 3):859–67. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–47. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 15.Monyer H, Brunashev N, Laurie DJ. Development and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1993;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 16.Feldman DE, Nicoll RA, Malenka RC. Synaptic plasticity at thalamocortical synapses in developing rat somatosensory cortex: LTP, LTD, and silent synapses. J Neurobiol. 1999 Oct;41(1):92–101. [PubMed] [Google Scholar]
- 17.Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995 May 25;375(6529):325–8. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Ari Y. Basic developmental rules and their implications for epilepsy in the immature brain. Epileptic Disord. 2006 Jun;8(2):91–102. [PubMed] [Google Scholar]
- 19.Harney SC, Rowan M, Anwyl R. Long-term depression of NMDA receptor-mediated synaptic transmission is dependent on activation of metabotropic glutamate receptors and is altered to long-term potentiation by low intracellular calcium buffering. J Neurosci. 2006 Jan 25;26(4):1128–32. doi: 10.1523/JNEUROSCI.2753-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malinow R. AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003 Apr 29;358(1432):707–14. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malenka RC. Synaptic plasticity and AMPA receptor trafficking. Ann N Y Acad Sci. 2003 Nov;1003:1–11. doi: 10.1196/annals.1300.001. [DOI] [PubMed] [Google Scholar]
- 22.Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005 Jun;76(2):99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Hu B, Nikolakopoulou AM, Cohen-Cory S. BDNF stabilizes synapses and maintains the structural complexity of optic axons in vivo. Development. 2005 Oct;132(19):4285–98. doi: 10.1242/dev.02017. [DOI] [PubMed] [Google Scholar]
- 24.Johnston MV, Alemi L, Harum KH. Learning, memory, and transcription factors. Pediatr Res. 2003 Mar;53(3):369–74. doi: 10.1203/01.PDR.0000049517.47493.E9. [DOI] [PubMed] [Google Scholar]
- 25.Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990 Aug 10;62(3):599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 26.Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002 Jan 31;415(6871):526–30. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- 27.Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005 Nov 8;15(21):1961–7. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 28.Lush ME, Li Y, Kwon CH, Chen J, Parada LF. Neurofibromin is required for barrel formation in the mouse somatosensory cortex. J Neurosci. 2008 Feb 13;28(7):1580–7. doi: 10.1523/JNEUROSCI.5236-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goh S, Weiss WA. Genotype-phenotype correlations in tuberous sclerosis: who and how to treat. Ann Neurol. 2006 Nov;60(5):505–7. doi: 10.1002/ana.20917. [DOI] [PubMed] [Google Scholar]
- 30.Paul E, Thiele E. Efficacy of sirolimus in treating tuberous sclerosis and lymphangioleiomyomatosis. N Engl J Med. 2008 Jan 10;358(2):190–2. doi: 10.1056/NEJMe0707153. [DOI] [PubMed] [Google Scholar]
- 31.Goorden SM, van Woerden GM, van der WL, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007 Dec;62(6):648–55. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- 32.Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O’Hara R, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Ann Neurol. 2008 Jan;63(1):40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997 May 13;94(10):5401–4. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bear MF, Dolen G, Osterweil E, Nagarajan N. Fragile X: translation in action. Neuropsychopharmacology. 2008 Jan;33(1):84–7. doi: 10.1038/sj.npp.1301610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002 May 28;99(11):7746–50. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007 Sep 25;104(39):15537–42. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson BM, Cox CL. Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci U S A. 2007 Feb 13;104(7):2454–9. doi: 10.1073/pnas.0610875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005 Mar 3;45(5):753–64. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 39.Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, et al. Correction of fragile X syndrome in mice. Neuron. 2007 Dec 20;56(6):955–62. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meredith RM, Holmgren CD, Weidum M, Burnashev N, Mansvelder HD. Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron. 2007 May 24;54(4):627–38. doi: 10.1016/j.neuron.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 41.Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, et al. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007 Oct 3;27(40):10685–94. doi: 10.1523/JNEUROSCI.2624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tessier CR, Broadie K. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development. 2008 Mar 5; doi: 10.1242/dev.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnston MV, Jeon OH, Pevsner J, Blue ME, Naidu S. Neurobiology of Rett syndrome: a genetic disorder of synapse development. Brain Dev. 2001 Dec;23(Suppl 1):S206–S213. doi: 10.1016/s0387-7604(01)00351-5. [DOI] [PubMed] [Google Scholar]
- 44.Blue ME, Naidu S, Johnston MV. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol. 1999 Apr;156(2):345–52. doi: 10.1006/exnr.1999.7030. [DOI] [PubMed] [Google Scholar]
- 45.Blue ME, Naidu S, Johnston MV. Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol. 1999 Apr;45(4):541–5. doi: 10.1002/1531-8249(199904)45:4<541::aid-ana21>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 46.Johnston MV, Blue ME, Naidu S. Rett syndrome and neuronal development. J Child Neurol. 2005 Sep;20(9):759–63. doi: 10.1177/08830738050200091101. [DOI] [PubMed] [Google Scholar]
- 47.Naidu S, Bibat G, Kratz L, Kelley RI, Pevsner J, Hoffman E, et al. Clinical variability in Rett syndrome. J Child Neurol. 2003 Oct;18(10):662–8. doi: 10.1177/08830738030180100801. [DOI] [PubMed] [Google Scholar]
- 48.Cohen DR, Matarazzo V, Palmer AM, Tu Y, Jeon OH, Pevsner J, et al. Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol Cell Neurosci. 2003 Apr;22(4):417–29. doi: 10.1016/s1044-7431(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 49.Mullaney BC, Johnston MV, Blue ME. Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience. 2004;123(4):939–49. doi: 10.1016/j.neuroscience.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 50.Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004 Nov;27(3):306–21. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 51.Ronnett GV, Leopold D, Cai X, Hoffbuhr KC, Moses L, Hoffman EP, et al. Olfactory biopsies demonstrate a defect in neuronal development in Rett’s syndrome. Ann Neurol. 2003 Aug;54(2):206–18. doi: 10.1002/ana.10633. [DOI] [PubMed] [Google Scholar]
- 52.Fukuda T, Itoh M, Ichikawa T, Washiyama K, Goto Y. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J Neuropathol Exp Neurol. 2005 Jun;64(6):537–44. doi: 10.1093/jnen/64.6.537. [DOI] [PubMed] [Google Scholar]
- 53.Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006 Jan 4;26(1):319–27. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006 Jan;21(1):217–27. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. 2008;18(3):294–309. doi: 10.1002/hipo.20389. [DOI] [PubMed] [Google Scholar]
- 56.Russell JC, Blue ME, Johnston MV, Naidu S, Hossain MA. Enhanced cell death in MeCP2 null cerebellar granule neurons exposed to excitotoxicity and hypoxia. Neuroscience. 2007 Dec 12;150(3):563–74. doi: 10.1016/j.neuroscience.2007.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smrt RD, Eaves-Egenes J, Barkho BZ, Santistevan NJ, Zhao C, Aimone JB, et al. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis. 2007 Jul;27(1):77–89. doi: 10.1016/j.nbd.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, et al. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006 Apr;129(Pt 4):887–98. doi: 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- 59.Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006 Oct 19;52(2):255–69. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005 Jun;19(12):18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 61.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006 Feb 2;49(3):341–8. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 62.Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007 Oct 3;27(40):10912–7. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnston MV. Excitotoxicity in neonatal hypoxia. Ment Retard Dev Disabil Res Rev. 2001;7(4):229–34. doi: 10.1002/mrdd.1032. [DOI] [PubMed] [Google Scholar]
- 64.Johnston MV, Trescher WH, Ishida A, Nakajima W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatric Research. 2001;49:735–741. doi: 10.1203/00006450-200106000-00003. Ref Type: Journal Full. [DOI] [PubMed] [Google Scholar]
- 65.Johnston MV, Hoon AH., Jr Cerebral palsy. Neuromolecular Med. 2006;8(4):435–50. doi: 10.1385/NMM:8:4:435. [DOI] [PubMed] [Google Scholar]
- 66.Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001 Nov;50(5):553–62. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002 Jan 15;22(2):455–63. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jensen FE. The role of glutamate receptor maturation in perinatal seizures and brain injury. Int J Dev Neurosci. 2002 Jun;20(35):339–47. doi: 10.1016/s0736-5748(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 69.Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, et al. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004 May 5;24(18):4412–20. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Folkerth RD. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005 Dec;20(12):940–9. doi: 10.1177/08830738050200120301. [DOI] [PubMed] [Google Scholar]
- 71.Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol. 2006 Jul 1;497(1):61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnston MV. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005 Jul;15(3):234–40. doi: 10.1111/j.1750-3639.2005.tb00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ishida A, Ishiwa S, Trescher WH, Nakajima W, Lange MS, Blue ME, et al. Delayed increase in neuronal nitric oxide synthase immunoreactivity in thalamus and other brain regions after hypoxic-ischemic injury in neonatal rats. Exp Neurol. 2001 Apr;168(2):323–33. doi: 10.1006/exnr.2000.7606. [DOI] [PubMed] [Google Scholar]
- 74.Baud O, Li J, Zhang Y, Neve RL, Volpe JJ, Rosenberg PA. Nitric oxide-induced cell death in developing oligodendrocytes is associated with mitochondrial dysfunction and apoptosis-inducing factor translocation. Eur J Neurosci. 2004 Oct;20(7):1713–26. doi: 10.1111/j.1460-9568.2004.03616.x. [DOI] [PubMed] [Google Scholar]
- 75.Mishra OP, Delivoria-Papadopoulos M. Nitric oxide-mediated Ca++-influx in neuronal nuclei and cortical synaptosomes of normoxic and hypoxic newborn piglets. Neurosci Lett. 2002 Jan 25;318(2):93–7. doi: 10.1016/s0304-3940(01)02484-3. [DOI] [PubMed] [Google Scholar]
- 76.Tsuji M, Higuchi Y, Shiraishi K, Kume T, Akaike A, Hattori H. Protective effect of aminoguanidine on hypoxic-ischemic brain damage and temporal profile of brain nitric oxide in neonatal rat. Pediatr Res. 2000 Jan;47(1):79–83. doi: 10.1203/00006450-200001000-00015. [DOI] [PubMed] [Google Scholar]
- 77.Blomgren K, Zhu C, Hallin U, Hagberg H. Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Biochem Biophys Res Commun. 2003 May 9;304(3):551–9. doi: 10.1016/s0006-291x(03)00628-4. [DOI] [PubMed] [Google Scholar]
- 78.Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci. 2000 Nov 1;20(21):7994–8004. doi: 10.1523/JNEUROSCI.20-21-07994.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Northington FJ, Ferriero DM, Flock DL, Martin LJ. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci. 2001 Mar 15;21(6):1931–8. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Payton KS, Sheldon RA, Mack DW, Zhu C, Blomgren K, Ferriero DM, et al. Antioxidant status alters levels of Fas-associated death domain-like IL-1B-converting enzyme inhibitory protein following neonatal hypoxia-ischemia. Dev Neurosci. 2007;29(45):403–11. doi: 10.1159/000105481. [DOI] [PubMed] [Google Scholar]
- 81.Stefanis L. Caspase-dependent and-independent neuronal death: two distinct pathways to neuronal injury. Neuroscientist. 2005 Feb;11(1):50–62. doi: 10.1177/1073858404271087. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Zhang X, Park TS, Gidday JM. Cerebral endothelial cell apoptosis after ischemia-reperfusion: role of PARP activation and AIF translocation. J Cereb Blood Flow Metab. 2005 Jul;25(7):868–77. doi: 10.1038/sj.jcbfm.9600081. [DOI] [PubMed] [Google Scholar]
- 83.Dawson VL, Dawson TM. Deadly conversations: nuclear-mitochondrial cross-talk. J Bioenerg Biomembr. 2004 Aug;36(4):287–94. doi: 10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- 84.Han BH, D’Costa A, Back SA, Parsadanian M, Patel S, Shah AR, et al. BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis. 2000 Feb;7(1):38–53. doi: 10.1006/nbdi.1999.0275. [DOI] [PubMed] [Google Scholar]
- 85.Johnston MV, Hagberg H. Sex and the pathogenesis of cerebral palsy. Dev Med Child Neurol. 2007 Jan;49(1):74–8. doi: 10.1017/s0012162207000199.x. [DOI] [PubMed] [Google Scholar]
- 86.Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, et al. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006 Feb;96(4):1016–27. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]
- 87.Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, et al. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004 Sep 10;279(37):38563–70. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- 88.Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004 Sep;90(5):1068–75. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- 89.McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005 Apr;25(4):502–12. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- 90.Li H, Pin S, Zeng Z, Wang MM, Andreasson KA, McCullough LD. Sex differences in cell death. Ann Neurol. 2005 Aug;58(2):317–21. doi: 10.1002/ana.20538. [DOI] [PubMed] [Google Scholar]
- 91.Johnston MV, Trescher WH, Ishida A, Nakajima W. Novel treatments after experimental brain injury. Semin Neonatol. 2000 Feb;5(1):75–86. doi: 10.1053/siny.1999.0116. [DOI] [PubMed] [Google Scholar]
- 92.Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, et al. Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol. 2001 Aug 15;62(4):401–5. doi: 10.1016/s0006-2952(01)00696-7. [DOI] [PubMed] [Google Scholar]
- 93.Tsuji M, Wilson MA, Lange MS, Johnston MV. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp Neurol. 2004 Sep;189(1):58–65. doi: 10.1016/j.expneurol.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 94.Nijboer CH, Groenendaal F, Kavelaars A, Hagberg HH, van BF, Heijnen CJ. Gender-specific neuroprotection by 2-iminobiotin after hypoxia-ischemia in the neonatal rat via a nitric oxide independent pathway. J Cereb Blood Flow Metab. 2007 Feb;27(2):282–92. doi: 10.1038/sj.jcbfm.9600342. [DOI] [PubMed] [Google Scholar]
- 95.Renolleau S, Fau S, Goyenvalle C, Joly LM, Chauvier D, Jacotot E, et al. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J Neurochem. 2007 Feb;100(4):1062–71. doi: 10.1111/j.1471-4159.2006.04269.x. [DOI] [PubMed] [Google Scholar]
- 96.Matsushita H, Johnston MV, Lange MS, Wilson MA. Protective effect of erythropoietin in neonatal hypoxic ischemia in mice. Neuroreport. 2003 Sep 15;14(13):1757–61. doi: 10.1097/00001756-200309150-00020. [DOI] [PubMed] [Google Scholar]
- 97.Wen TC, Rogido M, Peng H, Genetta T, Moore J, Sola A. Gender differences in long-term beneficial effects of erythropoietin given after neonatal stroke in postnatal day-7 rats. Neuroscience. 2006;139(3):803–11. doi: 10.1016/j.neuroscience.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 98.Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005 Feb 19;365(9460):663–70. doi: 10.1016/S0140-6736(05)17946-X. [DOI] [PubMed] [Google Scholar]
- 99.Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005 Oct 13;353(15):1574–84. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]