

Abstract
Spinal glial reaction and proinflammatory cytokine induction play an important role in the development of chronic pain states after tissue and nerve injury. The present study investigated the cellular and molecular mechanisms underlying descending facilitation of neuropathic pain with an emphasis on supraspinal glial–neuronal relationships. An early and transient reaction of microglia and prolonged reaction of astrocytes were found after chronic constriction injury (CCI) of the rat infraorbital nerve in the rostral ventromedial medulla (RVM), a major component of brainstem descending pain modulatory circuitry. There were prolonged elevations of cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) after CCI, and they were expressed in RVM astrocytes at 14 d after injury. Intra-RVM injection of microglial and astrocytic inhibitors attenuated mechanical hyperalgesia and allodynia at 3 and 14 d after CCI, respectively. Moreover, TNFR1 and IL-1R, receptors for TNF-α and IL-1β, respectively, were expressed primarily in RVM neurons exhibiting immunoreactivity to the NMDA receptor (NMDAR) subunit NR1. CCI increased TNFR1 and IL-1R levels and NR1 phosphorylation in the RVM. Neutralization of endogenous TNF-α and IL-1β in the RVM significantly reduced CCI-induced behavioral hypersensitivity and attenuated NR1 phosphorylation. Finally, intra-RVM administration of recombinant TNF-α or IL-1β upregulated NR1 phosphorylation and caused a reversible and NMDAR-dependent allodynia in normal rats, further suggesting that TNF-α and IL-1β couple glial hyperactivation with NMDAR function. These studies have addressed a novel contribution of supraspinal astrocytes and associated cytokines as well as central glial–neuronal interactions to the enhancement of descending facilitation of neuropathic pain.
Keywords: astrocyte, TNF-α, IL-1β, NMDA receptor, medulla, nerve injury
Introduction
The rostral ventromedial medulla (RVM) is an important component of the descending nociceptive system that projects to the spinal cord and trigeminal brainstem nuclei and constitutes a major mechanism in the control of pain transmission (Millan, 2002; Ren and Dubner, 2002; Fields et al., 2006). Recent studies indicate that hyperalgesia and allodynia in animal models of persistent pain are closely linked to long-lasting activation of descending modulatory circuits involving descending facilitation (Porreca et al., 2002; Dubner and Ren, 2004; Gebhart, 2004; Vanegas and Schaible, 2004), which significantly contributes to the development of persistent pain after peripheral inflammation (Urban and Gebhart, 1999; Wei et al., 1999; Guo et al., 2006) and nerve injury (Pertovaara et al., 1996; Kovelowski et al., 2000). Despite extensive studies on the role of RVM in descending pain modulation, the cellular mechanisms of its involvement in descending pain facilitation are poorly understood.
A considerable amount of evidence has demonstrated the existence of dynamic and bidirectional communication between glia and neurons at the synapse, suggesting that glia play an active role in regulation of synaptic transmission in the CNS (Fields and Stevens-Graham, 2002; Newman, 2003; Perea and Araque, 2006). Glial cells, primarily microglia and astrocytes, exhibit dynamic plasticity by converting from a relatively resting or quiescent state to a reactive or hyperactive state and appear to modulate neuronal activity. Interestingly, after hyperactivation, glia subsequently release cytokines at the spinal cord (DeLeo et al., 2004; Salter, 2004; Sommer and Kress, 2004; Marchand et al., 2005; Watkins et al., 2007) and spinal trigeminal nucleus (Piao et al., 2006; Guo et al., 2007), which may be implicated in central mechanisms of persistent pain (Ren and Dubner, 2008). Whether supraspinal glial hyperactivity and their interactions with neurons in the RVM circuitry constitute a mechanism underlying descending facilitation during the development of persistent pain has not been studied.
Here, we have developed a quantitative behavioral measure of mechanical hyperalgesia and allodynia associated with a rat model of orofacial painful neuropathy, chronic constriction injury of the infraorbital nerve (CCI-ION), to investigate the mechanisms of neuropathic pain with an emphasis on glial–neuronal interactions in the pain modulatory circuitry. We found a prolonged astrocytic reaction and increased expression of cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in astrocytes and their receptors in RVM neurons after nerve injury. Intra-RVM microinjection of glial inhibitors and neutralization of endogenous TNF-α and IL-1β significantly attenuated CCI-induced mechanical hyperalgesia and allodynia. We further found that CCI-induced enhancement of NMDA receptor (NMDAR) subunit NR1 phosphorylation in the RVM was significantly reversed by glial inhibitors or cytokine receptor antagonists. An elevated NMDAR subunit NR1 phosphorylation in the RVM and moderate behavioral hypersensitivity were also observed after microinjection of recombinant TNF-α and IL-1β in the RVM of normal rats, and were reversed by pretreatment with NMDAR antagonists. This is the first study showing that glial–neuronal interactions coupling proinflammatory cytokines to NMDAR function at the supraspinal level contribute to descending pain facilitation.
Materials and Methods
Animals.
Adult male Sprague Dawley rats weighing 175–350 g (Harlan) were used in all experiments. Rats were on a 12 h light/dark cycle and received food and water ad libitum. The experiments were approved by the Institutional Animal Care and Use Committee of the University of Maryland Dental School.
Trigeminal neuropathic pain.
A model of trigeminal neuropathic pain was made by chronic constriction injury to the unilateral infraorbital nerve (CCI-ION), which was performed based on the original description (Bennett and Xie, 1988) and via an intraoral approach described by Imamura et al. (1997). Animals were anesthetized with pentobarbital sodium (50 mg/kg, i.p.). Surgery was performed under an operation microscope. The head of the rat was supine and fixed on a table. A 1-cm-long incision was made along the gingivobuccal margin in the buccal mucosa, beginning immediately next to the first molar. The ION was freed from surrounding connective tissue by a glass rod and clearly visualized using a surgical microscope. At 3–4 mm from the nerve where its branches emerge from the infraorbital foramen, the ION was loosely tied with two chromic gut (4.0) ligatures, 2 mm apart. This caused minor constriction of the ION such that the superficial vasculature was minimally retarded. The wound was checked for hemostasis and the incision was closed using three 4.0 silk sutures. The sham-operated rats received only a unilateral nerve exposure without ligature. All surgical procedures were performed aseptically. In some cases, a long-lasting anesthetic agent 0.25% bupivacaine was injected at the incision sites before and after surgery to block local nociceptive inputs induced by acute tissue injury and then twice per day for another 2 d. Changes in gross behavior and body weight gain in CCI and sham rats were monitored throughout the study and were not significantly different when compared with that in naive rats.
Behavioral testing.
All behavioral tests were conducted under blind conditions as previously described (Ren, 1999). The rat was not restrained but habituated to stand on its hindpaws and lean against the experimenter's hand who was wearing a regular leather work glove. The habituation required no more than normal petting of the rat, and it was achieved within one-half an hour. A series of calibrated von Frey filaments with bending forces ranging from 9 mg to 118 g were slightly applied to the skin within the infraorbital territory, near the center of the vibrissal pad on hairy skin surrounding the mystacial vibrissae. These areas were stimulated on both sides: ipsilateral and contralateral to the side where surgery was performed. An active withdrawal of the head from the probing filament was defined as a response. Each von Frey filament was applied five times at intervals of a few seconds. The response frequencies [(number of responses/number of stimuli) × 100%] to a range of von Frey filament forces were determined, and a stimulus–response (S–R) curve was plotted. After a nonlinear regression analysis, an EF50 value, defined as the von Frey filament force (g) that produces a 50% response frequency, was derived from the S–R curve. We used EF50 value as a measure of mechanical sensitivity.
Intra-RVM microinjections.
As previously described (Guo et al., 2006), animals were anesthetized with 2–3% isoflurane in a gas mixture of 30% O2 balanced with nitrogen, and placed in a Kopf stereotaxic instrument. A midline incision was made after infiltration of lidocaine (2%) into the skin. A midline opening was made in the skull with a dental drill to insert a microinjection needle into the target site. The coordinates for the nucleus raphe magnus, the major structure of RVM, were as follows: 10.5 mm caudal to the bregma, midline, and 9.0 mm ventral to the surface of the cerebellum (Paxinos and Watson, 2005). To avoid penetration of the transverse sinus, the incisor bar was set at 4.7 mm below the horizontal plane passing through the interaural line. Animals were subsequently maintained at ∼1% halothane. Microinjections were performed by delivering drug solutions slowly over a 10 min period using a 0.5 μl Hamilton syringe with a 32 gauge needle. The needle was withdrawn 5 min after the completion of the injection, and the incision was sutured. All wound margins were covered with a local anesthetic ointment (Nupercainal; Rugby Laboratories). Different groups of animals were subjected to intra-RVM microinjection with a 0.5 μl volume solution of (1) glial metabolic inhibitors propentofylline (PPF) (1 fmol, 100 fmol, and 10 pmol; Sigma-Aldrich), fluorocitrate (FC) (1 and 100 fmol; Sigma-Aldrich), and minocycline (MC) (10 fmol and 1 pmol; Sigma-Aldrich); (2) cytokine receptor antagonists TNFRI/Fc (TNFRI antagonist; 50 fmol; R&D Systems) and IL-ra (Kineret; IL-1R antagonist; 3 pmol; Amgen); (3) recombinant rat TNF-α (rTNF-α; TNFR agonist; 120 fmol; R&D Systems) or IL-1β (rIL-1β; IL-1R agonist; 120 fmol; PeproTech); and (4) glutamate receptor antagonists MK801 (dizocilpine maleate) (noncompetitive NMDAR antagonist; 10 pmol; Sigma-Aldrich). All drugs except for fluorocitrate were dissolved or reconstituted in endotoxin-free sterile distilled water, aliquoted, and stored at −70°C. At the time of testing, the stored aliquot was thawed on ice and diluted in sterile 0.9% saline to a final concentration. The doses of these agents were carefully chosen by referring to the literature and adjusted by our pilot experiments. For example, in the RVM, we found that a dose ≤0.1 pmol is necessary to avoid an apparent effect of fluorocitrate on neurons (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) (S. Sweitzer et al., 2001; Raghavendra et al., 2003a,b; Ledeboer et al., 2005; Guo et al., 2006, 2007). The control rats underwent identical procedures with injection of the same volume (0.5 μl) of sterile 0.9% saline as vehicle treatment. The fluorocitrate solution was prepared as described by Paulsen et al. (1987). The vehicle solution was prepared in the same manner for separate control experiments, except that fluorocitrate was omitted.
Immunohistochemistry.
At different time points after nerve injury, rats were behaviorally tested to identify CCI-ION-induced mechanical allodynia, and then deeply anesthetized with pentobarbital and perfused transcardially with 200 ml of saline followed by 500 ml of cold (4°C) 0.1 m phosphate buffer containing 4% paraformaldehyde. The brainstem was removed, immersed in the same fixative overnight at 4°C, and transferred to 30% sucrose (w/v) in phosphate buffer for several days for cryoprotection. Thirty-micrometer-thick coronal sections of the brainstem were cut with a cryostat at −20°C. Free-floating tissue sections including RVM were incubated with relevant antibodies overnight. After washes, the sections were incubated with AffiniPure biotinylated secondary IgG (1:800; Jackson ImmunoResearch Laboratories) for 1 h. For fluorescence staining, the sections were incubated with streptavidin-Alexa Fluor 568 or 488 (1:600; Invitrogen) for 1 h. For peroxidase identification, the sections were reacted with avidin and biotinylated HRP complex (1:200; Vector Laboratories) for 1 h, and reacted with 0.025% diaminobenzidine (Sigma-Aldrich) and 0.003% hydrogen peroxide for 5–20 min. Immunostaining control studies were performed by omission of the primary or secondary antibodies, and by preabsorption with an excess (10 μg/ml) of the respective antigens. Double labeling was performed simultaneously with two primary antibodies obtained from different species. After overnight, the sections were incubated for 2 h in solutions containing species-specific secondary antibodies coupled to Alexa 568 or 488, respectively. After washes, all sections were mounted on gelatin-coated slides and coverslipped with Vectashield (Vector Laboratories). Images were collected sequentially using a Zeiss fluorescence microscope and a charge-coupled device camera controlled by SPOT software. If necessary, a Zeiss 510 MATA laser-scanning confocal microscope was further used. Adobe Photoshop (version CS) was used for image cropping and adjustment.
Western blot.
Naive and treated rats were anesthetized with 2% halothane and decapitated. The RVM tissues were removed as previously described (Guo et al., 2006) and homogenized in solubilization buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS, 1 mm Na3VO4, 1 U/ml aprotinin, 2 μg/ml leupetin, 2 μg/ml pepstatin A). The homogenate was centrifuged at 20,200 × g for 10 min at 4°C, and the supernatant was removed. The protein concentration was determined. Each sample contained proteins from one animal. The proteins (50 μg) were separated on a 7.5% SDS-PAGE gel and blotted to nitrocellulose membrane (GE Healthcare). The blot was incubated with the respective antibody overnight at 4°C. The membrane was washed with TBS and incubated for 1 h with anti-goat IgG horseradish peroxidase (HRP) (1:3000; Santa Cruz Biotechnology) in 5% milk/TBS. The immunoreactivity was detected using enhanced chemiluminescence (ECL) (GE Healthcare). The loading and blotting of equal amount of proteins were verified by reprobing the membrane with anti β-actin antiserum (Sigma-Aldrich). The ECL-exposed films were digitized, and densitometric quantification of immunoreactive bands was performed using U-SCAN-IT gel (version 4.3; Silk Scientific).
Antibodies.
The following antibodies were used for immunostaining and Western blot: rabbit or mouse anti-glial fibrillary acidic protein (GFAP) (astrocytic marker; 1: 1000; Dako), rabbit anti-S100β (for labeling astrocytic calcium-binding protein; 1:800; Fitzgerald), mouse anti-OX-42 (for labeling CD11b as microglial marker; 1:800; Serotec), rabbit anti-Iba-1 (for labeling microglial calcium-binding protein; 1:1000; Wako), mouse anti-NeuN (neuronal marker; 1:1000; Millipore Bioscience Research Reagents), goat anti-TNF-α (1:1000; R&D Systems), rabbit anti-IL-1β (1:2000; Millipore Bioscience Research Reagents), goat anti-TNFR1 (1:500; Santa Cruz Biotechnology), rabbit anti-IL-1R (1:500; Santa Cruz Biotechnology), mouse anti-NR1 (1:5000; Millipore), rabbit anti-P-ser896 NR1 (Sigma-Aldrich), and mouse anti-β-actin (Sigma-Aldrich).
Histological reconstruction.
The locations of microinjection sites in the RVM were determined by visualization of serial Nissl-stained tissue sections under a microscope. Rats with misplaced microinjection sties were excluded from the data analysis or considered as controls in some cases.
Data analysis.
Results were expressed as mean ± SEM. Statistical comparisons included Student's t test or one- or two-way ANOVA with the post hoc Scheffé F test in Western blot analysis or the Student–Newman–Keuls test in behavioral experiments (ANOVA with repeated measures). In all cases, p < 0.05 was considered to be statistically significant.
Results
Mechanical hyperalgesia and allodynia induced by trigeminal nerve injury
To probe a role of central glial–neuronal interaction in the development of persistent pain, we adapted and improved the CCI-ION model in the rat (Vos et al., 1994; Imamura et al., 1997). The ION is a pure sensory nerve, the largest branch of the maxillary division of the trigeminal nerve, and innervates the mystacial vibrissae, the hairy vibrissal pad, the upper lip, lateral nose and teeth, and mucosa of the upper jaw (Waite and Tracey, 1995). To reduce injury related to the surgical procedure and keep the facial skin intact, we performed the CCI-ION operation through an intraoral approach (Imamura et al., 1997). Although the testing of behavioral hyperalgesia and allodynia in spinal models of pain is straightforward, assessing nocifensive behavior of the trigeminal region is difficult. Moreover, in the CCI-ION model, only responses to noxious thermal stimulation (Imamura et al., 1997) or mechanical stimulation (Kitagawa et al., 2006) have been examined in restrained rats. To reduce the stress of rats in an experimental environment, we have developed an appropriate handling approach without restraint to assess the mechanical hyperalgesia and allodynia of the orofacial region in rats (Ren, 1999; Sugiyo et al., 2005). The response frequencies to a range of von Frey filament forces applied to the ION territory were determined and a S–R curve was plotted (Fig. 1A). An EF50 value was defined as the von Frey filament force (g) that produces a 50% response frequency. Compared with the baseline, the leftward shift of the S–R curve, resulting in a reduction of EF50, represents mechanical hyperalgesia and allodynia at 1 and 14 d after CCI-ION (Fig. 1A), because there was an increased response to suprathreshold stimuli and a decreased response threshold for nocifensive behavior. Compared with naive and sham-operated rats, there was a significant reduction of the EF50 values in the ipsilateral ION territory from 1 to 28 d after unilateral CCI-ION in rats (p < 0.001 vs sham group) (Fig. 1B). There were no changes in the contralateral EF50 values. A moderate and transient (for 1–3 d) reduction of the EF50 values was also seen in sham rats (p < 0.05 vs naive group), and was completely blocked by local anesthesia of the surrounding tissues with 0.25% bupivacaine (Moore, 1984) (Fig. 1B), suggesting that the response to the surgical procedure alone at 1–3 d is caused by input from local tissue after incision and inflammation. Thus, we have shown a long-lasting and stable hyperalgesia/allodynia in the CCI-ION rats and used this model to study the involvement of supraspinal glia/cytokines in neuropathic pain of the orofacial origin.
Figure 1.

Mechanical hyperalgesia/allodynia and time course-dependent hyperactivation of astrocytes in the RVM after CCI-ION. A, Examples of S–R function curves illustrating the intensity-dependent head withdrawal responses to mechanical stimuli. There is a significant leftward shift of the curve at 1 d (green) and 14 d (red) after CCI (p < 0.001; n = 9 rats) compared with the pre-CCI baseline (blue). The leftward shift of the S–R curves indicates an increased response to suprathreshold stimuli and a decrease in EF50 value, suggesting the presence of mechanical hyperalgesia and allodynia. B, Time course of mechanical hyperalgesia and allodynia as indicated by a reduction of the EF50 values. CCI-ION results in mechanical hyperalgesia and allodynia that persists over the 28 d observation period. The rats receiving a sham operation show a moderate reduction of the EF50 values that lasted for 3 d, which is totally blocked by local tissue anesthesia with injection of 0.25% bupivacaine (twice at 1–3 d) at the intraoral incision site in sham-operated rats. CCI-ION versus sham, ***p < 0.001; sham versus naive, #p < 0.05. C, Immunohistochemistry of GFAP in the RVM. a, Low power of a tissue section at the rostral medulla level. b, Enlarged RVM region corresponding to the small rectangle area in a. Enhanced expression of GFAP is observed at 3 d (c) and 14 d (d) after CCI, compared with GFAP immunostaining in naive animal (b). D, Immunostaining of S100β in the RVM. Increased S100β expression is observed at 3 d (b), 14 d (c), and 28 d (d) after CCI, compared with naive (a). E, Western blots illustrating CCI-induced increase in GFAP in the tissues punched out from the RVM. A representative blot is shown on top, and the mean protein levels are summarized below. Compared with naive and sham-operated (−) rats, the level of GFAP is selectively increased at the 14 and 28 d time points in CCI-ION (+) rats. The increase in GFAP at 1–3 d in sham-operated and CCI-ION rats is consistent with behavioral changes at these time points (B). β-Actin was used as a loading control. The asterisks indicate significant differences from the naive rats (*p < 0.05; n = 3 per time points). F, Western blots illustrating the effect of bupivacaine, a long-lasting local anesthetic, on the incision-induced increase in GFAP expression in the RVM at 3 d after sham operation. Bupivacaine (0.25%; 0.1 ml) is injected at the intraoral incision site twice in the first 3 d (+). Saline is injected as a vehicle control (−). Compared with naive rats, the GFAP expression is increased at 3 d after sham operation (*p < 0.05; n = 3 per group), which is completely abolished by the bupivacaine (n = 3). Scale bars: Ca, 0.5 mm; Cd (for Cb–d), Dd (for Da–d), 0.025 mm. NGC, Nucleus reticularis gigantocellularis; Py, pyramidal tract. Error bars indicate SEM.
Long-lasting astrocytic hyperactivation in the RVM after nerve injury
To examine whether nerve injury induced glial reaction in the RVM, we first observed changes in the expression of GFAP, a marker of reactive gliosis and proliferation of astrocytes, in RVM sections from naive rats and after CCI-ION (Fig. 1C). As shown in sample sections at 3 d (Fig. 1Cc) and at 14 d (Fig. 1Cd) after CCI-ION, increased GFAP immunoreactivity (IR) was seen in small cell bodies and their processes, compared with naive animals (Fig. 1Ca,b), suggesting prolonged hyperactivation of astrocytes in RVM after nerve injury. It is known that S100β is a Ca2+ binding protein that is produced primarily in astrocytes in the CNS. Thus, enhanced S100β expression was also used as a specific functional marker of astrocytes in the brain (Tanga et al., 2006). The pattern of enhanced expression of S100β was compared with GFAP expression by immunohistochemistry in the RVM of the CCI-ION (Fig. 1 Db–d) and naive rats (Fig. 1Da), beginning from 1 to 28 d, further confirming the long-lasting hyperactivation of astrocytes in the RVM after nerve injury. We then used Western blot to quantify the time course of changes of GFAP levels in the RVM tissue (Fig. 1E). Although increased GFAP expression was found in both sham and CCI rats at 1 and 3 d after surgery, the level of GFAP expression was consistently increased in the RVM at 7 d (by 234.3 ± 12.1%; p < 0.001; n = 4 rats), 14 d (by 231.70 ± 6.64%; p < 0.001; n = 4), 21 d (by 176.70 ± 18.70%; p < 0.05; n = 3), and 28 d (by 194.54 ± 8.30%; p < 0.001; n = 3) after CCI but not sham treatment (by 123.10 ± 7.38% at 7 d and 121.70 ± 5.64% at 14 d; p > 0.05; n = 4 per group) compared with the naive group (n = 3) (Fig. 1E). Consistent with a small and short-lasting behavioral hyperalgesia and allodynia at 1 and 3 d in the sham-operated rats (Fig. 1B), the transiently increased GFAP expression in the RVM of the sham-operated rats was found at the same time points (Fig. 1E). To distinguish whether these early effects in the CCI rats were partially attributable to the tissue incision during the oral surgery, we used local anesthesia of the surgical site to block the increased neural activity associated with tissue injury during the first 3 d in sham animals. Long-lasting anesthesia with 0.25% bupivacaine totally eliminated the increased GFAP expression at 3 d after sham operation (n = 3 per group) (Fig. 1F), suggesting that the earlier increase in GFAP expression observed in the RVM at 1 and 3 d after CCI-ION is partially caused by tissue injury. This observation is also consistent with a recent study (Obata et al., 2006) that hindpaw incision results in glial hyperactivation in the spinal dorsal horn, beginning within 1 d and reaching a maximum hyperactivation at 3 d after tissue injury.
In a parallel experiment, we extended our analysis to determine whether microglial reaction occurs by examining expression of microglial markers in the RVM (Fig. 2). Different from GFAP expression, CD11b expression in the RVM was primarily increased at 1 d (data not shown) and 3 d, but not 14 d and later, after CCI-ION compared with the naive condition (Fig. 2A). This early hyperactivation of microglia was also confirmed by immunostaining with a functional microglia marker, ionized calcium-binding adaptor molecule 1 (Iba1) (Fig. 2B). With Western blot analysis, an increase in CD11b expression in RVM tissues was only detected at early time points (1–3 d) in both CCI and sham-operated rats (p < 0.05; n = 3 per group), compared with naive animals (Fig. 2C). Local tissue anesthesia also completely blocked CD11b increase in the RVM from sham-operated rats at 3 d (n = 3 per group) (Fig. 2D), suggesting that tissue injury also caused microglial hyperactivation. These results indicate that nerve and tissue injury activate acutely (1–3 d) both astrocytes and microglia in the RVM, both of which are likely involved in supraspinal mechanisms underlying the generation of trigeminal neuropathic pain, consistent with previous studies on glial function in the spinal cord (DeLeo and Yezierski, 2001; Raghavendra et al., 2004; Ledeboer et al., 2006; Obata et al., 2006). In addition, nerve injury also induced a prolonged hyperactivation (at least for 28 d) of astrocytes in the RVM. The role of the long-term astrocytic reaction in the RVM during development of neuropathic pain was the focus of our subsequent studies below.
Figure 2.

Time course-dependent hyperactivation of microglia in the RVM after CCI. A, CD11b immunostaining in the RVM. a, Low power of a section at the rostral medulla level. b, Enlarged RVM region corresponding to the small rectangle area in a. CD11b expression is enhanced at 3 d (c) but not 14 d (d) after CCI-ION, compared with CD11b immunoreactivity in naive animal (b). B, Iba1 immunostaining in the RVM. Enhanced Iba1 expression is observed at 3 d (b) and declined to near control expression levels at 14 d (c) and 28 d (d) after CCI, compared with naive animal (a). C, Western blots illustrating CCI-induced short-lasting increase in CD11b in RVM tissues. Compared with naive rats, CD11b expression is temporarily increased at the 1–3 d time points but not at 14 and 28 d in both sham-operated (−) and CCI-ION (+) rats. The asterisks indicate significant differences from the naive rats (*p < 0.05; n = 3 per time points). D, Local tissue anesthesia (+), compared with saline injection (−), completely blocks sham operation-induced increase in CD11b in the RVM at 3 d, when compared with naive rats [sham (−) vs naive, p < 0.05; n = 3 per group]. Scale bars: Aa, 0.5 mm; Ad (for Ab–d), Bd (for Ba–d), 0.025 mm. NGC, Nucleus reticularis gigantocellularis. Error bars indicate SEM.
Consistent with previous studies (Piao et al., 2006; Guo et al., 2007), the time course-dependent upregulation of GFAP and CD11b expression was also observed after CCI-ION in the spinal trigeminal nucleus ipsilateral to the nerve injury (data not shown), a trigeminal site known to receive direct peripheral nociceptive input. Importantly, glial reaction in the RVM is specifically related to CCI-ION. The facial nucleus lateral to the RVM did not exhibit changes in GFAP and CD11b expression after CCI-ION (data not shown).
Selective inhibition of astrocytes by lower dose of glial toxin fluorocitrate
By its inhibition on aconitase, fluorocitrate interrupts the tricarboxylic acid (TCA) cycle and can be used as a mitochondrial inhibitor (Voloboueva et al., 2007). Fluorocitrate-induced glial inhibition results in a variety of metabolic effects including reduced ATP production and increased glucose metabolism (Fonnum et al., 1997; Hirose et al., 2007). Although not selectively affecting aconitase of different cellular origin per se, fluorocitrate primarily inhibits TCA cycle of astrocytes because of more avid uptake by astrocytes and has been used to assess the importance of glial cells for brain function in vivo (Fonnum et al., 1997; Zielke et al., 2007). However, fluorocitrate may also produce neuronal damage (Hornfeldt and Larson, 1990; Largo et al., 1996). Intrastriatal injection of 1 nmol of fluorocitrate produces ultrastructural alterations of astrocytes without affecting neurons, whereas a larger dose at 2 nmol also affects neuronal structures and metabolism (Paulsen et al., 1987).
To search for a safe range of doses for the selective effect of fluorocitrate on glial cells, we performed double staining for the neuronal marker, NeuN, with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), a label for cell apoptosis, or Fluoro-Jade, a marker of cell necrosis, in RVM sections after microinjection of the drug (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). When doses of fluorocitrate in the range between 1 fmol (2.2 ± 0.41 double-labeled cells/section; n = 4; p > 0.05) and 100 fmol (2.1 ± 0.27; n = 4; p > 0.05) (supplemental Fig. S1Ac,C, available at www.jneurosci.org as supplemental material) were used, there was no apparent cellular damage shown by Nissl (supplemental Fig. S1Aa, available at www.jneurosci.org as supplemental material) and TUNEL staining (supplemental Fig. S1Ab,c, available at www.jneurosci.org as supplemental material) except within the track of the microinjection needle and its nearby area compared with that seen in naive (0.25 ± 0.16 labeled cells/section; n = 4) (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material) and vehicle treatment (1.38 ± 0.18; n = 4) (supplemental Fig. S1Ab,C, available at www.jneurosci.org as supplemental material). This finding suggests that a few damaged cells may be induced by placement of the injection needle during the microinjection procedure but not by fluorocitrate. However, when a higher dose of fluorocitrate (10 pmol) was injected, TUNEL-labeled cells were significantly increased in the RVM (11.88 ± 1.26; n = 4; p < 0.001 vs vehicle group; n = 4) (supplemental Fig. S1Ad,C, available at www.jneurosci.org as supplemental material), with labeled cells in areas far away from the tissue track of the injection needle. Double immunostaining identified that all TUNEL-labeled cells were also positive to NeuN (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). These results suggest that a dose-dependent neuronal apoptosis occurred after microinjection of fluorocitrate into the RVM. Fluoro-Jade B staining was performed to examine whether fluorocitrate induces neuronal necrosis in the RVM. Many Fluoro-Jade-positive cells were located in the microinjection sites from rats injected with high (10 pmol; n = 4) but not low (100 fmol) dose of fluorocitrate (n = 4 per group) (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). No double-labeled cells for both Fluoro-Jade B and NeuN were found in the RVM, although Fluoro-Jade-positive cells and NeuN-immunoreactive neurons was also seen in the same RVM region after microinjection of fluorocitrate (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). This finding suggests that no significant necrosis of RVM neurons was apparent after microinjection of fluorocitrate. The Fluoro-Jade B-positive population may represent selective degeneration of RVM glial cells resulting from toxic effects of a high dose of fluorocitrate.
Neuronal damage has been found in the rat spinal dorsal horn after sciatic nerve injury (Polgár et al., 2005; Scholz et al., 2005). To test whether CCI-ION alone affected the extent of neuronal damage in the RVM, we also examined TUNEL and Fluoro-Jade staining in RVM sections from rats with the CCI or sham treatment at 7 and 14 d after surgery. In comparison with the naive animal, there were no significant differences in the number of TUNEL- or Fluoro-Jade-labeled cells in the RVM in the CCI- or sham-operated rats (p > 0.05; n = 4 per group) (data not shown), suggesting that there was no apparent neuronal damage in the RVM produced by the CCI-ION alone.
We then examined the effects of low dose of fluorocitrate on the glia cells in the RVM of CCI rats (supplemental Fig. S1E, available at www.jneurosci.org as supplemental material). We confirmed the lack of any neuronal apoptosis in the RVM sections from 3 d (n = 4 rats) (data not shown) and 14 d (n = 4) (supplemental Fig. S1Ed, available at www.jneurosci.org as supplemental material) CCI-treated rats after microinjection of low dose fluorocitrate (100 fmol) compared with vehicle (n = 3) (supplemental Fig. S1Ea, available at www.jneurosci.org as supplemental material). With regard to long GFAP turnover time, we examined changes in GFAP protein expression in RVM tissue at 6 h after astrocytic inhibition. Upregulation of GFAP expression in the RVM at 14 d after CCI (n = 4) (supplemental Fig. S1Eb, available at www.jneurosci.org as supplemental material) was decreased after microinjection of fluorocitrate (100 fmol) (n = 4) (supplemental Fig. S1Ee, available at www.jneurosci.org as supplemental material). In contrast, there was no effect of this dose of fluorocitrate on the elevated CD11b expression in the RVM sections at 3 d after CCI-treated rats (n = 4) (supplemental Fig. S1Ef, available at www.jneurosci.org as supplemental material) compared with vehicle injection (n = 3) (supplemental Fig. S1Ec, available at www.jneurosci.org as supplemental material). Western blot further confirmed that fluorocitrate (100 fmol) induced the selective inhibition of enhanced GFAP expression [91 ± 21.5% (FC; n = 3) vs 238 ± 45.9% (vehicle; n = 3); p < 0.05] (Fig. 3A) and S100β expression [83.3 ± 9.5% (FC; n = 3) vs 159.7 ± 17% (vehicle; n = 3); p < 0.001] (Fig. 3B) at 14 d after CCI in the RVM compared with vehicle treatment.
Figure 3.

Western blot analysis shows inhibition of fluorocitrate on glial hyperactivation and functions. A, B, Microinjection of fluorocitrate (FC+; 100 pmol) significantly inhibits enhanced expression of GFAP (A) and S100β (B) at 14 d after CCI-ION (p < 0.05; n = 3 per group) but does not affect their expression at 14 d after sham operation, compared with vehicle treatment (−). C, D, Fluorocitrate (100 pmol) significantly reverses upregulation of TNF-α (C) and IL-1β (D) at 14 d after CCI-ION (p < 0.05; n = 3 per group) but does not change their expression at 14 d after sham operation, compared with vehicle treatment (−). E, There are no effects of FC (100 pmol) on increases in CD11b expression at 3 d after sham operation and CCI-ION, compared with vehicle (Veh) (n = 3 per group). However, the microglial inhibitor MC (1 pmol) attenuates or abolishes CD11b increase in the RVM at 3 d after sham or CCI treatment. All asterisks indicate p < 0.05 when compared with the naive (n = 3 per group) in A–E. Error bars indicate SEM.
In order to determine whether astrocytic inhibition by fluorocitrate functionally reduces cytokine production in the RVM after CCI, we measured changes in TNF-α and IL-1β protein levels in the RVM. Fluorocitrate (100 fmol) totally prevented CCI-induced enhancement of TNF-α expression [86.3 ± 27.5% (FC; n = 3) vs 241.3 ± 30.1% (vehicle; n = 3); p < 0.001] (Fig. 3C) and IL-1β expression [109.7 ± 16.2% (FC; n = 3) vs 199 ± 25.4% (vehicle; n = 3); p < 0.01] (Fig. 3D) at 14 d in the RVM compared with vehicle treatment. However, this dose of fluorocitrate did not affect the basal GFAP (Fig. 3A) and S100β (Fig. 3B) and TNF-α and IL-1β protein levels in the RVM tissue at 14 d after a sham operation (Fig. 3C,D).
It was shown that intrathecal injection of fluorocitrate (0.1–1 nmol) reduced injury-induced elevation of CD11b expression in the spinal cord (Clark et al., 2007; Sun et al., 2008), questioning its preferential inhibition of astrocytic function. We examined the effect of fluorocitrate on microglial function in the RVM in our model and with the low dose used to study astrocytic function. The results showed that fluorocitrate (100 fmol) did not block tissue injury-induced enhancement of CD11b expression [159 ± 5% (FC; n = 3) vs 153 ± 3.93% (vehicle; n = 3); p > 0.05] (Fig. 3E) at 3 d in the RVM tissues of sham-operated rats, and also did not prevent nerve injury-induced elevation of CD11b expression [171.3 ± 22.1% (FC; n = 3) vs 168.3 ± 16.3% (vehicle; n = 3); p > 0.05] (Fig. 3E) in the RVM at 3 d after CCI. In contrast, the enhanced expression of CD11b in the RVM at 3 d after sham operation or CCI was attenuated or abolished by MC (1 pmol) at 4 h after the microinjection (n = 3 per group; p < 0.05) (Fig. 3E) compared with vehicle. Thus, our results suggest that low doses of fluorocitrate could be used to locally and effectively inhibit hyperactive astrocytes and their function without apparent neuronal degeneration and microglial disruption in the RVM.
Time course-dependent effects of glial inhibitors on allodynia
To identify participation of supraspinal glial hyperactivation in the cellular mechanisms underlying the initiation and maintenance of neuropathic pain, we tested the effects of selective disruption of glial metabolism by microinjection of single doses of glial inhibitors into the RVM on mechanical hyperalgesia and allodynia at early (3 d) and later time points (14 d) after CCI. Propentofylline has been known to depress both microglial and astrocytic activation, reduce proinflammatory cytokine release, and attenuate allodynia and hyperalgesia induced by nerve injury and inflammation when systemically or intrathecally injected (S. M. Sweitzer et al., 2001; Raghavendra et al., 2003a,b; Dorazil-Dudzik et al., 2004; Tawfik et al., 2008). The antiallodynic effect of propentofylline is associated with a reduction of reactive gliosis involving astroglia and microglia (S. Sweitzer et al., 2001; Raghavendra et al., 2003b; Sweitzer, 2006), which is consistent with a role of propentofylline as a nonselective glial modulator. Minocycline is a semisynthetic tetracycline antibiotic with a number of other properties distinct from its antimicrobial action including neuroprotection and antiinflammatory action (Elewa et al., 2006). It has been used to evaluate the involvement of microglia in the development of neuropathic pain, and has no direct action on astrocytes or neurons (Tikka et al., 2001; Raghavendra et al., 2003a; Ledeboer et al., 2005). Recent studies indicate that minocycline reduces microglial migration to cellular debris and decreases microglial Kv1.3 expression (Nutile-McMenemy et al., 2007).
We injected fluorocitrate, propentofylline, and minocycline into the RVM of the CCI and sham-operated rats at 3 and 14 d after surgery. As shown in Figure 4, all vehicle-treated CCI rats exhibited stable and strong hypersensitivity in behavioral tests (Fig. 4 A–C). A single dose of PPF (10 pmol) had no effect on EF50 values in the sham animals at 14 d after surgery (Fig. 4A, left). However, propentofylline (1 fmol, 100 fmol, and 10 pmol) significantly attenuated mechanical hyperalgesia and allodynia at 14 d after CCI in a dose-dependent manner, lasting for 4 h or longer (Fig. 4A, left). Similarly, at 3 d after nerve injury, a high dose of propentofylline (10 pmol) completely reversed hyperalgesia and allodynia at least for 6 h, and also transiently blocked moderate behavioral hypersensitivity in sham-treated animals at 3 d (Fig. 4A, right). Although it had no effect on EF50 values in the sham rats at 14 d, fluorocitrate (1 and 100 fmol) significantly attenuated mechanical hyperalgesia and allodynia to an extent similar to the high dose of propentofylline at 14 d after CCI, when compared with the vehicle-treated CCI rats (Fig. 4B, left). Interestingly, fluorocitrate at these doses did not reverse hyperalgesia and allodynia observed at 3 d after CCI (Fig. 4B, right). Fluorocitrate did produce a trend toward inhibition of behavioral hypersensitivity at 3 d in sham animals when compared with the vehicle-treated sham rats (Fig. 4B, right). In contrast, minocycline (1 pmol) produced inhibition of behavioral hyperalgesia and allodynia, which persisted over the 6 h observation period, at 3 d (Fig. 4C, right) but not 14 d (Fig. 4C, left) after CCI-ION. This dose of minocycline also transiently and significantly attenuated the moderate hyperalgesia and allodynia induced by sham operation at 3 d (Fig. 4C, right). These results support our hypothesis that the development of neuropathic pain behavior in CCI-ION rats involves glial hyperactivation in the RVM. In combination with the immunohistochemistry and Western blot results, our behavioral analysis further suggests that persistent astrocytic hyperactivation in the RVM contributes to mechanisms underlying the maintenance of neuropathic pain after nerve injury. These results also suggest that the initiation of behavioral hyperalgesia and allodynia in the CCI-ION rats primarily depends on cellular mechanisms relevant to microglial reaction in the RVM after nerve injury and tissue injury. It is known that minocycline interferes with the activity of matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9) (Machado et al., 2006), which are a family of zinc-dependent proteases responsible for extracellular matrix turnover and degradation of bioactive proteins. MMP-2 and MMP-9 have been found in dorsal root ganglion neurons and satellite cells and are involved in induction and maintenance of neuropathic pain through IL-1β cleavage and glial hyperactivation at the spinal cord (Kawasaki et al., 2008). Although the distribution of MMPs in the RVM and their regulation after nerve injury are not known, we cannot exclude the possible effect of minocycline on glial function via MMPs and other factors in the RVM.
Figure 4.

The different effect of glial inhibitors (propentofylline, fluorocitrate, and minocycline) on behavioral hyperalgesia/allodynia at early (3 d) and later (14 d) time points after CCI-ION. At posttreatment, the glial inhibitor propentofylline (A), the selective astrocytic inhibitor fluorocitrate (B), and the microglial inhibitor minocycline (C) are microinjected into the RVM at 3 d (right column) and 14 d (left column) after sham operation or CCI-ION. The dose-dependent effects of the glial inhibitors on behavioral hyperalgesia/allodynia are measured at 14 d after the surgery. An effective high dose of the inhibitor is then used for behavioral observation at the 3 d time point. Vehicles are microinjected as a control. A, PPF (1 fmol, 100 fmol, and 10 pmol) produces a dose-dependent attenuation of CCI-induced mechanical hyperalgesia and allodynia at 14 d, persistent for 4–24 h compared with vehicle microinjection in CCI rats (left). High-dose propentofylline (10 pmol) has no effect on EF50 values at 14 d in sham rats (left). This dose of propentofylline transiently blocks moderate hyperalgesia and allodynia at 3 d compared with vehicle in sham-operated rats (right), and also significantly abolishes mechanical allodynia at 3 d after CCI compared with vehicle in CCI rats (right). B, Two doses of FC (1 and 100 fmol) after microinjection into the RVM significantly attenuate hyperalgesia and allodynia similarly at least for 6 h, compared with vehicle treatment at 14 d in CCI rats (left). High dose of fluorocitrate (100 fmol) has no effect on EF50 values at 14 d in sham rats (left). However, fluorocitrate (100 fmol) does not prevent CCI-induced behavioral hypersensitivity to mechanical stimulation at 3 d after CCI or sham operation (right). C, In contrast, microinjection of MC (1 pmol) into the RVM significantly reduces hyperalgesia and allodynia for 6 h at 3 d after CCI-ION compared with vehicle treatment in CCI rats, and also transiently blocks sham operation-induced mechanical hypersensitivity (right). Lower dose of minocycline (10 fmol) induced a slight and short-lasting inhibition for CCI-induced allodynia (right). The two doses of minocycline have no effect on CCI-induced hyperalgesia and allodynia at 14 d (left). Also, there are no changes in EF50 values after minocycline (1 pmol) injection at 14 d after sham-operated rats. ***, ###p < 0.001; **p < 0.01; *, #,^ p < 0.05 versus CCI plus vehicle in left. ***p < 0.001, *p < 0.05, CCI plus drug versus CCI plus vehicle in right. #p < 0.05, sham plus drug versus sham plus vehicle in right. Error bars indicate SEM.
Enhanced expression of cytokines in RVM astrocytes after nerve injury
On hyperactivation, astrocytes and microglia increase expression and secretion of proinflammatory cytokines, such as TNF-α and IL-1β. These cytokines have been shown to increase synaptic efficacy in the hippocampus (Woolf and Salter, 2000; Beattie et al., 2002; Viviani et al., 2003; Perea and Araque, 2006, 2007) and contribute to neuronal hypersensitivity in spinal dorsal horn (DeLeo and Yezierski, 2001; Sommer and Kress, 2004; Marchand et al., 2005; Watkins et al., 2007). Hence we next examined whether astrocytic hyperactivation during the development of neuropathic pain is accompanied by an increase in cytokine levels in the RVM. Western blots showed that the expression of TNF-α and IL-1β in the RVM tissues was significantly upregulated by 58 and 62%, respectively, at 14 d after CCI compared with sham-operated animals (p < 0.05; n = 3 per group) (Fig. 5A). An increase of TNF-α and IL-1β immunostaining in the RVM cells was found after CCI-ION (n = 6) (Fig. 5B). To verify the source of cells expressing these cytokines, double labeling was performed and showed that both TNF-α IR and IL-1β IR were colocalized with GFAP IR but not NeuN IR in RVM cells at 14 d after CCI-ION (Fig. 5C,D). We did not find coexpression of the cytokines with CD11b (data not shown), possibly because of the very few hyperactivated microglia seen at 14 d. These results suggest that hyperactivated astrocytes are a primary source of the increased TNF-α and IL-1β in the RVM in the maintenance of neuropathic pain at 14 d after nerve injury.
Figure 5.

Enhanced expression of TNF-α and IL-1β in the RVM at 14 d after CCI-ION. A, Western blots illustrating that the levels of TNF-α (left) and IL-1β (right) are increased in the RVM in CCI-ION rats compared with sham-operated rats (*p < 0.05; n = 3 per group). The anti-TNF-α antibody identifies both the transmembrane TNF-α precursor (26 kDa) and matured TNF-α (17 kDa). Both TNF-α precursor and matured TNF-α exhibit an increase in the RVM at 14 d after CCI-ION. Error bars indicate SEM. B, Immunostaining of TNF-α and IL-1β in RVM. a, d, Low power of a tissue section at the rostral medulla level. b, e, Enlarged RVM region corresponds to the rectangle area in a and d, respectively. Compared with naive rats (b, e), there is an increase in TNF-α IR and IL-1β IR in RVM at 14 d after CCI-ION (c, f). The arrowheads indicate positively labeled cells. C, RVM tissue sections at 14 d after CCI are double stained for TNF-α (a, d; red) and GFAP (b; green) or NeuN (e; green). The arrowheads indicate positively labeled cells. Overlap of a and b reveals that the TNF-α IR is colocalized with GFAP IR in RVM cells (c; yellow, arrows), suggesting its presence in astrocytes. Overlap of d and e shows a lack of TNF-α in RVM neurons (f). D, RVM tissue sections were double stained for IL-1β (a, d; red) and GFAP (b; green) or NeuN (e; green). The arrowheads indicate positively labeled cells. Overlap of a and b reveals that the IL-1β-labeled cells are GFAP immunoreactive in RVM (c; yellow, arrows), suggesting its presence in astrocytes. Overlap of d and e shows a lack of IL-1β IR in these RVM neurons (f). Scale bars: Ba,d, 0.1 mm; Bb,c,e,f, 0.03 mm; C, D, 0.03 mm.
Increased expression of cytokine receptors in RVM neurons after nerve injury
As a possible mechanism of glial–neuronal interaction, we wanted to know whether cytokine receptors were distributed in RVM neurons as a link between astrocytes and neurons and if these receptors were upregulated in the RVM after trigeminal nerve injury. Immunostaining showed stronger expression of TNFR1, receptor for TNF-α, and IL-1RI, receptor for IL-1β, in the RVM at 14 d after CCI (Fig. 6Aa,b, Ca,b, respectively) compared with naive and sham-operated animals (data not shown). Double labeling indicated that TNFR1 IR and IL-1RI IR were primarily localized in RVM neurons (Fig. 6A,C). Western blot showed that the levels of TNFR1 and IL-1RI were significantly increased at 14 d after CCI when compared with naive and sham-operated rats (p < 0.05; n = 3 per group) (Fig. 6B,D). These data demonstrate enhanced expression of cytokine receptors for TNF-α for IL-1β in the RVM neurons during maintenance of neuropathic pain.
Figure 6.

Enhanced expression of cytokine receptors for TNF-α and IL-1β in RVM neurons at 14 d after CCI-ION. A, C, Immunostaining shows distribution of TNFR1 (A; red) and IL-1RI expression (B; red) in low power of a tissue section at the rostral medulla level (a) and enlarged RVM region (b) corresponding to the small rectangle area in a. The arrowheads in b and c indicate single-labeled neurons. Double labeling shows TNFR1-labeled (Ab) and IL-1RI-labeled cells (Cb) are immunoreactive to NeuN (Ac, Cc; green), suggesting their expression in the RVM neurons (Ad, Cd; arrows). B, D, Western blots illustrating that the levels of TNFR1 (B) and IL-1RI (D) are increased in the RVM tissues at 14 d in CCI-ION rats compared with sham-operated rats (p < 0.05; n = 3 per group). *p < 0.05 versus naive in B and D. Scale bars: Aa, Ca, 0.1 mm; Ab–d, Cb–d, 0.03 mm. Error bars indicate SEM.
Cytokine receptor activation is linked to phosphorylation of NMDARs and mechanical allodynia after nerve injury
We have shown that glutamate receptor subunit NMDARs are widely expressed in rat RVM neurons and are upregulated and phosphorylated in the RVM after tissue injury, contributing to thermal hyperalgesia and mechanical allodynia after peripheral inflammation (Guan et al., 2002; Guo et al., 2006). To provide morphological evidence that supports the interactions between glia/cytokines and neuronal glutamate receptors, we performed double immunostaining. The results showed that cytokine receptors TNFR1 and IL-1RI colocalized with NR1 expression, a principal component of NMDARs in RVM neurons at 14 d after CCI-ION (n = 4) (Fig. 7A,B), similar to the distribution pattern from untreated rats (data not shown). To examine whether CCI-ION also induces NMDAR activation, the phosphorylation levels of the NMDAR subunit NR1 were measured in RVM tissues. As shown in Figure 7C, the immunostaining against pNR1ser896 was clearly increased by twofold (p < 0.001; n = 3) at 14 d after CCI-ION compared with the sham-operated (data not shown) and naive rats (n = 3).
Figure 7.
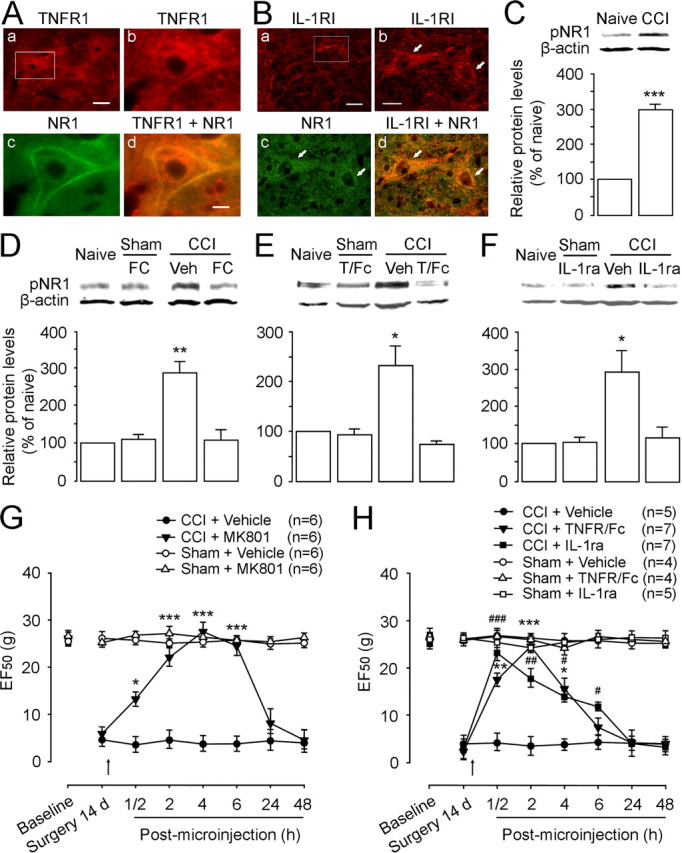
The effect of cytokine inhibitors on NMDAR subunit NR1 phosphorylation in the RVM and behavioral hyperalgesia and allodynia at 14 d after CCI-ION. A, B, Double immunostaining shows colocalization of TNFR1 (Ab; red) or IL-1RI (Bb; red) with NR1 (Ac, Bc; green) in RVM neurons. Overlay of b and c reveals double labeling of RVM neurons with TNFR1/NR1 (Ad; yellow-orange) or IL-1RI/NR1 (Bd; yellow-orange). Note colocalization of these receptors with NR1 in the cell membrane. C, Compared with naive rats, the level of pNR1ser896 is increased at 14 d after CCI-ION (p < 0.001; n = 3 per group). D, The increased pNR1ser896 is totally blocked by intra-RVM microinjection of astrocytic inhibitor FC (100 fmol) compared with naive and sham-operated rats (n = 3 per group). E, F, Neutralizing endogenous TNF-α and IL-1β in the RVM using TNFR1/Fc (T/Fc) (50 fmol) (E) and IL-1ra (3 pmol) (F), respectively, blocks CCI-induced increase in pNR1 expression in the RVM at 14 d after CCI-ION compared with sham treatment (p < 0.05; n = 3 per group). No effect of these inhibitors on basal pNR1 expression is observed in sham-operated rats compared with naive rats (n = 3). G, Microinjection of NMDAR channel blocker MK801 (10 pmol) into the RVM abolishes CCI-induced mechanical hyperalgesia and allodynia compared with vehicle (Veh) injection at 14 d after CCI-ION. H, No effect of intra-RVM TNFR1/Fc (50 fmol) and IL-1ra (3 pmol) on basal mechanical threshold is observed in sham-operated rats compared with naive rats. However, these doses of TNFR1/Fc and IL-1ra attenuate CCI-ION-induced hyperalgesia/allodynia for 4–6 h after microinjection compared with vehicle treatment in CCI rats. D–F, **p < 0.01; *p < 0.05 versus naive; G, H, ***, ###p < 0.001; ** , ##p < 0.01; *, #p < 0.05 versus CCI plus vehicle. Scale bars: Aa, Ba, 0.015 mm; Ab–d, Bb–d, 0.005 mm. Error bars indicate SEM.
To further explore whether astrocytic hyperactivation and consequent secretion of cytokines contributes to NMDAR activation, we examined the effect of local application of fluorocitrate on increased expression of pNR1 produced by nerve injury. Western blot showed that the increased pNR1ser896 was blocked by fluorocitrate (100 fmol) at 2 h after microinjection into the RVM at 14 d after CCI-ION (p < 0.01; n = 3) compared with vehicle treatment (n = 3) (Fig. 7D), whereas the same dose of fluorocitrate had no effect on pNR1 expression in sham rats (n = 3) (Fig. 7D). Moreover, the CCI-induced enhancement of pNR1 was also completely eliminated by the soluble anti-TNFR1 IgG, a biological sequester for endogenous TNF-α [TNFR/Fc (T/Fc); 50 fmol; n = 3; p < 0.05] (Fig. 7E) and IL-1ra, an IL-1RI antagonist (3 pmol; p < 0.05; n = 3) (Fig. 7F) at 2 h after intra-RVM injections, compared with vehicle injections (n = 3). We did not detect any effects of the immune agents on pNR1 expression in the RVM tissue in sham-operated rats (p > 0.05; n = 3 each group) (Fig. 7E,F), when compared with that in naive animals (n = 3). These results suggest that NMDAR activation in the RVM after CCI is facilitated by the activation of RVM astrocytes and the cytokine receptors for TNF-α and IL-1β during the maintenance of neuropathic pain.
To examine whether NMDARs and the cytokine receptors TNFR1 and IL-1RI contribute to CCI-induced mechanical hyperalgesia and allodynia, we injected the receptor antagonists into the RVM at 14 d after nerve injury. MK801 (10 pmol), an NMDAR channel blocker, did not affect baseline EF50 values in sham-treated rats (n = 6) (Fig. 7G); however, it significantly attenuated mechanical hyperalgesia and allodynia after CCI at least for 6 h (n = 6; p < 0.05) (Fig. 7G). The posttreatment with TNFR/Fc (50 fmol) or IL-1ra (3 pmol) in the RVM also temporally reversed behavioral hypersensitivity to a similar extent in the CCI rats but did not alter EF50 values recorded in sham rats (Fig. 7H). The microinjection of these compounds into the facial nucleus near the RVM in CCI-ION rats did not disrupt the development of behavioral hypersensitivity (data not shown), suggesting that the inhibitory effects of the receptor antagonists on CCI-induced hypersensitivity indeed occurred in the RVM but not the surrounding regions. Thus, we have demonstrated the involvement of RVM NMDAR activation in CCI-induced hyperalgesia/allodynia and its dependence on local astrocytic hyperactivation and cytokine release.
Intra-RVM exogenous TNF-α and IL-1β produce NMDAR activation and NMDAR-dependent descending facilitation
To test whether the cytokines directly increase NMDAR function and are implicated in descending pain facilitation, we microinjected recombinant rat TNF-α (rTNF-α) and IL-1β (rIL-1β) into the RVM of normal rats and further evaluated their effects on local pNR1 expression and mechanical nociception (Fig. 8). In comparison with vehicle, both exogenous rTNF-α (120 fmol) and rIL-1β (120 fmol) produced a significant upregulation of NR1 ser896 phosphorylation (by 1.8-fold and 1.71-fold, respectively; p < 0.05; n = 4 per group) at 2 h after microinjections (Fig. 8A). The cytokine-induced pNR1 increase was completely blocked by pretreatment with the NMDAR antagonist MK801 (10 pmol; p < 0.05; n = 4 per group) (Fig. 8A), suggesting that in addition to activating cytokine receptors at postsynaptic sites, these cytokines may enhance glutamate release from presynaptic terminals and glia. Finally, we examined the effects of exogenous cytokines on mechanical nociceptive thresholds (Fig. 8B,C). A single dose of rTNF-α (120 fmol) in the RVM produced behavioral hyperalgesia and allodynia, as indicated by a significant reduction of EF50 values (p < 0.05; n = 8) compared with vehicle injection (n = 5) (Fig. 8B). The rTNF-α-produced effect on EF50 began at 30 min and lasted for at least 4 h. Microinjection of rIL-1β (120 fmol) produced a similar descending facilitation to a lesser extent (Fig. 8C). The hyperalgesic/allodynic effects of these cytokines were completely blocked by pretreatment with MK801 (10 pmol; n = 5–8). MK801 alone did not affect EF50 values in normal rats (n = 5) (Fig. 8B). Thus, these results further confirm that the regulation of NMDAR activation by locally elevated cytokines TNF-α and IL-1β induces hyperexcitability in RVM neurons and contributes to NMDAR-dependent descending facilitation of hyperalgesia and allodynia.
Figure 8.

Intra-RVM rTNF-α and rIL-1β enhance pNR1 levels and produce descending pain facilitation in normal rats. A, Microinjection of rTNF-α (120 fmol) and rIL-1β (120 fmol) into the RVM enhances expression of pNR1ser896 in the RVM tissue compared with baseline levels in naive rats (p < 0.05; n = 4 per group). Pretreatment of an NMDAR channel blocker MK801 (+) (n = 4) completely prevents rTNF-α- and rIL-1β-induced increase in pNR1 compared with vehicle treatment (−). RVM tissues are collected at 2 h after microinjection of the recombinant agents into the RVM. B, Mechanical sensitivity of the skin is assessed after microinjection of rTNF-α. The behavioral hyperalgesia and allodynia develops and lasts for 4 h after microinjection of rTNF-α (120 fmol) as indicated by a reduction of EF50 values (p < 0.05). Pretreatment of MK801 (10 pmol) significantly abolishes rTNF-α-produced mechanical hypersensitivity compared with vehicle plus rTNF-α. C, Microinjection of rIL-1β (120 fmol) results in a significant reduction of EF50 values for 2 h (p < 0.05), which is prevented by pretreatment with MK801. *p < 0.05, rTNF-α or rIL-1β versus vehicle injection. Error bars indicate SEM.
Discussion
In the present study, we observed early microglial hyperactivation at 1–3 d after CCI-ION, followed by a prolonged astrocytic hyperactivation in the RVM lasting at least for 28 d, with a peak expression at 14 d. As expected, inhibition of local glial hyperactivation in the RVM using propentofylline significantly attenuated mechanical hyperalgesia/allodynia at both 3 and 14 d after nerve injury. Interestingly, microinjection of the microglial inhibitor minocycline in the RVM blocked CCI-induced hyperalgesia/allodynia at the early phase (3 d) but not the later phase (14 d). In contrast, fluorocitrate, when used at lower doses in the RVM, only attenuated behavioral hypersensitivity at the later phase. Consistent with our finding, studies with disruption of spinal microglial function by intrathecal injection or local application of minocycline suggest that spinal microglial hyperactivation is required for the initiation, but not the maintenance of nerve injury-induced hyperalgesia (Ledeboer et al., 2005) and evoked neuronal activity in the spinal dorsal horn (Owolabi and Saab, 2006). Similar to CCI-ION-induced glial reaction in the RVM and behavioral hypersensitivity, microglial hyperactivation preceding astrocytic hyperactivation is also observed in the spinal cord after inflammation (Raghavendra et al., 2004) and nerve injury (Kawasaki et al., 2008). In addition, recent evidence suggests that the prolonged reaction of astrocytes in the spinal cord plays an important role in maintaining neuropathic pain (Tanga et al., 2006; Zhuang et al., 2006; Kawasaki et al., 2008). Our data further support an important role of RVM microglia in the initiation phase and astrocytes in maintaining mechanical hyperalgesia/allodynia after nerve injury. Although supraspinal glial hyperactivation and cytokine expression had been found after injury (Raghavendra et al., 2004; Apkarian et al., 2006), no studies have investigated the possibility that they are involved in mechanisms underlying descending modulation of persistent pain. Thus, we provide the first evidence that a signaling sequence from glial hyperactivation, cytokine release, and glutamate receptor phosphorylation in the descending pain modulatory circuitry contributes to the cellular and molecular mechanisms of neuropathic pain.
On glial activation, cytokines including TNF-α and IL-1β are secreted from glial cells and modulate neuronal activity as chemical mediators between glia and neurons. Specifically, TNF-α and IL-1β signaling have been shown to facilitate central glutamate transmission and potentiate synaptic strength (Beattie et al., 2002; Viviani et al., 2003; Pickering et al., 2005; Yang et al., 2005). The current literature implicates an important role of TNF-α and IL-1β in the peripheral nerve (Zelenka et al., 2005), the spinal cord (Ferreira et al., 1988; Lindenlaub et al., 2000; S. M. Sweitzer et al., 2001; Ohtori et al., 2004), or the trigeminal nucleus (Guo et al., 2007) in the genesis or maintenance of persistent pain depending on locally enhanced NMDAR functions (Bursztajn et al., 2004; Guo et al., 2007; Zhang et al., 2008). Our study further shows that the sustained elevation of TNF-α and IL-1β in the RVM occurred at 14 d after CCI. More importantly, the use of antagonists for TNF-α receptor and IL-1β receptor neutralize the action of these endogenous cytokines in the RVM and greatly attenuate mechanical hyperalgesia and allodynia at 14 d after CCI. Therefore, these data suggest that secreted TNF-α and IL-1β on astrocytic hyperactivation in the RVM are involved in supraspinal mechanisms related to the maintenance of neuropathic pain.
NMDAR-containing neurons are widely distributed in the RVM, and NMDAR-dependent descending pain facilitation contributes to the development of hyperalgesia after injury (Guo et al., 2006). We have now shown colocalization of cytokine receptors TNFR1 and IL-1RI with the NMDAR subunit NR1 in RVM neurons. Neutralizing these cytokines also completely blocked CCI-induced NR1 phosphorylation. Furthermore, recombinant TNF-α and IL-1β induced robust increases in NR1 phosphorylation in the RVM. Thus, TNF-α and IL-1β associated with glial hyperactivation may affect NMDAR function, similar to other gliotransmitters including chemokines, ATP, and nitric oxide (Pickering et al., 2005; Perea and Araque, 2006, 2007). Coupling of cytokine receptors to the NMDAR may involve several steps. Indeed, recent evidence indicates that intracellular signaling pathways related to Src family tyrosine kinase, PKC (protein kinase C), extracellular signal-regulated kinases (ERKs), PSD-95 (postsynaptic density-95), phospholipase C, and phospholipase A2 contribute to IL-1β-induced NMDAR phosphorylation (Viviani et al., 2003, 2006; Guo et al., 2007). Activation of intracellular signaling pathways involving neuronal pERK and glial p38 mitogen-activated protein (MAP) kinases in dorsal root ganglion, spinal dorsal horn, and the RVM contribute importantly to synaptic plasticity in central sensitization and the development of persistent pain (Zhuang et al., 2005; Katsura et al., 2006; Wei et al., 2006; Imbe et al., 2007, 2008; Kawasaki et al., 2008). We hypothesize that the secreted TNF-α and IL-1β enhance phosphorylation of NMDARs by binding to their respective receptors expressed on NMDAR-containing neurons and trigger the intracellular signaling cascades. In addition, both TNF-α and IL-1β also potentiate presynaptic glutamate signaling (Beattie et al., 2002; Viviani et al., 2003; Pickering et al., 2005). Collectively, cytokine TNF-α and IL-1β signaling mediates communication from glial hyperactivation to neuronal hyperexcitability in the RVM by coincidently promoting NMDARs and amplifying glutamate signaling. Such supraspinal glial–cytokine–neuronal interactions may be critical for the development of descending pain facilitation of neuropathic pain. The role of the MAP kinase signaling cascades in RVM glial cells and neurons in the pathogenesis of neuropathic pain also requires further study.
The present study demonstrated that nerve injury induced upregulation of TNFR1 and IL-1RI and enhanced phosphorylation of NR1 in RVM, and that microinjection of TNF-α and IL-1β in the RVM of normal rats evoked NMDAR-dependent descending pain facilitation, suggesting that descending facilitation may originate from hyperactivity of RVM neurons that express these cytokine receptors and NMDARs. However, the physiological properties of these RVM neurons are unknown. In the RVM, three populations of neurons have been identified based on the correlation of their action potential firing rates with nocifensive responses (Fields et al., 1991). On-cells are thought to promote nociception, whereas off-cells to inhibit nociception. Although the role of neutral cells remains unknown, studies suggest that they might be involved in pain modulation after inflammation (Montagne-Clavel and Oliveras, 1994; Miki et al., 2002). We now know that there are parallel descending facilitatory and inhibitory systems modulating spinal nociceptive transmission (Millan, 2002; Gebhart, 2004; Dubner, 2006). After tissue and nerve injury, not only the enhanced descending facilitation parallels enhanced descending inhibition from the RVM, but also the net facilitatory may become dominant, resulting in behavioral hyperalgesia and allodynia (Porreca et al., 2002). Although lesions of cells expressing μ-opioid receptors with dermorphin–saporin conjugate in the RVM do reverse later maintenance of hyperalgesia (Porreca et al., 2001; Burgess et al., 2002), μ-opioid receptors are found not only on on-cells (Heinricher et al., 1992), but also are located on serotonergic cells (Kalyuzhny et al., 1996) that are considered to be neutral cells in the RVM. Evidence also indicates that the majority of spinally projecting serotonergic neurons in the RVM respond to μ-receptor agonists (Marinelli et al., 2002; Zhang et al., 2006). At present, it is still difficult to attribute the net effect of descending modulation to a single class of RVM neuronal activity.
It is important to address the finding that fluorocitrate may lead to neuronal damage after application with large doses into cell cultures (Hassel et al., 1995), brain slices (Stone et al., 1990), or in vivo brain tissue (Paulsen et al., 1987; Hornfeldt and Larson, 1990; Largo et al., 1996) and even induces animal seizures (Willoughby et al., 2003). One reason may be that astrocytic inhibition may lead to accumulation of extracellular glutamate in the CNS (Conti and Weinberg, 1999). We examined possible neuronal damage induced by microinjection of fluorocitrate, and observed that fluorocitrate caused neuronal apoptosis and glial necrosis in the RVM in a dose-dependent manner, which occurred only when high doses (10 pmol) were used. Consistent with the present study, Fonnum et al. (1997) have reported that low doses of fluorocitrate result in selective and reversible glial disruption in the striatum, without any ultrastructural evidence of neuronal damage at synaptic sites. In our experiment, the lower doses of fluorocitrate (100 fmol) did not cause cell damage and also had no effect on function of astrocytes in the RVM of normal animals; however, the lower doses completely abolished upregulation of GFAP, S100β, TNF-α, and IL-1β levels in the RVM and mechanical hypersensitivity at 14 d after CCI. These findings verify that fluorocitrate inhibits CCI-induced astrocytic hyperactivity at low doses after focal application in vivo. Interestingly, long-lasting hyperactivation of astrocytes paralleled an initial upregulation of glutamate transporter-1 (GLT-1) that is expressed predominantly in astrocytes in the spinal cord at 1–3 d, and downregulation of spinal GLT-1 at 7–14 d after nerve injury (Sung et al., 2003; Tawfik et al., 2008; Wang et al., 2008). Propentofylline prevented the decrease in astrocytic GLT-1 expression in the spinal cord at 14 d after nerve injury (Tawfik et al., 2008). Future studies are necessary to examine temporal changes in GLT-1 expression in the RVM after nerve injury and whether the glial inhibitors change astrocytic GLT-1 expression after CCI-ION.
Footnotes
This work was supported by National Institutes of Health Grants DE18573, 11964, and 15374.
References
- Apkarian AV, Lavarello S, Randolf A, Berra HH, Chialvo DR, Besedovsky HO, del Rey A. Expression of IL-1β in supraspinal brain regions in rats with neuropathic pain. Neurosci Lett. 2006;407:176–181. doi: 10.1016/j.neulet.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces abnormal pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–5136. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursztajn S, Rutkowski MD, Deleo JA. The role of the N-methyl-d-aspartate receptor NR1 subunit in peripheral nerve injury-induced mechanical allodynia, glial activation and chemokine expression in the mouse. Neuroscience. 2004;125:269–275. doi: 10.1016/j.neuroscience.2004.01.024. [DOI] [PubMed] [Google Scholar]
- Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. 2007;11:223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Conti F, Weinberg RJ. Shaping excitation at glutamatergic synapses. Trends Neurosci. 1999;22:451–458. doi: 10.1016/s0166-2236(99)01445-9. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- Dorazil-Dudzik M, Mika J, Schafer MK, Li Y, Obara I, Wordliczek J, Przewłocka B. The effects of local pentoxifylline and propentofylline treatment on formalin-induced pain and tumor necrosis factor-alpha messenger RNA levels in the inflamed tissue of the rat paw. Anesth Analg. 2004;98:1566–1573. doi: 10.1213/01.ANE.0000113235.88534.48. [DOI] [PubMed] [Google Scholar]
- Dubner R. Descending modulatory circuitry in the initiation and maintenance of neuropathic pain. In: Campbell JN, Basbaum AI, Dray A, Dubner R, Dworkin RH, Sang CN, editors. Emerging strategies for treatment of neuropathic pain. Seattle: IASP; 2006. pp. 123–138. [Google Scholar]
- Dubner R, Ren K. Brainstem mechanisms of persistent pain following injury. J Orofac Pain. 2004;18:299–305. [PubMed] [Google Scholar]
- Elewa HF, Hilali H, Hess DC, Machado LS, Fagan SC. Minocycline for short-term neuroprotection. Pharmacotherapy. 2006;26:515–521. doi: 10.1592/phco.26.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin-1 as a potent hyperalgesia agent antagonized by a tripeptide analogue. Nature. 1988;334:698–700. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI, Heinricher MM. Central nervous system mechanisms of pain modulation. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Ed 5. New York: Elsevier Churchill Livingstone; 2006. pp. 125–142. [Google Scholar]
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F, Johnsen A, Hassel B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia. 1997;21:106–113. [PubMed] [Google Scholar]
- Gebhart GF. Descending modulation of pain. Neurosci Biobehav Rev. 2004;27:729–737. doi: 10.1016/j.neubiorev.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Guan Y, Terayama R, Dubner R, Ren K. Plasticity in excitatory amino acid receptor-mediated descending pain modulation after inflammation. J Pharmacol Exp Ther. 2002;300:513–520. doi: 10.1124/jpet.300.2.513. [DOI] [PubMed] [Google Scholar]
- Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K. Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for pain facilitation. J Neurosci. 2006;26:126–137. doi: 10.1523/JNEUROSCI.3686-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Westergaard N, Schousboe A, Fonnum F. Metabolic differences between primary cultures of astrocytes and neurons from cerebellum and cerebral cortex. Effects of fluorocitrate. Neurochem Res. 1995;20:413–420. doi: 10.1007/BF00973096. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Fields HL. Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience. 1992;48:533–543. doi: 10.1016/0306-4522(92)90400-v. [DOI] [PubMed] [Google Scholar]
- Hirose S, Umetani Y, Amitani M, Hosoi R, Momosaki S, Hatazawa J, Gee A, Inoue O. Role of NMDA receptors in the increase of glucose metabolism in the rat brain induced by fluorocitrate. Neurosci Lett. 2007;415:259–263. doi: 10.1016/j.neulet.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Hornfeldt CS, Larson AA. Seizures induced by fluoroacetic acid and fluorocitrate may involve chelation of divalent cations in the spinal cord. Eur J Pharmacol. 1990;179:307–313. doi: 10.1016/0014-2999(90)90170-b. [DOI] [PubMed] [Google Scholar]
- Imamura Y, Kawamoto H, Nakanishi O. Characterization of heat-hyperalgesia in an experimental trigeminal neuropathy in rats. Exp Brain Res. 1997;116:97–103. doi: 10.1007/pl00005748. [DOI] [PubMed] [Google Scholar]
- Imbe H, Okamoto K, Aikawa F, Kimura A, Donishi T, Tamai Y, Iwai-Liao Y, Senba E. Effects of peripheral inflammation on activation of p38 mitogen-activated protein kinase in the rostral ventromedial medulla. Brain Res. 2007;1134:131–139. doi: 10.1016/j.brainres.2006.11.091. [DOI] [PubMed] [Google Scholar]
- Imbe H, Kimura A, Okamoto K, Donishi T, Aikawa F, Senba E, Tamai Y. Activation of ERK in the rostral ventromedial medulla is involved in hyperalgesia during peripheral inflammation. Brain Res. 2008;1187:103–110. doi: 10.1016/j.brainres.2007.10.075. [DOI] [PubMed] [Google Scholar]
- Kalyuzhny AE, Arvidsson U, Wu W, Wessendorf MW. Mu-opioid and delta-opioid receptors are expressed in brainstem antinociceptive circuits: studies using immunocytochemistry and retrograde tract-tracing. J Neurosci. 1996;16:6490–6503. doi: 10.1523/JNEUROSCI.16-20-06490.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa J, Takeda M, Suzuki I, Kadoi J, Tsuboi Y, Honda K, Matsumoto S, Nakagawa H, Tanabe A, Iwata K. Mechanisms involved in modulation of trigeminal primary afferent activity in rats with peripheral mononeuropathy. Eur J Neurosci. 2006;24:1976–1986. doi: 10.1111/j.1460-9568.2006.05065.x. [DOI] [PubMed] [Google Scholar]
- Kovelowski CJ, Ossipov MH, Sun H, Lai J, Malan TP, Porreca F. Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain. 2000;87:265–273. doi: 10.1016/S0304-3959(00)00290-6. [DOI] [PubMed] [Google Scholar]
- Largo C, Cuevas P, Somjen GG, Martín del Río R, Herreras O. The effect of depressing glial function in rat brain in situ on ion homeostasis, synaptic transmission, and neuron survival. J Neurosci. 1996;16:1219–1229. doi: 10.1523/JNEUROSCI.16-03-01219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Mahoney JH, Milligan ED, Martin D, Maier SF, Watkins LR. Spinal cord glia and interleukin-1 do not appear to mediate persistent allodynia induced by intramuscular acidic saline in rats. J Pain. 2006;7:757–767. doi: 10.1016/j.jpain.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Lindenlaub T, Teuteberg P, Hartung T, Sommer C. Effects of neutralizing antibodies to TNF-alpha on pain-related behavior and nerve regeneration in mice with chronic constriction injury. Brain Res. 2000;866:15–22. doi: 10.1016/s0006-8993(00)02190-9. [DOI] [PubMed] [Google Scholar]
- Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Schnell SA, Wessendorf MW, Christie MJ. Rostral ventromedial medulla neurons that project to the spinal cord express multiple opioid receptor phenotypes. J Neurosci. 2002;22:10847–10855. doi: 10.1523/JNEUROSCI.22-24-10847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki K, Zhou QQ, Guo W, Guan Y, Terayama R, Dubner R, Ren K. Changes in gene expression and neuronal phenotype in brainstem pain modulatory circuitry after inflammation. J Neurophysiol. 2002;87:750–760. doi: 10.1152/jn.00534.2001. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Montagne-Clavel J, Olivéras JL. Are ventromedial medulla neuronal properties modified by chronic peripheral inflammation? A single-unit study in the awake, freely moving polyarthritic rat. Brain Res. 1994;657:92–104. doi: 10.1016/0006-8993(94)90957-1. [DOI] [PubMed] [Google Scholar]
- Moore PA. Bupivacaine: a long-lasting local anesthetic for dentistry. Oral Surg Oral Med Oral Pathol. 1984;58:369–374. doi: 10.1016/0030-4220(84)90325-6. [DOI] [PubMed] [Google Scholar]
- Newman EA. New role for astrocytes: regulation of synaptic transmission. Trends Neurosci. 2003;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- Nutile-McMenemy N, Elfenbein A, Deleo JA. Minocycline decreases in vitro microglial motility, beta(1)-integrin, and Kv1.3 channel expression. J Neurochem. 2007;103:2035–2046. doi: 10.1111/j.1471-4159.2007.04889.x. [DOI] [PubMed] [Google Scholar]
- Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain. 2006;7:816–822. doi: 10.1016/j.jpain.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine. 2004;29:1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- Owolabi SA, Saab CY. Fractlkine and minocycline alter neuronal activity in the spinal cord dorsal horn. FEBS Lett. 2006;580:4306–4310. doi: 10.1016/j.febslet.2006.06.087. [DOI] [PubMed] [Google Scholar]
- Paulsen RE, Contestabile A, Villani L, Fonnum F. An in vivo model for studying function of brain tissue temporarily devoid of glial cell metabolism: the use of fluorocitrate. J Neurochem. 1987;48:1377–1385. doi: 10.1111/j.1471-4159.1987.tb05674.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Amsterdam: Elsevier; 2005. [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A. Synaptic information processing by astrocytes. J Physiol Paris. 2006;99:92–97. doi: 10.1016/j.jphysparis.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Wei H, Hämäläinen MM. Lidocaine in the rostroventromedial medulla and the periaqueductal gray attenuates allodynia in neuropathic rats. Neurosci Lett. 1996;218:127–130. doi: 10.1016/s0304-3940(96)13136-0. [DOI] [PubMed] [Google Scholar]
- Piao ZG, Cho IH, Park CK, Hong JP, Choi SY, Lee SJ, Lee S, Park K, Kim JS, Oh SB. Activation of glia and microglial p38 MAPK in medullary dorsal horn contributes to tactile hypersensitivity following trigeminal sensory nerve injury. Pain. 2006;121:219–231. doi: 10.1016/j.pain.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-α on glutamateergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Polgár E, Hughes DI, Arham AZ, Todd AJ. Loss of neurons from laminas I–III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. J Neurosci. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the μ-opioid receptor. J Neurosci. 2001;21:5281–5288. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003a;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, Rutkowski MD, DeLeo JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain. 2003b;104:655–664. doi: 10.1016/S0304-3959(03)00138-6. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Ren K. An improved method for assessing mechanical allodynia in the rat. Physiol Behav. 1999;67:711–716. doi: 10.1016/s0031-9384(99)00136-5. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6. doi: 10.1016/s0304-3959(02)00368-8. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anesthesiol. 2008;21:570–579. doi: 10.1097/ACO.0b013e32830edbdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW. Cellular neuroplasticity mechanisms mediating pain persistence. J Orofac Pain. 2004;18:318–324. [PubMed] [Google Scholar]
- Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Stone EA, Sessler FM, Liu WM. Glial localization of adenylate-cyclase-coupled beta-adrenoceptors in rat forebrain slices. Brain Res. 1990;530:295–300. doi: 10.1016/0006-8993(90)91298-u. [DOI] [PubMed] [Google Scholar]
- Sugiyo S, Takemura M, Dubner R, Ren K. Trigeminal transition zone-rostral ventromedial medulla connection and facilitation of orofacial hyperalgesia after masseter inflammation in rats. J Comp Neurol. 2005;495:510–523. doi: 10.1002/cne.20797. [DOI] [PubMed] [Google Scholar]
- Sun S, Cao H, Han M, Li TT, Zhao ZQ, Zhang YQ. Evidence for suppression of electroacupuncture on spinal glial activation and behavioral hypersensitivity in a rat model of monoarthritis. Brain Res Bull. 2008;75:83–93. doi: 10.1016/j.brainresbull.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001;297:1210–1217. [PubMed] [Google Scholar]
- Sweitzer SM, Pahl JL, DeLeo JA. Propentofylline attenuates vincristine-induced peripheral neuropathy in the rat. Neurosci Lett. 2006;400:258–261. doi: 10.1016/j.neulet.2006.02.058. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, Nutile-McMenemy N, Marks A, Deleo JA. Role of astrocytic S100beta in behavioral hypersensitivity in rodent models of neuropathic pain. Neuroscience. 2006;140:1003–1010. doi: 10.1016/j.neuroscience.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Tawfik VL, Regan MR, Haenggeli C, Lacroix-Fralish ML, Nutile-McMenemy N, Perez N, Rothstein JD, DeLeo JA. Propentofylline-induced astrocyte modulation leads to alterations in glial glutamate promoter activation following spinal nerve transection. Neuroscience. 2008;152:1086–1092. doi: 10.1016/j.neuroscience.2008.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7687–7692. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG. Descending control of persistent pain: inhibitory or facilitatory? Brain Res Rev. 2004;46:295–309. doi: 10.1016/j.brainresrev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M. Interleukin-1 beta released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J Biol Chem. 2006;281:30212–30222. doi: 10.1074/jbc.M602156200. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos BP, Strassman AM, Maciewicz RJ. Behavioral evidence of trigeminal neuropathic pain following chronic constriction injury to the rat's infraorbital nerve. J Neurosci. 1994;14:2708–2723. doi: 10.1523/JNEUROSCI.14-05-02708.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite PME, Tracey DJ. Trigeminal sensory system. In: Paxinos G, editor. The nervous system. San Diego: Academic; 1995. pp. 705–724. [Google Scholar]
- Wang W, Wang W, Wang Y, Huang J, Wu S, Li YQ. Temporal changes of astrocyte activation and glutamate transporter-1 expression in the spinal cord after spinal nerve ligation-induced neuropathic pain. Anat Rec (Hoboken) 2008;291:513–518. doi: 10.1002/ar.20673. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56:148–169. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Dubner R, Ren K. Nucleus reticularis gigantocellularis and nucleus raphe magnus in the brain stem exert opposite effects on behavioral hyperalgesia and spinal Fos protein expression after peripheral inflammation. Pain. 1999;80:127–141. doi: 10.1016/s0304-3959(98)00212-7. [DOI] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby JO, Mackenzie L, Broberg M, Thoren AE, Medvedev A, Sims NR, Nilsson M. Fluorocitrate-mediated astroglial dysfunction causes seizures. J Neurosci Res. 2003;74:160–166. doi: 10.1002/jnr.10743. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 2005;1034:172–179. doi: 10.1016/j.brainres.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Zelenka M, Schäfers M, Sommer C. Intraneural injection of interleukin-1β and tumor necrosis factor-α into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005;116:257–263. doi: 10.1016/j.pain.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Zhang L, Sykes KT, Buhler AV, Hammond DL. Electrophysiological heterogeneity of spinally projecting serotonergic and nonserotonergic neurons in the rostral ventromedial medulla. J Neurophysiol. 2006;95:1853–1863. doi: 10.1152/jn.00883.2005. [DOI] [PubMed] [Google Scholar]
- Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, Berman BM, Lao L. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135:232–239. doi: 10.1016/j.pain.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke HR, Zielke CL, Baab PJ, Tildon JT. Effect of fluorocitrate on cerebral oxidation of lactate and glucose in freely moving rats. J Neurochem. 2007;101:9–16. doi: 10.1111/j.1471-4159.2006.04335.x. [DOI] [PubMed] [Google Scholar]