

Abstract
Cocaine addiction is characterized by an impaired ability to develop adaptive behaviors that can compete with cocaine seeking, implying a deficit in the ability to induce plasticity in cortico-accumbens circuitry critical for regulating motivated behavior. RWe found that rats withdrawn from cocaine self-administration had a marked in vivo deficit in the ability to develop long-term potentation (LTP) and depression (LTD) in the nucleus accumbens core subregion following stimulation of prefrontal cortex. N-acetylcysteine treatment prevents relapse in animal models and craving in humans by activating cystine-glutamate exchange and thereby stimulating extrasynaptic metabotropic glutamate receptors (mGluR). N-acetylcysteine treatment restored the ability to induce LTP and LTD by indirectly stimulating mGluR2/3 and mGluR5, respectively. Cocaine self-administration induces metaplasticity that inhibits the further induction of synaptic plasticity, and this impairment can be reversed by N-acetylcysteine, a drug that also prevents relapse.
INTRODUCTION
Cocaine addiction is a learned behavior characterized by compulsive drug seeking and high vulnerability to relapse, even after periods of prolonged abstinence1. The impaired ability of cocaine addicts to regulate drug seeking is thought to be rooted in long term neuroadaptations in prefrontal glutamatergic input to basal ganglia motor circuitry, and animal studies have brought particular focus to glutamatergic innervation of the core compartment of the nucleus accumbens (NAcore) by prelimbic prefrontal cortex (PFC)2. Potentially important cocaine-induced cellular adaptations in glutamatergic input to the NAcore have been identified, including changes in presynaptic regulation of glutamate release2, dendritic spine morphology3 and postsynaptic proteins that regulate spine morphology4 and integrate glutamate signals5. On the other hand, there is an absence of long-term depression (LTD) after chronic cocaine6, suggesting cocaine-induced metaplasticity. Metaplasticity is the change in the ability to generate synaptic plasticity, where a priming activity (e.g. chronic cocaine administration) alters the capacity of a subsequent high (HFS) or low frequency stimulation (LFS) protocol to induce subsequent neuroplasticity, such as long-term potentiation (LTP) or long term depression (LTD)7. Thus, tissue slices were used to demonstrate that prolonged withdrawal from self-administered cocaine impaired the induction of LTD at excitatory synapses in NAcore medium spiny neurons (MSN)6. Lending behavioral relevance to the observation, blocking LTD at MSN glutamatergic synapses inhibits amphetamine induced locomotor sensitization, an animal model of psychostimulant-induced plasticity8. Based upon these data, it has been proposed that chronic cocaine depresses PFC-accumbens synapses, thereby occluding further synaptic depression after low frequency stimulation (LFS) protocols9. This view is also supported by the finding that chronic methamphetamine induces chronic presynaptic depression at the excitatory striatal synapses10.
A pre-existing LTD-like state at the PFC-accumbens synapses in animals withdrawn from chronic cocaine is challenged by recent reports showing increased surface expression of AMPA receptors, increased ratio of AMPA to NMDA synaptic currents, increased EPSCs frequency and amplitude5,11; all of which suggest LTP-like potentiation at these synapses. In this study we hypothesized that after prolonged withdrawal from chronic cocaine self administration, cocaine-induced metaplasticity at the excitatory synapses in the NAcore attenuates further potentiation as well as depression by HFS and LFS, respectively. In addition, if cocaine-induced metaplasticity is relevant to relapse vulnerability, a drug that inhibits relapse and normalizes synaptic glutamate transmission between the PFC and NAcore should reverse the observed cocaine induced metaplasticity. N-acetylcysteine reduces relapse in the reinstatement animal model of drug-seeking as well as conditioned reactivity to drug cues in cocaine addicts, and reverses several cocaine-induced neuroadaptations in glutamate transmission12-16. Thus, we hypothesized that N-acetylcysteine (NAC) would reverse cocaine-induced metaplasticity, and restore the ability to induce LTP and LTD.
RESULTS
Field Potentials Evoked in the NAcore are field EPSPs
To characterize cocaine-induced metaplasticity, in vivo recordings of extracellular field potentials were evoked in the NAcore by stimulating the PFC in anesthetized rats extinguished from cocaine self-administration for at least 3 weeks (Fig. 1a). We employed an in vivo protocol because of the important role identified for PFC afferents to the NAcore in animal models of relapse and neuroimaging studies in addicts2, 17. Also, this preparation permits isolation of PFC from amygdala, hippocampal or thalamic glutamatergic afferents to the NAcore, thereby allowing a circuit-level analysis of cocaine-induced metaplasticity.
Figure 1. Characterization of nucleus accumbens field potentials evoked from the prefrontal cortex.

a-b, Illustration of the experimental protocol showing the stimulation electrode in the PFC and recording electrode in the NAcore. Stimulation of the PFC was targeted to the ventral prelimbic cortex (PrL; circle). The field potentials attained maximum amplitude when stimulation in PFC was delivered 3.5 mm ventral to the surface of the brain. Representative traces (average of 10 consecutive field potentials) are shown at different depths of stimulation in the PFC; arrow indicates the stimulation artifact. Current ejection of pontamine blue spot marked the recording site in the NAcore. ac- anterior commissure, Cg-cingulate cortex, IL- infralimbic cortex, NAshell- accumbens shell. c, Left, sample trace demonstrating that recorded fields are sensitive to CNQX (100 μM) and TTX (10 μM), but independent of GABAergic currents (bicuculline, Bic, 100 μM). Right, representative traces (average of 5 consecutive field potentials) before (1) and after (2) CNQX and a subtraction of the 2 traces (1-2) equivalent to the AMPA receptor dependent current. d, Effect of different drugs on field amplitude measured as an average of 5 min bins, immediately before and 25 min after drug perfusion. CNQX (100 μM, p=0.002), bicuculline (100 μM), picrotoxin (1.0 mM), TTX (10 μM, p= 0.029), compared to baseline using a Mann-Whitney test. n is shown in the bars. In all figures, scale bars correspond to 0.2 mV, 10 msec, and error bars indicate mean ± s.e.m.
Since previous characterizations of PFC evoked field potentials in the NAcore were performed in tissue slices, we first pharmacologically and anatomically characterized the field potentials recorded in vivo (Fig. 1). Field potentials restricted to the dorsomedial NAcore (Fig. 1b-c) showed large amplitude, simple short latency sink currents comparable to the well-characterized fields recorded in the dendritic CA1 area of the hippocampus. This probably reflects a relatively organized, non-random spatial orientation of MSN dendritic fields in this quadrant of the NAcore18. Outside of this region of the NAcore, field potentials were generally lower in amplitude and more complex in appearance. Corresponding to the previously reported topography of PFC innvervation of the accumbens19, field potential amplitude recorded in the NAcore was maximal when the ventral prelimbic PFC was stimulated (Fig. 1b). The field potentials were reversibly blocked by the administration of the glutamate receptor antagonist CNQX or the voltage-dependent Na+ channel blocker TTX into the NAcore via a microdialysis probe placed 0.5-1.0 mm from the recording electrode (Fig. 1c-d). The short latency positive peak (Fig. 1c) previously reported in NAcore slices after CNQX treatment20 was abolished by TTX perfusion into the PFC, but not into the NAc (Fig S1), indicating that this peak arises from volume conduction at the site of stimulation. In contrast with CNQX or TTX, reverse dialysis of the GABAA receptor antagonists picrotoxin or bicuculline were without effect on field amplitude (Fig. 1c-d), even though prolonged perfusion caused large amplitude, negative potentials independent of PFC stimulation (Fig. S3). This pharmacological profile suggests that the field potentials measured in this study arise primarily from glutamatergic EPSPs, and are minimally contaminated by population spikes, since GABAergic receptor antagonists did not alter the field appearance or amplitude21. In addition, the field potential did not fail when high frequency stimulation (50 Hz) was administered (Fig. S3) and the onset latency (<9 msec) of the fEPSP approximates monosynaptic action potential latencies evoked in the NAcore by in vivo PFC stimulation (11.3 msec22 and 14 msec23). Taken together, these data are consistent with the field potentials being fEPSPs comprised predominantly of monosynaptic glutamatergic responses.
Withdrawal from Cocaine Potentiates Baseline fEPSPs
Rats were trained to self-administer cocaine, and placed in extinction training for at least 3 weeks prior to recording field potentials (Fig. 2a). Potentiation of field amplitude was measured in cocaine relative to control rats, as estimated from current-voltage curves (Fig. 2b). This is consistent with behavioral, biochemical, imaging, and electrophysiological data all showing robust increases in various parameters indicative of excitatory transmission3,5,11,24, and indicates a pre-existing LTP-like state in the accumbens of animals withdrawn from cocaine. However, at very high currents (>1mA), the field amplitude was not different between the cocaine and control animals (Fig. 2b). At high current intensities, the fEPSP amplitude was often complicated by a second component (Fig. S4), indicating recruitment of a different population of synapses, probably multisynaptic25.
Figure 2. Field potentials in the NAcore reveal potentiation of the input-output curve in animals trained to self-administer cocaine.

a, Active vs. inactive lever pressing throughout self-administration training days (SA; 10 days), and extinction (E; 21 days), n=42. A two-way repeated measures ANOVA was used to determine that active lever pressing was greater than inactive lever pressing over last 3 days of SA, F(1, 164) = 15.40, p<0.001. b, Input-output curves from rats extinguished from self-administered cocaine vs. control animals. A two-way ANOVA was used to show that the input-output curve for cocaine was different from the control group (group F(1,374)=14.43, p< 0.001). Inset shows representative evoked field potentials for an input-output curve (0.3-1.5 mA) in a cocaine trained animal. All data shown as mean ± s.e.m.
Withdrawal from Cocaine Attenuates LTP
To further examine the status of PFC to NAcore glutamatergic synapses, HFS (2 bursts, at 50 Hz for 2 sec) was used to induce LTP in control animals (Fig. 3a). This HFS protocol failed to elicit LTP in cocaine rats (Fig. 3a). Varying the stimulation frequencies, 30 Hz, 50 Hz, 100 Hz (Fig. 3b), or number of bursts (Fig. S5) also failed to induce LTP in cocaine subjects. In control subjects, only 50 Hz HFS resulted in LTP (Fig. 3b). Although HFS using 100 Hz did not induce LTP in control rats (Fig. 3b), a diverse range of individual responses was observed (4:8 were >25% potentiated) similar to HFS results in slices26. It is possible that extinction training may have contributed to the impairment of LTP in cocaine-trained animals. Therefore, rats were trained to self-administer cocaine but kept in their home cages (abstinence) for 3 weeks rather than undergoing extinction training. Akin to extinguished animals, LTP could not be induced in abstinent cocaine rats (Fig. S6). Taken together, these results suggest that the potentiation of PFC-accumbens synapses after chronic cocaine revealed in the input-output curve (Fig. 2b) may be occluding further potentiation7.
Figure 3. LTP is impaired in rats trained to self-administer cocaine.

a, Upper panel: Representative experiment inducing LTP in a control rat after HFS (50 Hz). HFS was delivered at the arrow. Insets show an average of 5 consecutive fEPSP before (1) and after (2) HFS. Middle panel: Representative experiment of LTP failure in a cocaine rat after HFS (50 Hz). Lower panel: HFS (50 Hz) failed to induce LTP in the cocaine (n=13) compared to control animals (n= 8 yoked saline, n= 8 naive). A two-way repeated measures ANOVA revealed a significant effect of cocaine treatment: F(1,1458)= 11.05, p= 0.003. b, Upper and middle panels: LTP was induced in control animals only at 50 HZ (30Hz, 50Hz, 100Hz, group F(2,247)= 4.094, p= 0.042, time F(19,247)= 2.23, p= 0.003, and LTP in the cocaine animals could not be induced by any stimulation protocol (30Hz, 50Hz, 100Hz). Lower panel: Percentage potentiation from baseline of 30, 50, and 100 Hz HFS in cocaine and control goups. Each bar represents an average of the 5 min time bin immediately before (baseline) and 15-20 min after HFS. n is shown in the bars. A one-way ANOVA revealed effect of frequency only in control rats. F(3,31)= 6.243, p< 0.001. Data shown as mean ± s.e.m.
* p< 0.05 compared to baseline using a Bonferronni post hoc analysis
Withdrawal from Cocaine Attenuates LTD
While the basal potentiation of fEPSPs predicts an occlusion of LTP, it also predicts facilitation of LTD7,27. However, while LTD was readily induced in control animals by a low frequency stimulation protocol of 3 bursts at 5 Hz for 3 min each, LTD was significantly impaired in rats trained to self-administer cocaine (Fig. 4a). Akin to previous LTD experiments in accumbens slices28,29, LTD measured by fEPSPs in vivo was mGluR5 dependent (Fig. S7). These results are consistent with the previous work in NAcore slices where LTD is blocked after prolonged withdrawal from chronic cocaine6. The attenuated LTD was overcome in the cocaine group by employing a 5-burst instead of the 3-burst LFS induction protocol, and no further depotentiation could be produced using a 7- or 9- burst protocol (Fig. 4b). In control rats, the 3-, 5-, 7- and 9-burst protocols elicited equivalent LTD (Fig. 4b). In addition, as with the 3-burst protocol, blocking mGluR5 with MPEP blocked 5-burst induced LTD (Fig. S7), indicating that a similar mechanism governs the 3- and 5-burst induced LTD. Therefore, the LTD-impairment was not an occlusion phenomenon due to a preexisting state of LTD in the cocaine subjects.
Figure 4. LTD is impaired after chronic cocaine, but can be rescued using stronger LTD protocols.
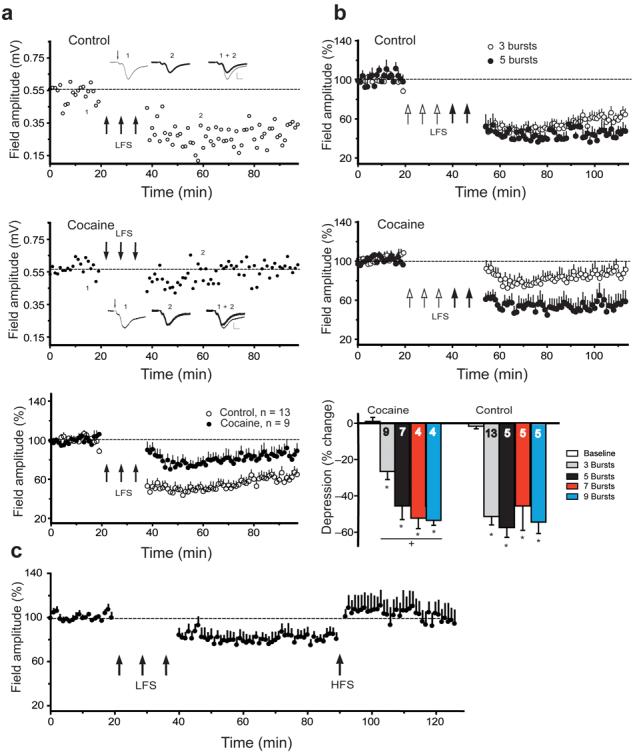
a, Upper panel: Representative experiment of LTD in a control rat after LFS. LFS was delivered at the arrows. Insets show an average of 5 consecutive fEPSP before (1) and after (2) LFS. Middle panel: Representative experiment of LTD impairment in a cocaine rat after LFS. Lower panel: LTD is impaired in the cocaine rats compared to controls (3 bursts LFS protocol) (n= 3 yoked saline, n= 10 naive) using a 2-way repeated measures ANOVA, F(1,1180)= 15.21, p= 0.001. LFS was delivered at the arrows, and the dotted line corresponds to the average amplitude before LFS. b, Upper and middle panels: In control animals, 5 and 3 bursts were not different; in cocaine rats, 5 bursts LFS resulted in robust LTD compared to 3 bursts (F(1,826)= 7.95, p = 0.014). Lower panel: Percentage depression from baseline of 3, 5, 7 or 9 bursts in cocaine and control groups. Each bar represents an average of 5 min time bin immediately before (baseline) and 15-20 min after LFS. n is shown in the bar and data are shown as means ± s.e.m. A one way ANOVA revealed significant LTD in both cocaine F(4,43)= 36.88, p< 0.001 and control subjects F(4,45)= 36.56, p< 0.001. c, Cocaine withdrawn subjects underwent 3-burst LFS to induce LTD, followed by HFS to induce LTP-like potentiation (F(7,87)= 3.72, p< 0.001).
* p< 0.05, compared to baseline using a Bonferroni multiple comparisons.
+ p< 0.05, comparing 5 to 3 bursts
A final experiment was conducted to evaluate the hypothesis that the PFC-accumbens synapses exist in a potentiated state after withdrawal from cocaine self-administration. Synapses were first depotentiated in chronic cocaine animals using the 3-burst LTD protocol, and in this partially depotentiated state it was possible to induce LTP (Fig. 4c), supporting the likelihood that the induction of LTP was masked in cocaine-withdrawn subjects by the synapses being in a pre-existing potentiated state. As well, these data pose the interesting possibility that restoring the capacity to induce LTD may contribute to restoring the ability to induce LTP.
N-acetylcysteine (NAC) Restores LTP and LTD
Down-regulation of the cystine-glutamate exchanger and signaling through group II metabotropic glutamate receptors (mGluR2/3) are associated with withdrawal from chronic cocaine administration, and restoration of cystine-glutamate exchange or tone onto mGluR2/3 prevents relapse in the reinstatement model of cocaine-12,15,30-32, as well as heroin-seeking13,33. The effect of NAC has been linked to the following sequence of events initiated by activating cystine-glutamate exchange and ultimately reducing synaptic release of glutamate by PFC afferents into the NAcore2,34, including 1) increasing nonsynaptic extracellular glutamate15, 2) thereby increasing tone on release regulating mGluR2/3 presynaptic autoreceptors14, and hence 3) reducing synaptic glutamate release12,14,15. Given these data showing the capacity of NAC to normalize extrasynaptic glutamate and attenuate synaptic glutamate release from the PFC into the NAcore, as well as the ability to inhibit drug seeking, we next determined if NAC administration could reverse the cocaine induced metaplasticity and restore the capacity of PFC stimulation to induce LTP and LTD in the NAcore. Rats were administered NAC (100 mg/kg ip) 2.5 hours before measuring field potentials (Fig. 5a). This pretreatment period was used because 2-3 hrs are required for NAC treatment to elevate extracellular glutamate in the accumbens of chronic cocaine withdrawn animals12,15. After generating baseline input-output curves, LTP or LTD protocols were invoked as described above. NAC pretreatment shifted the potentiated basal input-output curve in the cocaine animals to the right (depotentiated), making the curves between cocaine and control animals superimposable (Fig. 5b). NAC also restored the capacity to induce both LTP and LTD in cocaine rats without affecting the induction of LTP or LTD in control subjects (Fig. 5c-d).
Figure 5. N-acetylcysteine restores the capacity to induce both LTP and LTD.
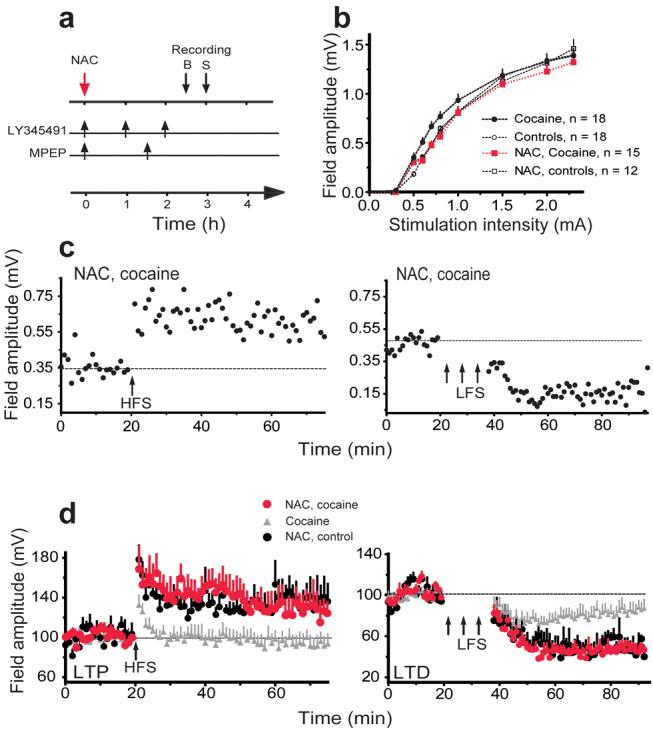
a, Experimental paradigm: NAC (100 mg/kg, ip) was injected 2.5 hrs before collecting baseline data (B), and 3 hrs prior to inducing LTP or LTD (S). LY345491 (1 mg/kg, ip) was administered with NAC and 2 times thereafter (arrows), and MPEP (10 mg/kg, ip) was administered with NAC and once thereafter. b, Current-voltage relationship for different groups showing that NAC normalized the input-output curve in cocaine animals without affecting control rats. (2-way ANOVA, treatment F(3,489) =8.321, p< 0.001). c-d, Representative experiments (c) and average of all experiments (d) showing that NAC restored the ability to induce LTP in cocaine rats (NAC, cocaine n=8) resulting in equivalent potentiation to LTP in control rats injected with NAC (NAC, control, n=6). NAC also restored the ability to induce LTD in cocaine rats (NAC, cocaine n=5) resulting in equivalent depression to LTD in control rats injected with NAC (NAC, control, n=4). The cocaine group from figure 2a is shown for comparison (grey). A two-way ANOVA comparing NAC, cocaine with NAC, control revealed no effect of group or interaction. All data are shown as mean ± s.e.m.
Restoration of LTP and LTD Depends Upon mGluRs
The capacity of NAC to regulate synaptic glutamate release and inhibit cocaine-seeking requires stimulating extrasynaptic mGluR2/314. Accordingly, the mGluR dependence of NAC-induced restoration of LTP and LTD was characterized. The mGluR2/3 antagonist LY341495 (1mg/kg, ip) was administered in parallel with NAC (100 mg/kg, ip) using a treatment regimen previously shown to block the ability of NAC to inhibit cocaine-seeking (Fig. 5a)14. LY341495 abolished the ability of NAC to restore LTP in animals trained to self-administer cocaine (Fig. 6a). In contrast, did not alter the NAC restoration of LTD (Fig. 6a). These data are consistent with the restoration of normal transynaptic glutamate levels by NAC stimulating mGluR2/3 presynaptic autoreceptors to decrease synaptic glutamate release probability35,36 and thereby depotentiating the excitatory synapses in cocaine animals (e.g. normalizing the input-output curve by NAC as shown in Fig. 5b). Previous studies in CA1 and dentate hippocampal neurons have shown that blocking mGluR2/3 inhibits the ability of HFS to induce LTP37,38, and LY341495 pretreatment also inhibits LTP induction in MSNs from control animals (Fig. S8).
Figure 6. Metabotropic glutamate receptors mediate NAC restoration of plasticity.

a, LY345491 (1 mg/kg, ip) prevented NAC-induced restoration of LTP (treatment F(1,374)= 6.10, p= 0.031), but did not block NAC restoration of LTD. See figure 5a for treatment protocol. b, The mGluR5 antagonist MPEP (10 mg/kg, ip) blocked the NAC restoration of impaired LTD in cocaine rats (treatment F(1,540)= 8.93, p =0.014), but did not block NAC restoration of LTP. c, Percentage change from baseline in control and cocaine animals after NAC and LY345491 or MPEP. Each bar represents an average of 5 min time bin immediately before (baseline) and 15-20 min after LFS or HFS. One-way ANOVA: LTP F(4,69)= 8.77, p< 0.001; LTD F(4,58)= 40.31, p< 0.001. n for each treatment group is shown in the bars, and data are shown as mean ± s.e.m.
* p< 0.05, compared with baseline using a Bonferroni multiple comparisons.
+ p< 0.05, compared with NAC in cocaine animals (NAC+cocaine)
Given the lack of effect by mGluR2/3 blockade on NAC restoration of LTD, and the well-established involvement of mGluR5 stimulation in LTD induction in the accumbens (Fig. S7)28,29, animals were treated with the mGluR5 antagonist MPEP (2 injections of 10mg/kg, ip; given with NAC and 1.5 hrs later; see Fig. 5a). MPEP blocked the capacity of NAC to restore LTD in cocaine animals, but did not affect the NAC-induced restoration of LTP (Fig. 6b). Thus, akin to the restoration of LTP resulting from stimulating mGluR2/3, NAC-induced restoration of LTD likely arises from restoring extrasynaptic glutamate and physiological tone to mGluR5 which re-establishes a critical mechanism for inducing mGluR5-dependent LTD. Figure 6c summarizes the effects of NAC and mGluR antagonists on LTP and LTD.
mGluR5 stimulation by NAC does not inhibit cocaine-seeking
Stimulating mGluR2/3 receptors has previously been shown to inhibit relapse to cocaine seeking in animal models31,32. Moreover, the capacity of NAC to inhibit cocaine-induced reinstatement of cocaine-seeking is prevented by pretreatment with an mGluR2/3 antagonist14. Based upon these data it has been concluded that the glutamate released by NAC-induced activation of cystine-glutamate exchange stimulates mGluR2/3 to presynaptically inhibit synaptic glutamate release and thereby reduce cocaine-seeking. Consequently, it is possible that the mGluR2/3-dependent restoration of LTP by NAC may contribute the inhibition of cocaine-seeking. In contrast, the existing data support an opposite conclusion for stimulating mGluR5. Thus, blockade of mGluR5 reduces cocaine-seeking in animal models employing conditioned place preference or the reinstatement of lever pressing39,40. Accordingly, experiments were conducted to further examine the role of mGluR5 using the reinstatement model of cocaine-seeking. Animals were trained to self-administer cocaine and following at least 2 weeks of extinction training were administered either the mGluR5 antagonist MPEP (10 mg/kg, ip) or the positive allosteric modulator CDPPB (3 or 10 mg/kg, ip). Neither of these treatments reinstated cocaine-seeking when administered alone (Fig. 7a). While pretreatment with CDPPB (10 mg/kg, ip) did not alter cocaine-seeking elicited by an acute cocaine injection (10 mg/kg, ip), pretreatment with MPEP (10 mg/kg, ip) inhibited cocaine-seeking (Fig. 7b). Given that CDPPB is a positive allosteric modulator41 and extracellular glutamate is reduced by chronic cocaine administration15, it is possible that a lack of glutamatergic tone on mGluR5 may have diminished any effect of CDPPB administration on cocaine-seeking. In order to partially restore glutamatergic tone, a relatively low dose of NAC (33 mg/kg, ip) was administered along with CDPPB using the treatment protocol illustrated in Fig 7c. This dose of NAC resulted in a partial reduction in cocaine-seeking, and co-administration of CDPPB reversed the NAC-induced inhibition (Fig. 7c). Taken together, the data in Fig 7 indicate that stimulation of mGluR5 by NAC-induced release of glutamate may promote relapse. Thus, blockade of mGluR5 inhibited cocaine-induced lever pressing while the positive allosteric modulator countermanded NAC induced reduction in cocaine-seeking.
Figure 7. The regulation of drug-seeking by the mGluR5 antagonist (MPEP) and positive allosteric modulator (CDPPB).

a, Neither compound induces lever pressing when administered alone. b, When administered 20 min prior to a cocaine priming injection (10 mg/kg, ip) MPEP inhibited cocaine-induced lever pressing (F(3,31)= 4.76, p= 0.011). c, The administration of NAC inhibits cocaine-induced lever pressing and this effect of NAC was abolished by pretreatment with CDPPB (F(2,32)= 16.04, p< 0.001). Self-administration refers to the mean ± sem active lever presses over the last 2 days of cocaine training. In panels b and c all animals received all treatments in random order separated by a 72 hr intertrial interval. n for each treatment group is shown in the bars, and data are shown as mean ± s.e.m.
* p< 0.05, compared to extinction using a Bonferroni multiple comparisons
+ p< 0.05, comparing NAC + Vehicle with NAC + CDPPB
DISCUSSION
The capacity of excitatory synapses in the cortico-striatal projection to undergo neuroplasticity is important for both establishing well-learned habits and for allowing well-learned behaviors to be updated according to new information imposed by a changing environment42. Addiction has been characterized as an inability to modify or stop drug-seeking habits in spite of environmental information indicating the behavior is maladaptive2. Critical to modifying striatal habit behavior is that the sensory information integrated in the PFC has access to the striatum via the nucleus accumbens. By employing in vivo field potentials the present study demonstrated that self-administered cocaine induces metaplasticity that impairs the ability of PFC stimulation to produce LTP or LTD in nucleus accumbens MSNs, and that N-acetylcysteine reverses cocaine induced-metaplasticity allowing the induction of both LTP and LTD.
The impairment in LTP likely results from the fact that LTP was occluded by a pre-existing state of LTP-like potentiation, as evidenced by the input-output curve being shifted to the left (Fig. 2b), as well as the ability to potentiate the PFC-accumbens synapses after depotentiation (Fig. 4c). A variety of other measurements are also consistent with chronic cocaine inducing LTP-like changes and enhanced glutamate responsiveness in accumbens MSN's, including increased AMPA/NMDA ratio of synaptic currents11, increased surface expression of AMPA receptors5, and enhanced behavioral responsiveness to microinjected AMPA24,43. However, the simultaneous loss of LTD cannot be explained by the occlusion of synaptic grading, and indicates that cocaine self-administration induces additional metaplasticity that reduces the capacity to elicit LTD via a mechanism distinct from LTP-related metaplasticity.
The relevance of cocaine induced-metaplasticity to the behavioral pathology of addiction is indicated not only by the fact that the impairment is occurring in a cortico-striatal pathway critical for regulating adaptive behavior2, but also by the fact that treatment with NAC, a drug known to inhibit cocaine seeking in the reinstatement model of addiction15 and drug cue reactivity in cocaine addicts16, restored the ability to induce LTP and LTD. This raises the question of a causal relationship between synaptic potentiation and the susceptibility to relapse. The reinstatement of drug-seeking by cue or drug depends upon potentiated PFC glutamate release into the NAcore34,44 and potentiated postsynaptic glutamate receptor signaling5. A variety of data indicate that NAC inhibits drug seeking by activating the cystine-glutamate exchanger, thereby increasing extrasynaptic glutamate and stimulating mGluR2/3 presynaptic autoreceptors to reduce synaptic glutamate release probability2. Moreover, direct pharmacological agonist stimulation of mGluR2/3 inhibits cocaine seeking31,32. Given the strong link between mGluR2/3 regulation of both synaptic glutamate release and drug seeking, the capacity of mGluR2/3 antagonist to inhibit NAC restoration of LTP is consistent with the possibility that normalizing the ability of PFC-NAcore synapses to become potentiated is ameliorative in the relapse to drug seeking.
In contrast to LTP, the restoration of LTD was linked to NAC-induced restoration of the tone on mGluR5. Akin to mGluR2/3, mGluR5 receptors are predominantly perisynaptic45 and thus accessible by the increased extracellular glutamate produced after NAC stimulation of cystine-glutamate exchange. In contrast to mGluR2/3, mGluR5 receptors are primarily postsynaptic45 and mGluR5 stimulation is well characterized to facilitate the induction of LTD in corticostriatal synapses28. Accordingly, blocking mGluR5 with MPEP prevented NAC from restoring LTD. However, contrary to an mGluR2/3 antagonist preventing NAC from reducing drug-seeking, the effect of NAC was reversed by promoting mGluR5 stimulation with a positive allosteric modulator. This is consistent with the present and previous experiments showing that inhibiting mGluR5 or mGluR5 gene deletion reduces the expression of many behaviors elicited by chronic cocaine administration, including the reinstatement of cocaine seeking, conditioned reward and locomotor sensitization39,46. These findings indicate that while the diminished glutamatergic tone on mGluR2/3 caused by down-regulated cystine-glutamate exchange promotes cocaine-seeking, reduced tone on mGluR5 has an opposing action. Consequently, the impairment in LTD may be protective and limit the intensity of drug-seeking.
The data in this report indicate that metaplasticity induced by cocaine self-administration markedly impairs the ability to induce LTP and LTD in PFC glutamatergic afferents to NAcore. The LTP impairment arises from synaptic occlusion while the abridged LTD can be attributed to reduced stimulation of mGluR5. By restoring tone onto mGluR2/3 and mGluR5 receptors, NAC was able to reverse the metaplasticity established by cocaine self-administration and restore LTP and LTD, respectively. Given the efficacy of NAC restoration of tone onto mGluR2/3 at inhibiting cocaine- and heroin-seeking,31,32 these data reveal a critical link between cocaine seeking and cocaine-induced metaplasticity that is exemplified by a loss of LTP in PFC to NAcore glutamatergic synapses. Moreover, considering the presumed importance of bidirectional neuroplasticity in acquiring new behaviors47, it is interesting to speculate that cocaine-induced metaplasticity may reflect the well known difficulty addicts experience in acquiring new behaviors to compete with drug seeking.
Methods
Animal housing and surgery
All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, and all procedures were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. One week after arrival, male Sprague-Dawley rats (250g) were anesthetized with ketamine HCl (87.5 mg/kg Ketaset, Fort Dodge Animal Health) and xylazine (5 mg/kg Rompum, Bayer), and implanted with intravenous catheters. The catheters were flushed daily with cefazolin (0.2 ml of 0.1 gm/ml) and heparin (0.2 ml of 100 IU) to prevent infection and maintain catheter patency. Rats recovered for a week before behavioral training15.
Self-administration and extinction procedures
Rats were trained to self administer cocaine in a standard operant chamber with 2 retractable levers. The self-administration regimen consisted of 10 days self-administration at >10 infusions/session. Daily sessions lasted 2 hrs, with an active lever press resulting in 0.2 mg in 0.05 ml cocaine infusion over 3 seconds, while inactive lever presses were of no consequence. Each infusion was followed by a 20 second timeout during which lever presses did not result in further cocaine infusions. Cocaine self-administering rats were often paired with yoked saline controls. Extinction procedures began 24 hr after the animal met the acquisition criterion, and lasted for at least three weeks. Lever presses were inconsequential throughout the extinction training.
Reinstatement of cocaine-seeking
In some animals, at 3 weeks after extinction training the capacity of various treatments to reinstate lever pressing was examined. Prior to examining the effect of compounds on cocaine-induced drug-seeking, animals were administered vehicle (20% w/v 2-hydroxypropyl-β-cyclodextrin), the mGluR5 positive allosteric modulator (CDPPB; 3 or 10 mg/kg, ip), or MPEP (10 mg/kg, ip) prior to being placed into the operant box to determine if the compounds by themselves affected drug-seeking. Up to 3 trials were conducted in each animal separated by a minimum 72 hr intertrial interval. In the first cocaine-priming experiment, all animals were injected in random order with vehicle, CDPPB (10 mg/kg, ip) or MPEP (10 mg/kg, ip) 20 min prior to administering a cocaine priming injection (10 mg/kg, ip). In the second group of rats, each animal was pretreated with NAC (33 mg/kg, ip) plus vehicle or CDPPB (10 mg/kg, ip) as shown in figure 7c. Within each cocaine-priming experiment, multiple treatments were administered in random sequence, separated by a minimum 72 hr intertrial interval during which animals continued with daily extinction training, and a maximum of 3 reinstatement trials was used in each animal.
Electrophysiological recordings
Extracellular field potentials were recorded after 3 weeks of extinction. Rats were anesthetized with urethane (1.5 g/kg, i.p.), and mounted in a stereotaxic apparatus (Narishige, Tokyo, Japan). Subcutaneous atropine methylbromide (0.3mg/kg) was used to minimize secretions and improve ventilation as needed. The animal's body temperature was maintained using an FHC temperature control system. Concentric bipolar stimulating electrodes (Rhodes medical Instruments) were placed in the ventral prelimbic medial prefrontal cortex (anteroposterior (AP) +3.0mm; mediolateral (ML) +0.6mm; dorsoventral (DV) −3.3mm from brain surface). Glass recording electrodes (warner instruments) were pulled using a Narishige PE-2 puller (1-2 mega ohm). Filling solution consisted of 0.5M sodium acetate with 2% pontamine sky blue. Recording electrodes were aimed at the dorsomedial region of the nucleus accumbens (AP +1.8mm; ML +1.3 to 1.5mm; DV −5.5 to −6.2mm from brain surface) (Paxinos and Watson, 6th edition). The brain was stabilized with agar and baseline measurements were obtained one hour after the surgery. Data collection hardware is described in detail elsewhere 48. Briefly, extracellular field potentials were amplified by NPI Instruments (Tamm, Germany) SEC-05LX amplifier, and the data band-pass filtered at 300Hz, then digitized by a National Instruments PCM-C1016E4 board (Austin, Texas) feeding into a computer. Custom Labview Software (Lee Campbell, Salk Institute, La Jolla, Ca.) was used for data collection and analysis. Data were collected every 30 sec, at a 10kHz sampling frequency, and then averaged every 1 minute. Pulse width was set to 0.3 msec and basal stimulation intensity corresponded to (30-40)% of minimum current intensity that evoked a maximum field response for LTP experiments, and (40-50)% for LTD experiments. Baseline data were collected for 20-30 min prior to the induction protocol (LTP or LTD). Field potential amplitude was measured as the difference between the mean of a 2-4 msec window prior to the stimulation artifact and the mean of a 1msec window around 15 msec following the stimulation artifact (corresponding to the negative peak of the field potential; see figure S4). Data were then normalized to baseline. The LTP protocol involved titanic stimulation at the minimum current intensity that evoked a maximum field response (from an input-output curve) using 2 bursts of 100 pulses at 50 Hz (2 sec.) 49, with 20 sec inter-burst interval. The LTD protocol involved stimulation at the minimum current intensity that evoked a maximum response using 3 trains of 900 pulses at 5 Hz (3min), with a 5 min. inter-train interval6.
Reverse dialysis with electrophysiology
Microdialysis probes were used to deliver drugs locally into the nucleus accumbens. Probes were constructed with inlet and outlet fused silica tubing, inserted into a semipermeable membrane with 2 mm active membrane length 15. After cranial surgery, probes were lowered (at 6 degrees mediolateral) into the accumbens (2.0 mm lateral to midline, 2.0 mm anterior to bregma, and 7.0 mm ventral to brain surface), over a period of 15-20 min using a customized motor system. Dialysis buffer (aCSF: 5 mM glucose, 2.5 mM KCl, 140 mM NaCl, 1.4 mM CaCl2, 1.2 mM MgCl2, and 0.15% PBS, pH 7.4) was perfused at a rate of 2 μl/min for 2 hours to allow the preparation to stabilize. Recording and stimulating electrodes were then advanced into the NAcore and PFC, respectively. After optimizating the recording site (Fig. 1b), the preparation was allowed to recover for an additional hour before any recordings were obtained. aCSF was continuously perfused. The tip of the electrode was 0.5-1.0 mm from the membrane of the microdialysis probe. A similar procedure has shown not to affect the electrophysiological properties of striatal neurons in vivo50. Baseline field potentials were recorded for 30 minutes, and the drugs were infused. A CMA switch was used to change between the aCSF and the different drugs used. To ensure the perfused drug reached the recording site, CNQX was always perfused first, and the time required for the field potential to disappear (5-10 min) was measured. CNQX was then washed out of the dialysis probe until field amplitude returned to baseline and additional drugs perfused for 30-40 min each, with a washout occurring between the reverse dialysis of each subsequent drug.
Groups
The control group consisted of yoked saline rats as well as naïve rats. Data from these animals were pooled because they did not show any difference in the capacity of LFS or HFS to induce LTD or LTP, respectively (Fig. S8). Withdrawal group rats went through 10 days of self administration followed by nearly 3 weeks of extinction.
Drugs
N-acetylcysteine (33 or 100 mg/kg), LY341495 (1 mg/kg), 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CPDDB; 3 or 10 mg/kg) and MPEP (10mg/kg, 2 injections) were administered i.p. See figure 5a for drug treatment protocols.
Histology
Pontamine sky blue (2%, 50 μA negative current for 5 min) was used to mark the recording site at the end of each recording session. Animals were transcardially perfused with 10% formalin, and then decapitated. Brains were later sectioned with a vibratome and the slices stained with cresyl violet.
Analysis
The data were analyzed using a two-way repeated measures ANOVA (without including the baseline data points since data were normalized to the baseline) in order to compare the whole data sets. F values are reported in the figure legends only when significant (p< 0.05). For selected time bins comparisons, a one-way ANOVA was used, and when more than 2 groups were compared a Bonferroni multiple comparison post hoc comparison was employed (SPSS 16.0 Software for Windows;SPSS Inc., Chicago, IL. or Graphpad prism). Statistical significance was set at a p ≤0.05. A Kolmogrov-Smirnov test was used to ascertain a normal distribution, and a Mann-Whitney test employed to compare between groups in figure 1d.
Supplementary Material
Acknowledgements
We thank Dr Floh Thiels, University of Pittsburgh, for her invaluable advice on field recording. Research in this report was supported by USPHS grants DA03906, DA12513, DA015369, and DA024355.
References
- 1.Mendelson JH, Mello NK. Management of cocaine abuse and dependence. N Engl J Med. 1996;334:965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- 2.Kalivas PW, O'Brien C. Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology. 2008;33:166–180. doi: 10.1038/sj.npp.1301564. [DOI] [PubMed] [Google Scholar]
- 3.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 4.Toda S, Shen HW, Peters J, Cagle S, Kalivas PW. Cocaine increases actin cycling: effects in the reinstatement model of drug seeking. J Neurosci. 2006;26:1579–1587. doi: 10.1523/JNEUROSCI.4132-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conrad KL, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- 7.Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 8.Brebner K, et al. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- 9.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 10.Bamford NS, et al. Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron. 2008;58:89–103. doi: 10.1016/j.neuron.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madayag A, et al. Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci. 2007:13968–13976. doi: 10.1523/JNEUROSCI.2808-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou W, Kalivas PW. N-Acetylcysteine Reduces Extinction Responding and Induces Enduring Reductions in Cue- and Heroin-Induced Drug-Seeking. Biological Psychiatry. 2008;63:338–340. doi: 10.1016/j.biopsych.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker DA, et al. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 16.LaRowe SD, et al. Is cocaine desire reduced by N-acetylcysteine? Am J Psychiatry. 2007;164:1115–1117. doi: 10.1176/ajp.2007.164.7.1115. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein RA, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Dongen YC, Mailly P, Thierry AM, Groenewegen HJ, Deniau JM. Three-dimensional organization of dendrites and local axon collaterals of shell and core medium-sized spiny projection neurons of the rat nucleus accumbens. Brain Struct Funct. 2008;213:129–147. doi: 10.1007/s00429-008-0173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in rat: An anterograde tract-tracing study with Phaseolus vulgais leucoagglutinin. J.Comp.Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- 20.Nicola SM, Kombian SB, Malenka RC. Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci. 1996;16:1591–1604. doi: 10.1523/JNEUROSCI.16-05-01591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pennartz CM, Boeijinga PH, Lopes da Silva FH. Locally evoked potentials in slices of the rat nucleus accumbens: NMDA and non-NMDA receptor mediated components and modulation by GABA. Brain Res. 1990;529:30–41. doi: 10.1016/0006-8993(90)90808-o. [DOI] [PubMed] [Google Scholar]
- 22.Montaron MF, Deniau JM, Menetrey A, Glowinski J, Thierry AM. Prefrontal cortex inputs of the nucleus accumbens-nigro-thalamic circuit. Neuroscience. 1996;71:371–382. doi: 10.1016/0306-4522(95)00455-6. [DOI] [PubMed] [Google Scholar]
- 23.Finch DM. Neurophysiology of converging synaptic inputs from the rat prefrontal cortex, amygdala, midline thalamus, and hippocampal formation onto single neurons of the caudate/putamen and nucleus accumbens. Hippocampus. 1996;6:495–512. doi: 10.1002/(SICI)1098-1063(1996)6:5<495::AID-HIPO3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 24.Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J.Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rozas C, et al. Developmental inhibitory gate controls the relay of activity to the superficial layers of the visual cortex. J Neurosci. 2001;21:6791–6801. doi: 10.1523/JNEUROSCI.21-17-06791.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pennartz CMA, Ameerun RF, Groenewegen HJ, Lopes da Silva FH. Synaptic plasticity in an in vitro slice preparation of the rat nucleus accumbens. Eur.J.Neurosci. 1993;5:107–117. doi: 10.1111/j.1460-9568.1993.tb00475.x. [DOI] [PubMed] [Google Scholar]
- 27.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 28.Robbe D, Bockaert J, Manzoni OJ. Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci. 2002;16:2231–2235. doi: 10.1046/j.1460-9568.2002.02273.x. [DOI] [PubMed] [Google Scholar]
- 29.Fourgeaud L, et al. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–6945. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kau KS, et al. Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience. 2008;155:530–537. doi: 10.1016/j.neuroscience.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baptista MA, Martin-Fardon R, Weiss F. Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement versus primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J Neurosci. 2004;24:4723–4727. doi: 10.1523/JNEUROSCI.0176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters J, Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology (Berl) 2006;186:143–149. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- 33.Bossert JM, Gray SM, Lu L, Shaham Y. Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology. 2006;31:2197–2209. doi: 10.1038/sj.npp.1300977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grueter BA, Winder DG. Group II and III metabotropic glutamate receptors suppress excitatory synaptic transmission in the dorsolateral bed nucleus of the stria terminalis. Neuropsychopharmacology. 2005;30:1302–1311. doi: 10.1038/sj.npp.1300672. [DOI] [PubMed] [Google Scholar]
- 36.Losonczy A, Somogyi P, Nusser Z. Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J Neurophysiol. 2003;89:1910–1919. doi: 10.1152/jn.00842.2002. [DOI] [PubMed] [Google Scholar]
- 37.Wu J, Rowan MJ, Anwyl R. An NMDAR-independent LTP mediated by group II metabotropic glutamate receptors and p42/44 MAP kinase in the dentate gyrus in vitro. Neuropharmacology. 2004;46:311–317. doi: 10.1016/j.neuropharm.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 38.Grover LM, Yan C. Evidence for involvement of group II/III metabotropic glutamate receptors in NMDA receptor-independent long-term potentiation in area CA1 of rat hippocampus. J Neurophysiol. 1999;82:2956–2969. doi: 10.1152/jn.1999.82.6.2956. [DOI] [PubMed] [Google Scholar]
- 39.Backstrom P, Hyytia P. Ionotropic and metabotropic glutamate receptor antagonism attenuates cue-induced cocaine seeking. Neuropsychopharmacology. 2006;31:778–786. doi: 10.1038/sj.npp.1300845. [DOI] [PubMed] [Google Scholar]
- 40.McGeehan AJ, Olive MF. The mGluR5 antagonist MPEP reduces the conditioned rewarding effects of cocaine but not other drugs of abuse. Synapse. 2003;47:240–242. doi: 10.1002/syn.10166. [DOI] [PubMed] [Google Scholar]
- 41.Lindsley CW, et al. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides that potentiate receptor function in vivo. J Med Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 42.Yin HH, Knowlton BJ, Balleine BW. Inactivation of dorsolateral striatum enhances sensitivity to changes in the action-outcome contingency in instrumental conditioning. Behav Brain Res. 2006;166:189–196. doi: 10.1016/j.bbr.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 43.Suto N, Tanabe LM, Austin JD, Creekmore E, Vezina P. Previous exposure to VTA amphetamine enhances cocaine self-administration under a progressive ratio schedule in an NMDA, AMPA/kainate, and metabotropic glutamate receptor-dependent manner. Neuropsychopharmacology. 2003;28:629–639. doi: 10.1038/sj.npp.1300075. [DOI] [PubMed] [Google Scholar]
- 44.LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitrano DA, Smith Y. Comparative analysis of the subcellular and subsynaptic localization of mGluR1a and mGluR5 metabotropic glutamate receptors in the shell and core of the nucleus accumbens in rat and monkey. J Comp Neurol. 2007;500:788–806. doi: 10.1002/cne.21214. [DOI] [PubMed] [Google Scholar]
- 46.Chiamulera C, et al. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nature Neuroscience. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 47.De Roo M, Klauser P, Garcia PM, Poglia L, Muller D. Spine dynamics and synapse remodeling during LTP and memory processes. Prog Brain Res. 2008;169:199–207. doi: 10.1016/S0079-6123(07)00011-8. [DOI] [PubMed] [Google Scholar]
- 48.Lavin A, et al. Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci. 2005;25:5013–5023. doi: 10.1523/JNEUROSCI.0557-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goto Y, Grace AA. Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron. 2005;47:255–266. doi: 10.1016/j.neuron.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 50.West AR, Moore H, Grace AA. Direct examination of local regulation of membrane activity in striatal and prefrontal cortical neurons in vivo using simultaneous intracellular recording and microdialysis. J Pharmacol Exp Ther. 2002;301:867–877. doi: 10.1124/jpet.301.3.867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.