

Abstract
The Death Inducing Signaling Complex (DISC) formed by Fas receptor, FADD and caspase-8 is a pivotal trigger of apoptosis1-3. The Fas/FADD DISC represents a receptor platform, which once assembled initiates the induction of programmed cell death. A highly oligomeric network of homotypic protein interactions comprised of the death domains (DD) of Fas and FADD is at the center of DISC formation4, 5. Thus characterising the mechanistic basis for the Fas/FADD interaction is paramount for understanding DISC signaling but has remained enigmatic largely due to a lack of structural data. We have successfully formed and isolated the Fas/FADD DD complex and here we report the 2.7 Å crystal structure. The complex shows a tetrameric arrangement of four FADD DDs bound to four Fas DDs. We show that an opening of the Fas DD exposes the FADD binding site and simultaneously generates a Fas/Fas bridge. The result is a regulatory Fas/FADD complex bridge governed by weak protein:protein interactions revealing a model where the complex functions as a mechanistic switch. This switch prevents accidental DISC assembly, yet allows for highly processive DISC formation and clustering upon a sufficient stimulus. Thus besides depicting a previously unknown mode of death domain interactions, these results further uncover a mechanism for receptor signaling solely by oligomerization and clustering events.
As typical for oligomeric signaling platforms the DISC acts as a cellular switch, which exists in the “off” position in the absence of a stimulus followed by oligomerization of its constituents to form the active (“on” position) oligomeric platform6. In the case of the Fas/FADD DISC the apoptotic signal per se is the binding of Fas ligand (FasL), which in a basic view (Supplementary Fig. 1a) leads to recruitment of FADD via death domain (DD) interactions. FADD in turn recruits caspase-8 through death effector domain (DED) interactions leading to activation of this apical caspase1, 2, 7. However, from a cell signaling point of view this string of events remains on a descriptive level, since the binding partners do not (and should not) interact in the absence of a sufficient stimulus. Productive DISC formation is only observed when ligand binding occurs in a permissive environment, such as predisposition of Fas in membrane rafts, and is furthermore characterized by the formation of highly oligomeric DISC clusters8-14. Since the DD interaction of Fas and FADD is at the heart of the DISC interaction network (Supplementary Fig. 1b), the question arises about how a simple DD interaction mediates such complexity? Other signaling platforms, for example the Apoptosome or Inflammasomes15, 16, are proposed to elegantly exploit a nucleotide dependent regulation to properly signal oligomerization as opposed to the seemingly simple death domain interaction of Fas and FADD.
In addition to mutations implicated in disease states, a variety of mutants have been generated in order to define interfaces in the Fas/FADD complex (Supplementary Table 2; reviewed in 5, 14). However, despite this information and effort, the nature of the primary Fas/FADD interaction and its implication on the actual mechanism for formation of the Fas/FADD DD network remains elusive14. In our study we were able to gain insight into this mechanism by elucidating the crystal structure of the Fas/FADD DD complex from which we conclude a model explaining how observed contacts are mediated by a primary Fas/FADD complex, which acts as a sensitive switch governing DISC formation. This extends and completes our view of death domains, which initiated from rigid defined domain complexes such as the constitutive 1:1 complex seen in the Apaf-1/caspase-9 CARD/CARD interaction, to current views which include plasticity and asymmetry of complexes in the death domain superfamily5 (see also Supplementary Discussion). Thus the model presented here can furthermore serve as a template for other signaling platforms lacking enzymatic components which are solely mediated by oligomerisation and clustering events. Notorious for its elusiveness and solubility difficulties14, we succeeded in producing a soluble Fas/FADD complex when E. coli lysates of recombinantly expressed Fas DD and FADD DD were combined prior to purification. After purification we optimized conditions to obtain crystals of the complex and solved the 2.7 Å resolution structure. Crystals indexed in the hexagonal space group P61 with two tetrameric assemblies each comprising four Fas DD and four FADD DD in the asymmetric unit (Fig. 1, Supplementary Table 1). The tetrameric arrangement in fact represents a dimer of two Fas/FADD DD complex dimers. In this arrangement Fas provides all contacts (see also Supplementary Discussion). All residues of the Fas DD are well defined in the electron density. FADD is defined from G93 to G191 with the region around residues 107−149 showing more diffuse density due to a high degree of overall motion caused by a lack of crystal contacts (Supplementary Fig. 1c).
Figure 1. Overall structure of the Fas/FADD DD complex.

(left) The structure shows a tetrameric arrangement of Fas/FADD DD complexes. Contacts between the complexes are solely mediated by Fas molecules. (right) Cartoon representation of the Fas/FADD complex structure. Color coding: one Fas/FADD DD complex is displayed in green (Fas) and blue (FADD) ribbons, while the remaining three complexes are colored red, magenta and blue (surface representation).
Inspection of the interfaces shows that the actual Fas/FADD complex is formed by helix one and six of the FADD DD through employment of a hydrophobic patch surrounded by polar residues (Fig. 2a, d). Unlike the FADD DD, the Fas DD in the complex deviates from the typical DD fold17-19. Comparison with the solution structure of the isolated Fas DD20 reveals that Fas in the complex has undergone an opening in which helix six has shifted and fused with helix five to form a long helix we term the stem helix (Fig. 2b, c). In addition to Fas opening, a new helix at the C-terminus of Fas dubbed the C-helix is observed (Fig. 2a). The main interaction interface on Fas is formed by hydrophobic residues that become available only upon Fas opening (Fig. 2d). Thus the opening of Fas discovered in the crystal structure is a crucial event in its interaction with FADD. Significantly, other FADD interacting death receptors show similar residues as those observed in Fas at the complex interface (see Supplementary Fig. 4c and Discussion).
Figure 2. Fas/FADD DD complex: Fas/FADD and Fas/Fas interactions are dependent on Fas opening.

a, Primary Fas/FADD DD complex. FADD (blue) adopts the characteristic death domain fold with helices one and six participating in the main interaction site. In Fas (green) only helices one to four approximately adopt a death domain-like fold, while a long helix (stem helix) is found in place of helices five and six, which together with helix one provides the main interaction residues for FADD binding. Additionally a helix at the C-terminus of Fas (C-helix) is observed. b, Conformational change in Fas. Comparison of the structure of unbound Fas DD (closed form in orange; pdb entry: 1DDF) and Fas in the Fas/FADD complex. Due to formation of the stem helix residues of helix five and six shift significantly. Additionally, the rearrangement of helix six exposes part of the hydrophobic core of Fas. c, Cartoon illustration of Fas opening. d, Primary Fas/FADD interface. View onto interfaces governing primary complex formation. Surface representation shows complementary hydrophobic patches (yellow) on FADD and Fas surrounded by polar residues (magenta). The hydrophobic interface on Fas becomes exposed upon Fas opening. e, Fas-Dimer unit. Another consequence of Fas opening is the formation of Fas-dimer units, which interact via the stem and C-helix. (right) Cartoon representation of the dimer. f, The Fas/FADD DD complex is weak. Dilution experiment of the isolated Fas/FADD complex shows cooperative dissociation of the complex below concentrations as high as 50 μM (plot derived from quantitative SDS-PAGE analysis of Fas-DD retained on Ni-NTA resin from various Fas-DD/FADD-DD-His6 complex dilutions).
Broader investigation of the Fas/FADD interface divulges the disperse nature of the interaction surface, which lacks the presence of defined and focused interaction sites often referred to as hot spots21-23. This phenomena is used in regulatory complexes characterized by weak binding despite sufficiently large interaction surfaces22, 24, 25. In addition to the lack of hot spots, a loss of compact tertiary structure due to the opening of Fas further enhances flexibility of the system and thus additionally weakens the complex. Besides providing the primary interface for binding of FADD, a second consequence of the opening of Fas is to allow for interaction with another open Fas molecule forming a Fas/Fas bridge. The predominant Fas/Fas association observed in the structure is formed by stem-helices and C-helices of the two Fas molecules building a Fas/Fas dimer (Fig. 2e), which compared to the tetramer, better approximates structural requirements in the context of the natural membrane association (see Supplementary Discussion). Thus opening of Fas provides both the basis for FADD binding and the formation of a regulatory bridge between Fas molecules. Due to the difficult nature of the Fas/FADD complex, biochemical analysis is limited (Supplementary Fig. 2a), but indeed confirms a weak primary Fas/FADD complex which dissociates at concentrations as high as 50 μM (Fig. 2f, Supplementary Fig. 2d). Mutational analysis of the Fas/FADD interface reveals predominately mild phenotypes (Supplementary Fig. 3a-c, and data not shown), in line with previous observations for complexes lacking binding hot spots such as IFNalpha2/IFNAR126.
In solution, the Fas/FADD DD complex indeed shows a tetrameric behavior (Supplementary Fig. 2b-d), which is further corroborated by previously reported results of a solubility enhanced complex which also pointed to a tetrameric arrangement27. Taken together these results establish that the observed tetramer in the crystal structure reflects the Fas/FADD DD arrangement present in solution and is not a result of crystal formation. In addition the tendency of the tetramer to form higher oligomers can be observed (Supplementary Fig. 2 c, d).
To obtain further insight into the binding of FADD, we overlaid the structure of full length FADD18 onto the DD of FADD in the primary complex (Fig. 3a). The drastic conformational change in Fas, which is essential to bind FADD, is also accompanied by a change in the FADD death domain. The C-terminal helix of FADD has to shift in order to avoid a steric clash with the newly formed C-helix of Fas, substantiating results which attribute a negative role in cell death induction to this region of Fas (Fig, 3a; Supplementary Fig. 3d)28. This observation is even more crucial in the context of full length FADD, since the FADD C-terminus is located at the interface of the DED and DD. Along this line, an overall conformational rearrangement, namely the relative position of the DED and the DD of FADD, can also be concluded to expose caspase-8 binding residues determined in earlier studies (summarized in 5, 18). This change is readily enabled by the interdomain linker, which is composed exclusively of Ala, Gly and Pro residues. Indeed our studies found that when full length FADD is incubated with Fas a reduced initial complex formation can be observed, underlining a conformational change (Fig. 3b), while longer incubation of the two proteins lead to the formation of protein clusters (Fig. 3c).
Figure 3. The Fas/FADD bridge in the DISC: binding of full length FADD and the key role of Fas opening in vivo.

a, Overlay of the structure of full length FADD* (pdb entry: 2gf5; orange) onto the Fas/FADD complex structure. The last helix of the DD of unbound FADD (red dot) shifts to avoid clashing with the newly formed Fas C-helix in the Fas/FADD complex. b, Conformational change in full length FADD. Proteins were expressed separately, and His-tagged versions of the DD of FADD, or full length FADD, were mixed with untagged Fas. Ni-NTA chromatography demonstrates that full length FADD shows reduced initial binding to the Fas DD, when compared to the FADD DD protein. c, While initial binding of full length FADD to Fas DD is reduced, prolonged incubation leads to the formation of DISC-like structures. Incubation of both proteins overnight led to the formation of ring-like structures with a strong tendency to form clusters as determined using electron microscopy. Displayed are single ring-like structures and clusters from several images. Due to their strong tendency to self-adhere no consistent monolayer for thorough evaluation could be generated to date. d/e, Propagating Fas opening results in hyperactive Fas. (d) Location of Ile313 in closed (unbound, orange) and open (complex, green) form of Fas. (e) Huh7 cells transfected with Fas I313D show elevated cell death, assessed by Annexin V reactivity, compared to Fas wt upon stimulation with Fas Antibody (left, standard deviations, n=3), FasL (middle), and also in the absence of a stimulus (right). Equal cell surface Fas expression was confirmed by FACS and immunoblot (data not shown). *Full length FADD refers to the well characterized FADD F25Y mutant (see Supplementary Methods)
Details revealed in the Fas/FADD death domain structure show that Fas opening is key to both FADD binding and formation of a Fas/Fas bridge. To validate the relevance of Fas opening in vivo, we sought to create a mutation that interfered with the closed form of Fas and propagated Fas opening. If Fas opening is a prerequisite for DISC formation, this forced opening of Fas should produce a hyperactive phenotype. By comparing the structures of closed and open Fas we identified Ile313 as a candidate. Ile313 is part of the hydrophobic core and resides in helix 6 of the closed form and is thus at the center of the opening event. At the same time it does not directly participate in the Fas/FADD interaction or the observed Fas/Fas dimer in the complex structure (Fig. 3d, Supplementary Fig. 4a). Indeed mutation of Ile313 to Asp resulted in significantly enhanced killing. Enhanced cell death by Fas I313D compared to wild type could be observed when cells were stimulated with Fas antibody or FasL and even in the absence of a stimulus, further establishing the central role of Fas opening (Fig. 3e).
Based on the analysis of the Fas/FADD structure which displays key regulatory features of DISC formation, and taking into account previous findings, we conclude a model of how receptor signaling in the DISC occurs solely through clustering events. In this model the opening of Fas is central to form the Fas/Fas bridge and to recruit FADD. The Fas/Fas bridge and FADD binding are governed by weak protein:protein interactions only stabilized upon processive clustering, thus representing the actual regulatory switch governing DISC formation (Fig. 4). Until now, the dominant model for DISC formation proposed that trimeric Fas ligand cross-linked units of preformed trimeric Fas in local membrane rafts8-14, leading to higher order arrangements. This model is substantiated by mutations in the Fas DD (Supplementary Table 2; reviewed in 5, 14) that are not located at the primary Fas/FADD interface, but rather adjacently on the globular units of open Fas observed here. While illustrating the overall process, this initial model did not provide a mechanism for the actual clustering of Fas DDs within the cell and most importantly the relationship between Fas clustering and FADD binding, both of which are explained in our model. In this model only when a sufficient number of Fas molecules are in close proximity, as is expected upon Fas ligand binding, can the open forms of Fas become stabilized (Fig. 4a) since the newly formed stem helices can interact with each other. An interlinked consequence of this interaction is that Fas molecules become connected through this sensitive bridge - setting the stage for a chain reaction. Now the formation of Fas bridges linking the Fas DD trimeric units, which interact via the globular portion of open Fas, directly result in rapid and processive clustering on the inside of the cell membrane (Fig. 4b). Also, FADD is now able to bind the open Fas molecules, key for DISC signaling, and additionally increasing stabilization of the Fas/Fas bridges thereby further fostering DISC formation and clustering (Fig. 4b).
Figure 4. Model of DISC formation: mechanism of receptor signaling through clustering.
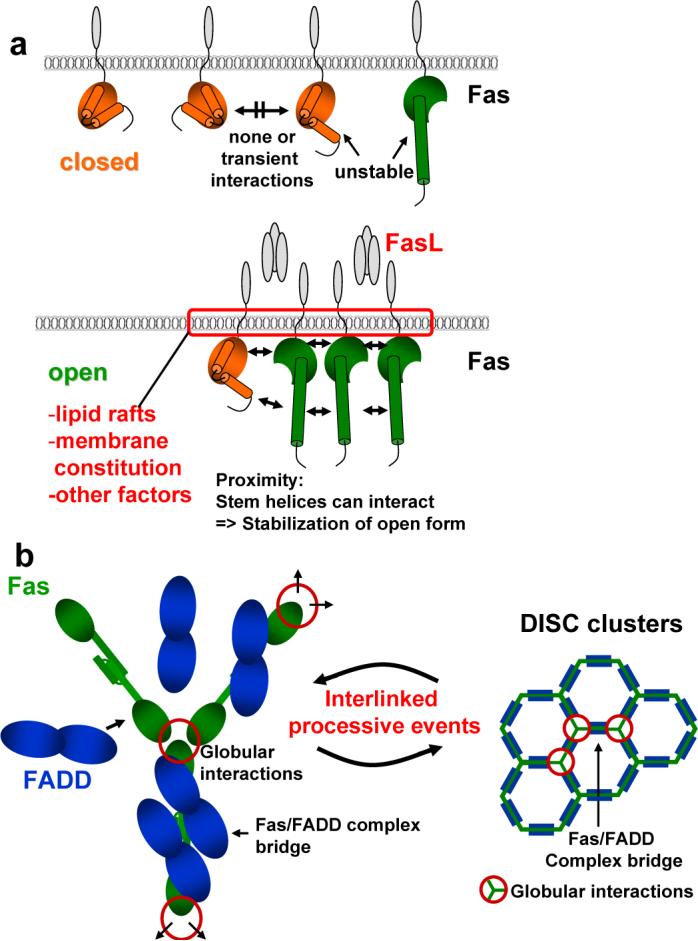
Schematic illustrating DISC formation. a, Model for Fas opening. Like in any two state model, it can be assumed that the closed and open form of Fas exist in equilibrium. In the absence of an apoptotic signal the equilibrium between closed and open forms of Fas strongly favors the closed form while open forms are unstable. Upon an apoptotic stimulus the equilibrium shifts to favor the open form of Fas. Fas molecules are brought together by Fas ligand in a permissive environment dependent on several factors including lipid rafts and membrane constitution. The close proximity of Fas DDs now allows for stem helices to interact leading to stabilization of the open form. b, Opening and formation of the Fas/Fas bridge links trimeric DD units defined in earlier studies (Supplementary Table 2), which are formed by globular regions of the open Fas. FADD can now bind to the open Fas molecules further stabilizing the bridge. The consequence is processive interlinked DISC formation and clustering in which open Fas molecules interacting via their globular domains are linked by a multitude of weak Fas/FADD bridges leading to overall stable DISC clusters. This permits activation of caspase-8, presumable by proximity enforced dimerization30, and induction of Apoptosis.
In summary the proximity induced stabilization of open Fas, in combination with the formation of the weak bridge, allows timing and strict regulation of DISC formation solely by clustering as opposed to, for example, a nucleotide exchange mediated oligomerisation as in the apoptosome29.
METHODS SUMMARY
The Fas/FADD death domain complex was obtained by mixing crude E. coli lysates from recombinantly expressed proteins at high concentration. Large amounts of pure, soluble and crystallization grade complex could be obtained after Ni-affinity and ion-exchange chromatography. Crystallization could be achieved under acidic conditions, which after several rounds of optimization resulted in diffracting crystals, one to 2.7 Å resolution. Initial phases were obtained using a Pt-derivative crystal followed by standard structure solution using the native data.
Supplementary Material
Acknowledgements
We thank Scott Snipas for protein sequencing and technical assistance, Dr. John Reed for kindly providing Fas cDNA and Dr. Andrey Bobkov for the AUC. This work was supported by P30 CA030199 cancer center grant and R01AA017238 to SJR, PO1CA69381 to GSS, and MCEXT-033534 to RS. Data measured at beamline X29 of the National Synchrotron Light Source was also supported by Biological and Environmental Research DOE, and National Center for Research Resources NIH. Earlier stages of the work were supported by a LLS scholarship to SJR. SJR is currently a V Foundation scholar.
Footnotes
Full Methods and associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information accompanies the paper on www.nature.com/nature.
References
- 1.Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999;11:255–60. doi: 10.1016/s0955-0674(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 2.Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–41. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 4.Fesik SW. Insights into programmed cell death through structural biology. Cell. 2000;103:273–82. doi: 10.1016/s0092-8674(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 5.Park HH, et al. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Festjens N, Cornelis S, Lamkanfi M, Vandenabeele P. Caspase-containing complexes in the regulation of cell death and inflammation. Biol Chem. 2006;387:1005–16. doi: 10.1515/BC.2006.124. [DOI] [PubMed] [Google Scholar]
- 7.Tibbetts MD, Zheng L, Lenardo MJ. The death effector domain protein family: regulators of cellular homeostasis. Nat Immunol. 2003;4:404–9. doi: 10.1038/ni0503-404. [DOI] [PubMed] [Google Scholar]
- 8.Algeciras-Schimnich A, et al. Molecular ordering of the initial signaling events of CD95. Mol Cell Biol. 2002;22:207–20. doi: 10.1128/MCB.22.1.207-220.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feig C, Tchikov V, Schutze S, Peter ME. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. Embo J. 2007;26:221–31. doi: 10.1038/sj.emboj.7601460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lavrik IN, et al. Analysis of CD95 threshold signaling: triggering of CD95 (FAS/APO-1) at low concentrations primarily results in survival signaling. J Biol Chem. 2007;282:13664–71. doi: 10.1074/jbc.M700434200. [DOI] [PubMed] [Google Scholar]
- 11.Muppidi JR, Siegel RM. Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat Immunol. 2004;5:182–9. doi: 10.1038/ni1024. [DOI] [PubMed] [Google Scholar]
- 12.O'Reilly LA, et al. Modifications and intracellular trafficking of FADD/MORT1 and caspase-8 after stimulation of T lymphocytes. Cell Death Differ. 2004;11:724–36. doi: 10.1038/sj.cdd.4401408. [DOI] [PubMed] [Google Scholar]
- 13.Siegel RM, et al. SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol. 2004;167:735–44. doi: 10.1083/jcb.200406101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner MH, Wu C, Walsh CM. Emerging roles for the death adaptor FADD in death receptor avidity and cell cycle regulation. Cell Cycle. 2006;5:2332–8. doi: 10.4161/cc.5.20.3385. [DOI] [PubMed] [Google Scholar]
- 15.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–22. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 17.Berglund H, et al. The three-dimensional solution structure and dynamic properties of the human FADD death domain. J Mol Biol. 2000;302:171–88. doi: 10.1006/jmbi.2000.4011. [DOI] [PubMed] [Google Scholar]
- 18.Carrington PE, et al. The structure of FADD and its mode of interaction with procaspase-8. Mol Cell. 2006;22:599–610. doi: 10.1016/j.molcel.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 19.Jeong EJ, et al. The solution structure of FADD death domain. Structural basis of death domain interactions of Fas and FADD. J Biol Chem. 1999;274:16337–42. doi: 10.1074/jbc.274.23.16337. [DOI] [PubMed] [Google Scholar]
- 20.Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature. 1996;384:638–41. doi: 10.1038/384638a0. [DOI] [PubMed] [Google Scholar]
- 21.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 22.Reichmann D, Rahat O, Cohen M, Neuvirth H, Schreiber G. The molecular architecture of protein-protein binding sites. Curr Opin Struct Biol. 2007;17:67–76. doi: 10.1016/j.sbi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 24.Huxford T, et al. Solvent exposed non-contacting amino acids play a critical role in NF-kappaB/IkappaBalpha complex formation. J Mol Biol. 2002;324:587–97. doi: 10.1016/s0022-2836(02)01149-x. [DOI] [PubMed] [Google Scholar]
- 25.Janin J, Miller S, Chothia C. Surface, subunit interfaces and interior of oligomeric proteins. J Mol Biol. 1988;204:155–64. doi: 10.1016/0022-2836(88)90606-7. [DOI] [PubMed] [Google Scholar]
- 26.Roisman LC, Jaitin DA, Baker DP, Schreiber G. Mutational analysis of the IFNAR1 binding site on IFNalpha2 reveals the architecture of a weak ligand-receptor binding-site. J Mol Biol. 2005;353:271–81. doi: 10.1016/j.jmb.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 27.Ferguson BJ, et al. Biophysical and cell-based evidence for differential interactions between the death domains of CD95/Fas and FADD. Cell Death Differ. 2007;14:1717–9. doi: 10.1038/sj.cdd.4402191. [DOI] [PubMed] [Google Scholar]
- 28.Itoh N, Nagata S. A novel protein domain required for apoptosis. Mutational analysis of human Fas antigen. J Biol Chem. 1993;268:10932–7. [PubMed] [Google Scholar]
- 29.Kim HE, Du F, Fang M, Wang X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci U S A. 2005;102:17545–50. doi: 10.1073/pnas.0507900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boatright KM, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–41. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.