

Abstract
With the numerous reports of new technologies and biomarkers reported in the literature, it may be surprising that there are not an equal number of new products available to the clinical diagnostic laboratory. Powerful potential tools such as protein microarrays and MS patterns have been extensively published yet commercialization and acceptance of these technologies has yet to happen. The reasons for this are a combination of industry risk avoidance, academic focus on discovery, and a lack of appreciation for the high standards and regulation that surrounds the clinical diagnostic laboratory. The development and validation of a new technology or biomarker ensures that a test is reproducible, controllable, and has a defined accuracy and clinical predictive result but this information is only obtained through somewhat mundane but necessary experimental work. The use of design of experiment principles helps to define material parameters to ensure performance. The organization and documentation of this work through a quality system is both mandated and practical. All of this must be done before a test can reach the market with the safety and effectiveness review of regulatory agencies.
Keywords: Biomarker development, FDA filing
1 Introduction
The naïve view of “translational research” is that once a technology or biomarker has been discovered, all it needs is validation through a clinical trial and it is ready to be deployed for use in patients. This has led to false hope, incredible hype by the media but disappointment on the part of patients because many promising technologies fail to materialize within the predicted timeframes [1]. The main reason for the discrepancy between perception and reality is that safety and effectiveness must be assured, and this is far beyond the simple characterization of a test or therapy. It has been recognized that translational research is important, and indeed the Journal of Laboratory and Clinical Medicine changed its name to Translational Research in 2006 [2, 3]. However, the processes of delivering discoveries from discovery to patient benefit seem to be vague beyond the academic publication of findings [3]. Having an additional research group confirm findings has been the measure of reproducibility but this can neither prove nor confirm reproducibility without a detailed investigation of the limits of the procedure reagents and any associated instrumentation. The measure of academic success is still publication [4] while the measure of industrial success is generation of products. Between the innovation factor needed for publication and the risk-aversion of industry lies the area of largely unfunded and unappreciated development. It is this area that gives the ability of discoveries to move from discovery to a validated and clinically useful technology or biomarker.
2 Rigor
There is usually the most excitement when a new potential biomarker or technology is discovered and first published. For example, there have been numerous potential biomarkers and even combinations of biomarkers that have been described for ovarian cancer detection [5-8]. While some of these have been known for several years, none have yet been approved by the FDA for use as an aid in diagnosing ovarian cancer. Biomarkers such as osteopontin and prolactin are well defined in terms of testing procedures yet their use in ovarian cancer diagnosis and monitoring has not been confirmed. It has been proposed that candidate biomarkers are eliminated by a variety of factors while in the pipeline [9]. In much the same way as drug candidates are eliminated, biomarkers are eliminated during the process of screening with ever increasing numbers of samples when the sensitivity and specificity become more clearly defined. This has certainly been the case with ovarian cancer markers to date. The only biomarker that has been used in the clinical setting is the CA125 assay. While it has been approved for several years its use is approved only for managing disease, not as a diagnostic tool.
Development of any biomarker or technology requires an understanding of the limits of each process, reagent, instrument, software, or associated materials and a mechanism to control for these through specifications and quality measures [10]. A pipeline for the development of a biomarker or a new technology into a diagnostic can be found in Fig. 1. This shows the steps that are taken in the stabilization, characterization, and development of specifications for the control of a new device which is different than that of a pharmaceutical [11]. The general approach to starting with a new biomarker using a defined technology is usually a reproducibility study. Here, known antigen or control material would be used in a design of experiments (DOEs) study where signal intensity or S/N could be used as the response. A DOE is a structured, organized method for determining the relationship between factors (Xs) affecting a process and the output of that process (Y) (http://www.isixsigma.com/dictionary/Design_of_Experi ments_-_DOE-41.htm). Careful planning and execution of the study is necessary in order to get results that are meaningful. The planning of the study and analysis of the results is often done using commercially available software. Through these studies, interactions with different factors can be detected that would otherwise go undetected if only one variable were tested and others were held constant [12]. Once this is completed, the system needs adaptation to clinical samples with the process being repeated. Gradually, there will be a host of experiments done do determine specifications with limits for everything from reagent buffers to instrument settings. All of these will be defined in controlled documents and reviewed before any formal trial is done to validate the marker.
Figure 1.

Diagnostic product pipeline building up to release to the clinical laboratory. Formal design control begins at the second stage.
New technologies add an additional layer of complexity. This is especially true of technologies that involve multivariate index assays for which the FDA has issued a guidance document (http://www.fda.gov/cdrh/oivd/guidance/1610.html). Technologies such as microarrays tend to be modifications of existing techniques and therefore have a base of knowledge and information that needs to be provided. The complexity of a microarray is the development of each individual “assay” and then the incorporation of multiple assays into a single array. Control over array manufacturing and final performance of tests is critical. These issues have been tackled and are outlined in the FDA guidance document for multivariate index assays.
Newer techniques such as MS patterns have no current development history. These were first reported in 2002 [13-15] and were greeted with both great expectations and skepticism [16, 17]. At least part of the reasons for some researchers not being able to reproduce results were the lack of understanding of the technology and the factors that could influence robustness. What was needed was an understanding of the test principle, the limits of reagents, instruments, procedures, and software that could result in technology failure or drift. Some of the high level work was done to elucidate the test principle [18]. A second study demonstrated reproducibility but through controlling factors tightly rather than understanding and defining failure limits for all aspects of the technology [19]. While this approach did result in a demonstration of reproducibility, the methods used were not practical for deployment to the clinical laboratory setting where robustness must accompany reproducibility.
More recent studies on reproducibility have raised even greater challenges in bringing these technologies to the clinical laboratory. One study described the use of high resolution SELDI to discover a pattern that would be useful in the early detection of breast cancer [20]. In this study, serum from patients from clinics in Italy were collected, processed, and frozen. They were then shipped to a single laboratory for the analysis by SELDI and a rigorous pattern selection method was used to generate a pattern that recognized early stage I breast cancer. The same clinics then collected further samples over a 14 month period and these were submitted as unknown samples. The sensitivity and specificity of the pattern held up over this relatively long time period. However, it required highly developed procedures, robotics to perform the test and an environmentally controlled laboratory (both temperature and humidity) in order to achieve these results. Thus, the method was reproducible but impractical to deploy as a routine method. Furthermore, the samples were all collected and processed at a limited number of sites thereby reducing the possible preanalytical variation that could occur.
The possibility of misleading results caused by such sample bias has been documented in recent publications. McLerran et al. [21, 22] described a study to confirm a proteomic pattern as a potential early detection system for prostate cancer. The results of their study indicated that, although the controlled the laboratory aspects of the test, the pattern did not reliably detect prostate cancer [22]. They then described what they saw as the root of the problem – sample bias. Their cancer samples were older and were handled differently from their normal samples [21]. Although sample bias has been described before, it was noted that the issues with new technology such as proteomic patterns are likely to be greater because of the simultaneous measurement of multiple forms of proteins and peptide fragments [23]. This has led to the suggestion that more familiar technologies or hybrid technologies be used as a bridge in order to avoid the pitfalls of these complex and unknown issues [24, 25]. The suggestion is that proteomic patterns be utilized as discovery tools but the translation to the clinical laboratory be done utilizing immunoassays or immuno MS techniques where issues such as sample bias would be better understood and where laboratory to laboratory reproducibility issues would not be subject to such rigorous controlled conditions.
As with any new technology there are a host of other unknown or undefined factors that can influence results. These include not only factors such as sample procurement and handling, but also analytical methods, reagents, and postanalytical factors including software development and validation [26]. Table 1 summarizes some of these factors but this list will be expanded depending on the actual instrumentation, technique, and analytical software that are to be used. While this example of mass spectrometric patterns show an impressive list of factors to be defined, an equally long list can also be generated for microarrays, gene arrays, and other new technologies that have been discovered [27, 28].
Table 1.
Recommended practices for clinical applications of protein profiling by MALDI TOF MS
| Preanalytical | Evaluate optimum patient preparation |
| Identify optimum procedures for specimen collection and processing | |
| Analyze specimen stability | |
| Develop criteria for specimen acceptability | |
| Analytical | Prepare calibrators for mass, resolution, and detector sensitivity |
| Use internal standards | |
| Automate specimen preparation | |
| Optimize methods to yield highest possible signals for peaks of interest | |
| Identify sequences of peaks of interest | |
| Develop calibration materials for components of interest | |
| QC: prepare/identify at least two concentrations of control material | |
| Evaluate reproducibility (precision) | |
| Evaluate LOD and linearity | |
| Evaluate reference intervals | |
| Evaluate interferences such as hemolysis, lipemia, renal failure, acute-phase responses | |
| Develop materials or programs for external comparison/proficiency testing of analyzers | |
| Postanalytical | Analyze each spectrum to identify peaks before applying diagnostic algorithms |
| Develop criteria for the acceptability of each spectrum based on peak characteristics | |
| Use peaks rather than raw data as the basis for diagnostic analysis | |
| Use caution in interpretation of peaks with m/z<1200 | |
| Select peaks with high intensities and sample stability for diagnosis | |
| Select approximately equal numbers of peaks that increase and decrease in intensity as diagnostic discriminators | |
| In developing a training set for diagnosis, careful clinical classification of patients is essential | |
| Clinical validity depends on having a typical rather than highly selected population of patients | |
| The number of training specimens should be at least ten times the number of measured values | |
| Any clinical application should use a fixed training set and algorithm for analysis | |
| Any analysis should provide a numerical value | |
| Diagnostic performance should be evaluated with ROC curves to select cutoffs | |
| A sensitivity analysis should be performed of the necessary precision for accurate diagnostic performance | |
| There should be QC procedures for daily verification of software performance |
Adapted from Hortin [26].
3 Regulation
All in vitro diagnostic devices are regulated Office of In Vitro Diagnostic device Evaluation and Safety (OIVD) under the Food, Drug, and Cosmetic Act [29]. There are two basic classifications for products that are commercialized: products approved under a Premarket Approval (PMA) and those marketed after receiving Premarket Notification (510(k)). The term “FDA Approval” is often wrongly used for the correct term “FDA notification” when referring to a premarket notification or 510(k). The FDA has recognized significant challenges in the review of new biomarkers and technology. As a result they have responded with a number of regulatory tools and processes to encourage communication between developers and the agency to provide the most rapid pathway possible without compromising safety and effectiveness as shown in Fig. 2 [30]. The rules governing how to notify FDA of intent to market have been reviewed as well in order to provide clarity to complex technologies. For example, a draft guidance document was issued outlining multivariate index assays (http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?FR=820.30) that provide information for those technologies and assays that involve a panel approach such as the recently cleared Mammaprint® test, protein microarrays, and MS patterns.
Figure 2.

The approval process for devices under the 510(k) mechanism. From http://www.fda.gov/cdrh/ode/parad510.html (1998).
The FDA regulations are heavily focused on product labeling (http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?fr=801). It is this part of the code that mandates the device's intended use statement that clearly states the limitations of what a device's performance characteristics are in the validation trials. For example, it is often thought that tumor markers are used to screen for disease. In fact, with the exception of PSA, markers are only to be used for the monitoring of diagnosed disease. The clinical trials used in devices are specifically designed to support the intended use statement and the results must demonstrate that the device performs as described. The performance characteristics of the device may not confirm a desired use such as screening because of poor sensitivity and specificity. It is therefore not supported by either the manufacturer or the FDA for that use and the intended use statement must reflect only what has been validated.
The intended use also drives and defines the development activities surrounding a device. The development of a device is subject to oversight by the FDA and this is outlined in 21CFR820.30 under design control (http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?FR=820.30). While the principles of a quality system and design control are well understood by manufacturers, these are often overlooked by or unknown to basic researchers. It is the elements of design control that dictate the solidifying of a test or technology design specifications (design input) and the review of data to confirm that the performance meets these requirements (design output). An example of the workflow for design control can be found in Fig. 3. The entire process can be easily summed up; you say what you are going to do, you do it, and you prove that you did it. The process need not be burdensome and can assure that the technology is understood, controlled, defined, and validated.
Figure 3.
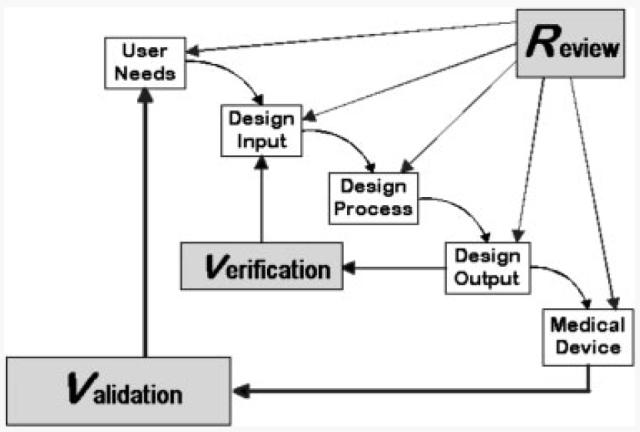
Design review process as outlined by FDA (http://www.fda.gov/cdrh/comp/designgd.html).
Tests that are developed, manufactured, validated, and performed by an individual laboratory are not subject to FDA notification. However, the laboratory must comply with the Clinical Laboratory Improvement Amendments of 1988 (CLIA88). The test or technology must have records of validation and control measures must be in place. However, critical active ingredients used in the test may be required to have been produced under a Quality System as described above and must be listed with the FDA. These are called analyte-specific reagents (ASR's). In addition, this test may neither be run by additional laboratories nor sold to other laboratories without FDA notification.
Two other classes of devices are Research Use Only Devices (RUO's) and Investigational Use Only devices (IUO). RUO devices are the class that most new technologies and biomarkers fall into at the publication stage. The tests are only to be used to confirm a hypothesis or new intended use and cannot be used in any way as a reporting result to clinicians. While data from these studies may be used for publication purposes, the data cannot be used for diagnostic purposes. The second group, IUO devices, is used to gather data to support an intended use for a device that will be filed with the FDA. These devices have been through the development phase and are characterized with the performance data to be gathered in a formal trial. Initial performance has been completed but the final data will confirm and support the biomarker or device's use as a diagnostic. The regulatory rules for informed consent and review by an institutional review board apply in this situation [29].
The regulation and required information for filing a new technology or biomarker with the FDA may seem to be overwhelming at times. The FDA has recognized this and has responded with an initiative, the Critical Path, designed to help ensure that the regulatory process proceeds in a timely fashion [30, 31]. An update on this initiative published in 2006 can be found at http://www.fda.gov/oc/initiatives/criti calpath/reports/opp_report.pdf. Regulations for new technologies and biomarkers are in place both from the FDA and from CLIA88. These are to ensure that devices are safe and effective before they can be used in the clinical diagnostic laboratory setting. While some may view these regulations as keeping devices away from patients, they are in place to prevent a rush to use before they are understood and thoroughly characterized.
4 Reality
New technology and biomarkers or panels of biomarkers for diagnosis of diseases such as cancer are published on a regular basis. Many of these are radical in design, concept, or new protein discovery. The reality is that few make it through the rigors of development, validation, and verification and into the commercial market. There have been attempts to develop tests without the expense and time required for validation through the regulatory process but these have been unsuccessful [32]. Such was the case with the Roche Ampli-Chip™ where the manufacturer believed this to be an ASR not requiring premarket notification. In the end, the FDA classified this as a class II device and it was eventually filed (http://www.fda.gov/cdrh/pdf4/k042259.pdf).
The recent clearance of the MammaPrint in June of 2007 has heralded a new era in diagnostic testing (http://www.fda.gov/cdrh/pdf4/k042259.pdf). This profiling test was first described in the literature in 2002 [33] and showed the promise of diagnostic tool to predict outcome of disease in breast cancer. The company, Agendia BV, did the proper development to ensure that the test was reproducible and robust before performing the validation studies necessary to file with the FDA [34]. The test was engineered from an array containing approximately 25 000 60-mer oligonucleotides to a more limited custom array with 1900 60-mer probes including the 70 prognostic classifier genes that could be used in a high throughput test and the results of this effort was published in 2006. The following are examples of the type of studies done to successfully deliver this test to the clinical laboratory. The test results of the newly engineered test were compared to the originally reported results with good correlation (Fig. 4). Reproducibility was demonstrated in a two experiment studies with a correlation of 0.995 (Fig. 5) and the robustness was demonstrated in a 12 months study of classification of three samples (Fig. 6). Then the clinical studies were performed to complete the information needed for notification and a summary of this can be found at http://www.fda.gov/cdrh/pdf7/K070675.pdf. Furthermore, only licensed facilities are currently permitted to run the test with the manufacturer qualifying these sites. Currently in US, only the Molecular Profiling Institute in Phoenix can use the test. This ensures that this complex but important test is run properly especially in the first year of testing.
Figure 4.
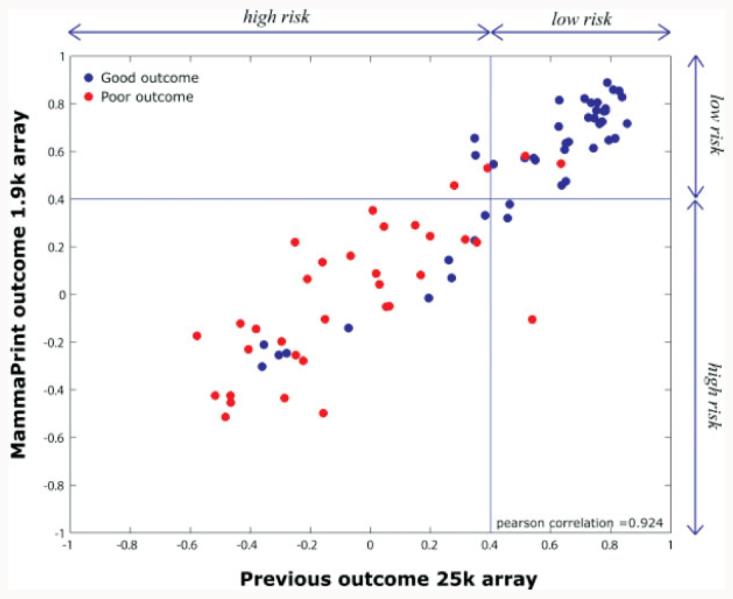
Comparison of results of original study with research array and the developed MammaPrint array. From ref. [34].
Figure 5.
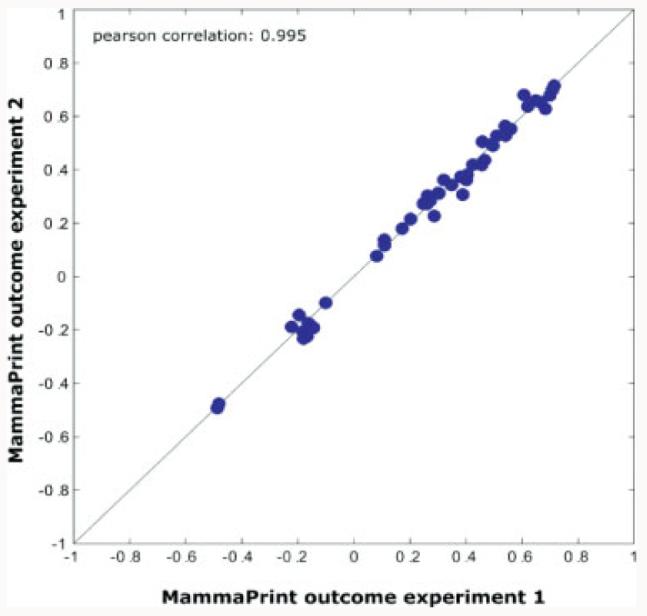
Comparison of outcomes using two different MammaPrint experiments and a set of samples. The correlation demonstrates a correlation of 0.995 over a good range of outcomes. From ref. [34].
Figure 6.
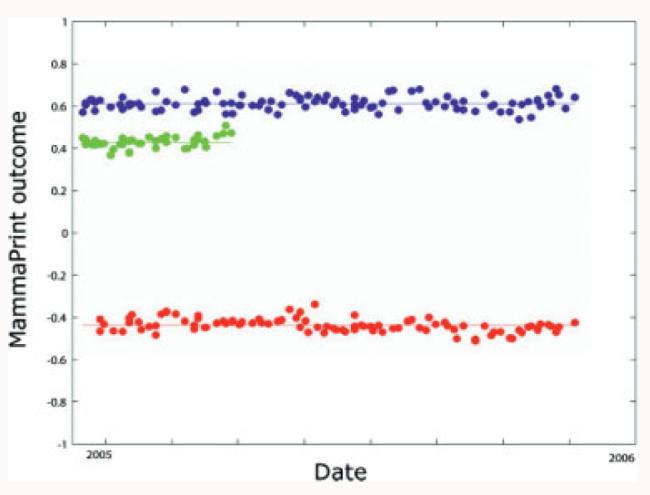
Results of three samples run on several time points over a 12 month period. The outcomes are demonstrated to be reproducible over the period. From ref. [34].
The road to a reproducible and sufficiently robust test for a single site laboratory setting has been long. Indeed, many of the problems that are now being encountered with proteomic technologies and protein microarray platforms have also been obstacles to genomic technologies. Despite considerable investment of both time and money, only three tests (MammaPrint, Oncotype DX and Avirara DX H/I) are in use and only one (MammaPrint) has been FDA cleared. All three of these tests are done in limited laboratory settings and are not available for use in routine clinical laboratories [35]. This speaks to the issues that surround the limited robustness and need for extensive quality control not feasible in routine laboratories. Presumably continued development of these technologies will result in the more widespread dissemination of these tests.
Despite the thorough and expensive process to develop and validate a technology or biomarker, success resulting in positive patient outcome is dependent on acceptance in the clinical laboratory and the medical community. In a recent article, the American Society of Clinical Oncology reviewed the performance of the MammaPrint test and concluded that this profiling did classify patients with very good or very poor prognosis. However, they also concluded that the results of additional studies would be required before they could definitively recommend how to use this assay in clinical practice [36].
New technologies and biomarkers are being discovered and reported with ever increasing frequency. The complexity of the technology in some cases helps to uncover new potential biomarkers. Once reported, the road to a fully developed and validated clinical test is long. The often overlooked development phase must be completed before any new technology or biomarker can be determined to be clinically relevant or of no value. Without this, it is possible to waste time and effort in a clinical trial and discard a good potential test or be mislead into concluding that a potential test is useful. Regulations are in place to ensure that tests are properly developed and validated. The review process is being adapted to address the issues that are raised by some of these new tests. This will help to make the path to the clinical laboratory and patient benefit as rapid as possible without compromising safety or effectiveness.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This Research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
The author has declared no conflict of interest.
5 References
- 1.Check E. Proteomics and cancer: Running before we can walk? Nature. 2004;429:496–497. doi: 10.1038/429496a. [DOI] [PubMed] [Google Scholar]
- [2.Geraghty J. Adenomatous polyposis coli and translational medicine. Lancet. 1996;348:422. doi: 10.1016/S0140-6736(05)64535-7. [DOI] [PubMed] [Google Scholar]
- 3.Crist TB, Schafer AI, Walsh RA. Translating basic discoveries into better health care: The APM's recommendations for improving translational research. Am. J. Med. 2004;116:431–434. doi: 10.1016/j.amjmed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence PA. The mismeasurement of science. Curr. Biol. 2007;17:R583–R585. doi: 10.1016/j.cub.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Kim JH, Skates SJ, Uede T, Wong KK, et al. Osteopontin as a potential diagnostic biomarker for ovarian cancer. JAMA. 2002;287:1671–1679. doi: 10.1001/jama.287.13.1671. [DOI] [PubMed] [Google Scholar]
- 6.Gorelik E, Landsittel DP, Marrangoni AM, Modugno F, et al. Multiplexed immunobead-based cytokine profiling for early detection of ovarian cancer. Cancer Epidemiol. Biomarkers. Prev. 2005;14:981–987. doi: 10.1158/1055-9965.EPI-04-0404. [DOI] [PubMed] [Google Scholar]
- 7.Mor G, Visintin I, Lai Y, Zhao H, et al. Serum protein markers for early detection of ovarian cancer. Proc. Natl. Acad. Sci. USA. 2005;102:7677–7682. doi: 10.1073/pnas.0502178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z, Bast RC, Jr., Yu Y, Li J, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004;64:5882–5890. doi: 10.1158/0008-5472.CAN-04-0746. [DOI] [PubMed] [Google Scholar]
- 9.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006;24:971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 10.Krouwer JS. Assay Development and Evaluation – A Manufacturer's Perspective. AACC Press; Washington, DC: 2002. [Google Scholar]
- 11.Phillips KA, Van Bebber S, Issa AM. Diagnostics and biomarker development: Priming the pipeline. Nat. Rev. Drug Discov. 2006;5:463–469. doi: 10.1038/nrd2033. [DOI] [PubMed] [Google Scholar]
- 12.Lim M, Ye H, Panoskaltsis N, Drakakis EM, et al. Intelligent bioprocessing for haemotopoietic cell cultures using monitoring and design of experiments. Biotechnol. Adv. 2007;25:353–368. doi: 10.1016/j.biotechadv.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 14.Petricoin EF, III, Ornstein DK, Paweletz CP, Ardekani A, et al. Serum proteomic patterns for detection of prostate cancer. J. Natl. Cancer Inst. 2002;94:1576–1578. doi: 10.1093/jnci/94.20.1576. [DOI] [PubMed] [Google Scholar]
- 15.Kozak KR, Amneus MW, Pusey SM, Su F, et al. Identification of biomarkers for ovarian cancer using strong anion-exchange ProteinChips: Potential use in diagnosis and prognosis. Proc. Natl. Acad. Sci. USA. 2003;100:12343–12348. doi: 10.1073/pnas.2033602100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baggerly KA, Morris JS, Coombes KR. Reproducibility of SELDI-TOF protein patterns in serum: Comparing datasets from different experiments. Bioinformatics. 2004;20:777–785. doi: 10.1093/bioinformatics/btg484. [DOI] [PubMed] [Google Scholar]
- 17.Diamandis EP. Analysis of serum proteomic patterns for early cancer diagnosis: Drawing attention to potential problems. J. Natl. Cancer. Inst. 2004;96:353–356. doi: 10.1093/jnci/djh056. [DOI] [PubMed] [Google Scholar]
- 18.Mehta AI, Ross S, Lowenthal MS, Fusaro V, et al. Biomarker amplification by serum carrier protein binding. Dis. Markers. 2003;19:1–10. doi: 10.1155/2003/104879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semmes OJ, Feng Z, Adam BL, Banez LL, et al. Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer: I. Assessment of platform reproducibility. Clin. Chem. 2005;51:102–112. doi: 10.1373/clinchem.2004.038950. [DOI] [PubMed] [Google Scholar]
- 20.Belluco C, Petricoin EF, Mammano E, Facchiano F, et al. Serum proteomic analysis identifies a highly sensitive and specific discriminatory pattern in stage 1 breast cancer. Ann. Surg. Oncol. 2007;9:2470–2476. doi: 10.1245/s10434-007-9354-3. [DOI] [PubMed] [Google Scholar]
- 21.McLerran D, Grizzle WE, Feng Z, Bigbee WL, et al. Analytical validation of serum proteomic profiling for diagnosis of prostate cancer: Sources of sample bias. Clin. Chem. 2008;54:44–52. doi: 10.1373/clinchem.2007.091470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLerran D, Grizzle WE, Feng Z, Thompson IM, et al. SELDI-TOF MS whole serum proteomic profiling with IMAC surface does not reliably detect prostate cancer. Clin. Chem. 2008;54:53–60. doi: 10.1373/clinchem.2007.091496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banks RE. Preanalytical influences in clinical proteomic studies: Raising awareness of fundamental issues in sample banking. Clin. Chem. 2008;54:6–7. doi: 10.1373/clinchem.2007.097667. [DOI] [PubMed] [Google Scholar]
- 24.Kohn EC, Azad N, Annunziata C, Dhamoon AS, Whiteley G. Proteomics as a tool for biomarker discovery. Dis. Markers. 2007;23:411–417. doi: 10.1155/2007/967023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liotta LA, Petricoin EF. Putting the “bio” back into biomarkers: Orienting proteomic discovery toward biology and away from the measurement platform. Clin. Chem. 2008;54:3–5. doi: 10.1373/clinchem.2007.097659. [DOI] [PubMed] [Google Scholar]
- 26.Hortin GL. Can mass spectrometric protein profiling meet desired standards of clinical laboratory practice? Clin. Chem. 2005;51:3–5. doi: 10.1373/clinchem.2004.043281. [DOI] [PubMed] [Google Scholar]
- 27.Shi L, Tong W, Fang H, Scherf U, et al. Cross-platform comparability of microarray technology: Intra-platform consistency and appropriate data analysis procedures are essential. BMC Bioinformatics. 2005;6:S12. doi: 10.1186/1471-2105-6-S2-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuscoe JC, Tong W, Shi L. QA/QC issues to aid regulatory acceptance of microarray gene expression data. Environ. Mol. Mutagen. 2007;48:349–353. doi: 10.1002/em.20293. [DOI] [PubMed] [Google Scholar]
- 29.Mansfield E, O'Leary TJ, Gutman SI. Food and Drug Administration regulation of in vitro diagnostic devices. J. Mol. Diagn. 2005;7:2–7. doi: 10.1016/S1525-1578(10)60002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hackett JL, Gutman SI. Introduction to the Food and Drug Administration (FDA) regulatory process. J. Proteome Res. 2005;4:1110–1113. doi: 10.1021/pr050059a. [DOI] [PubMed] [Google Scholar]
- 31.Aziz KJ. The FDA's Critical Path Initiative for Medical Products. J. Clin. Ligand Assay. 2006;29:171–176. [Google Scholar]
- 32.Kling J. Roche's microarray tests US FDA's diagnostic policy. Nat. Biotechnol. 2003;21:959–960. doi: 10.1038/nbt0903-959. [DOI] [PubMed] [Google Scholar]
- 33.van't Veer LJ, Dai H, van de Vijver MJ, He YD, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 34.Glas AM, Floore A, Delahaye LJ, Witteveen AT, et al. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genom. 2006;7:278. doi: 10.1186/1471-2164-7-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchionni L, Wilson RF, Wolff AC, Marinopoulos S, et al. Systematic review: Gene expression profiling assays in early-stage breast cancer. Ann. Intern. Med. 2008;148:358–369. doi: 10.7326/0003-4819-148-5-200803040-00208. [DOI] [PubMed] [Google Scholar]
- 36.Harris L, Fritsche H, Mennel R, Norton L, et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. 2007;25:5287–5312. doi: 10.1200/JCO.2007.14.2364. [DOI] [PubMed] [Google Scholar]