

Abstract
The traceless Staudinger ligation can be mediated by phosphinothiols under physiological conditions. Proximal positive charges are necessary to achieve that transformation, presumably because those charges discourage protonation of the key iminophosphorane intermediate. Here, a series of cationic phosphinothiols is used to probe Coulombic effects on the traceless Staudinger ligations in aqueous buffers. The reagent bis(m-N,N-dimethylaminomethylphenyl)phosphinomethanethiol (3) is found to be superior to others, both in its ability to mediate traceless Staudinger ligations in water and in the efficiency of its synthesis.
1. Introduction
The advent of methodology for the chemoselective ligation of peptide fragments has made proteins accessible targets for chemical synthesis. Many proteins have already been assembled from synthetic peptides using prior capture strategies. The most utilized method of peptide ligation is “native chemical ligation”—the coupling of a peptide (or protein) containing a C-terminal thioester with another peptide containing an N-terminal cysteine residue.1 An extension of native chemical ligation, “expressed protein ligation”, employs an engineered intein to access a polypeptide containing the C-terminal thioester.2 Although this strategy for protein thioester formation has expanded the utility of native chemical ligation for protein semisynthesis, it retains the intrinsic requirement of a cysteine residue at the ligation juncture.
Emerging strategies for protein assembly avoid the need for a cysteine residue at the ligation site.3 The traceless Staudinger ligation is one such strategy (Scheme 1).4 This method is based on the Staudinger reduction, wherein a phosphine reduces an azide via an iminophosphorane intermediate. The iminophosphorane can be acylated to yield, ultimately, an amide. An important feature of this reaction is its potential to mediate the nonengrammic ligation of peptides at virtually any residue.5 The Staudinger ligation is chemoselective (especially regarding the relatively nonreactive azide),6 retains stereochemistry,7 and has been used in the orthogonal assembly of a protein,8 and for the site-specific immobilization of peptides and proteins to a surface.9
Scheme 1.

Putative mechanism of the traceless Staudinger ligation.
Recently, we reported on the development of water-soluble phosphinothiols that can mediate the traceless Staudinger ligation in water in moderate yields.10 The most efficacious such reagent, bis(p-N,N-dimethylaminoethylphenyl)phosphinomethanethiol (1) (Figure 1), can also evoke transthioesterification to generate phosphinothioesters at the C-terminus of proteins generated by expressed protein ligation.
Figure 1.

Bis(p-N,N-dimethylaminoethylphenyl)phosphinomethanethiol (1), a reagent that mediates the traceless Staudinger ligation in water.10
Water-soluble phosphinothiol 1 was designed to have N,N-dimethylamino groups that could not only impart water solubility but also (when protonated) serve to discourage protonation of the iminophosphorane nitrogen, which leads to hydrolysis of the P–N bond prior to S→N acyl transfer (Scheme 1). Indeed, we have shown that positive charges are favored over negative charges in mediating traceless Staudinger ligations in water.10 Herein, we consider further the importance of Coulombic effects on aqueous Staudinger ligations and report on markedly improved reagents.
2. Results and discussion
The positively-charged amino groups of phosphinothiol 1 are eight bonds away from the key iminophosphorane nitrogen in Scheme 1. Even at this distance, however, through-space Coulombic effects can affect pKa values. For example, CH3(CH2)8NH3+ has a pKa of 10.65.11 The addition of a positive charge, as in +H3N(CH2)8NH3+, lowers the pKa to 10.10. Likewise, CH3(CH2)5NH3+ and +H3N(CH2)5NH3+ have pKa values of 10.63 and 9.74, respectively.11 Long-range Coulombic effects were also evident in our attempts to mediate the traceless Staudinger ligation in water.10 Based on these considerations, we reasoned that favorable Coulombic effects would be enhanced by increasing the proximity of the positive charges.
We put forth compounds 2 and 3 to test our reasoning (Figure 2). In these compounds, the N,N-dimethylamino group is closer to the reaction center by its being in the meta rather than the para position of the aryl ring (2 and 3), and its having a methylene rather than an ethylene linker (3). The N,N-dimethylaminoethyl substituent in the meta position is situated seven bonds away from the key iminophosphorane nitrogen, compared to the eight-bond distance of 1. The meta N,N-dimethylaminomethyl substituent in phosphinothiol 3 is even closer, at a distance of six bonds. The ortho position was not explored, as we feared that ortho substituents would add steric encumbrance to the reaction center, and that the protonated N,N-dimethylamino group could act as an efficient intramolecular catalyst for deleterious protonation of the iminophosphorane nitrogen.
Figure 2.

Target phosphinothiols with enhanced Coulombic effects for aqueous traceless Staudinger ligations.
We also reasoned that the efficacy of the reagent in mediating the traceless Staudinger ligation in water would be enhanced by additional cationic charges. The amplified Coulombic effects could have a more pronounced effect on the protonation state of the iminophosphorane nitrogen. Phosphinothiol 4, which has four rather than two meta N,N-dimethylaminomethyl groups, was thus another target for synthesis and analysis (Figure 2).
2.1 Synthesis of phosphinothiols 2–4
In phosphinothiols 1–4, tertiary amino groups, rather than primary or secondary ones, were chosen to obviate intramolecular S→N acyl transfer in ensuing thioesters. Due to the presence of nitrogen, phosphorus, and sulfur in the phosphinothiol, several orthogonal protection/deprotection steps were necessary to effect its synthesis. Accordingly, the route to 1 consisted of nine linear steps.10 The purification of most intermediates was possible by chromatography using simple silica plugs, as the reactions were efficient and essentially “spot-to-spot” conversions. Moreover, each reaction in the route was readily scalable. Nonetheless, the time required to generate fully-deprotected 1 from commercial starting materials was approximately one week. We sought to sought to obtain an efficacious reagent in a shorter time.
Phosphinothiols 2–4 were synthesized by analogous synthetic routes (Scheme 2). Although (3-bromobenzyl)dimethylamine (9) was available from commercial sources, bromide 6 was accessible from 3-bromophenethyl alcohol through the displacement of its mesylate in 5 with dimethylamine. Bromide 8 was synthesized from bromination of 5-bromo-m-xylene with N-bromosuccinimide and catalytic AIBN to give 7, followed by benzylic displacement with dimethylamine.12 Double Grignard addition of the respective aryl bromides with diethylphosphite gave the bis-adducts 10–12. Bromide 8 must be dried stringently under high vacuum, and the addition of a few drops of 1,2-dibromoethane was also advantageous for facilitating Grignard initiation. Reduction with DIBAl-H and subsequent protection with borane gave phosphine-borane complexes 13–15. The protected phosphinothioester intermediates 17–19 were synthesized in a convergent manner utilizing the easily-prepared bromide 16,13 which had been employed in the convergent synthesis of (diphenylphosphino)methanethiol.7
Scheme 2.

Reagents and conditions: (a) MsCl, NEt3, CH2Cl2 (b) HNMe2, THF (c) NBS, cat. AIBN, CCl4 (d) HNMe2, benzene (e) Mg(s), THF (f) (EtO)2P(O)H, THF (g) DIBAl-H, CH2Cl2 (h) BH3·SMe2, CH2Cl2 (i) NaH, BrCH2SAc (16), DMF (j) DABCO, toluene, 40 °C (k) NaOH, MeOH (l) 4 N HCl/dioxane.
Phosphine-borane complexes 17–19 are stable to prolonged storage without decomposition. Removal of the borane groups with DABCO, deprotection of the thiol in basic methanol, and subsequent acidification with 4 N HCl/dioxane gave the chloride salts of phosphinothiols 2–4, which are easy-to-handle, odor-free white solids that are soluble at >1 M in 0.4 M sodium phosphate buffer (pH 7.8).
It was found that the synthesis of compound 2 was comparable to its para counterpart 1. On the contrary, the efficiency in the synthesis of benzylic compound 3 was dramatically enhanced, as most transformations were completed in 20 min with excellent yields. Although the overall yield of 4 was low at 5%, the overall yields of 2 and 3 were 15 and 41%, respectively. Thus, the yield of 3 is markedly greater than that for phosphinothiol 1 (17%).10 The commercial availability of (3-bromobenzyl)dimethylamine (9) and shorter reaction times reduced the synthetic route to a water-soluble phosphinothiol from nine (1) to five (3) transformations, increased the overall yield from 17 to 41%, and reduced the time period required for the synthesis from 7 to 2½ days.
2.2 Evaluation of phosphinothiols 2–4
To assess our reasoning on Coulombic effects on the traceless Staudinger ligation in water, we used phosphinothiols 2–4 to effect a model dipeptide coupling with 13C-labeled azido-glycine 2710 (Figure 3). Phosphinothioesters 23–26 were synthesized by reaction of the respective phosphinothiol (1–4) with AcGlyOPfP in the presence of DIEA. Equimolar amounts of the resulting phosphinothioester with 13C-labeled azido-glycine 27 were used in each reaction, and yields were determined by quantitative 13C NMR spectroscopy in sodium phosphate buffers of varying pH. As with phosphinothioester 23 (derived from 1), phosphinothioesters 24–26 provided amide 28 with yields dependent on the pH of the reaction mixture, peaking at pH ~8.0. At each pH, however, the ligation yield with 25 was superior to that with phosphinothiol 23, 24 and 26, as expected from a consideration of the proximity of Coulombic interactions.
Figure 3. Comparison of amide yield in traceless Staudinger ligations mediated by phosphinothiols 1–4 in aqueous buffer.
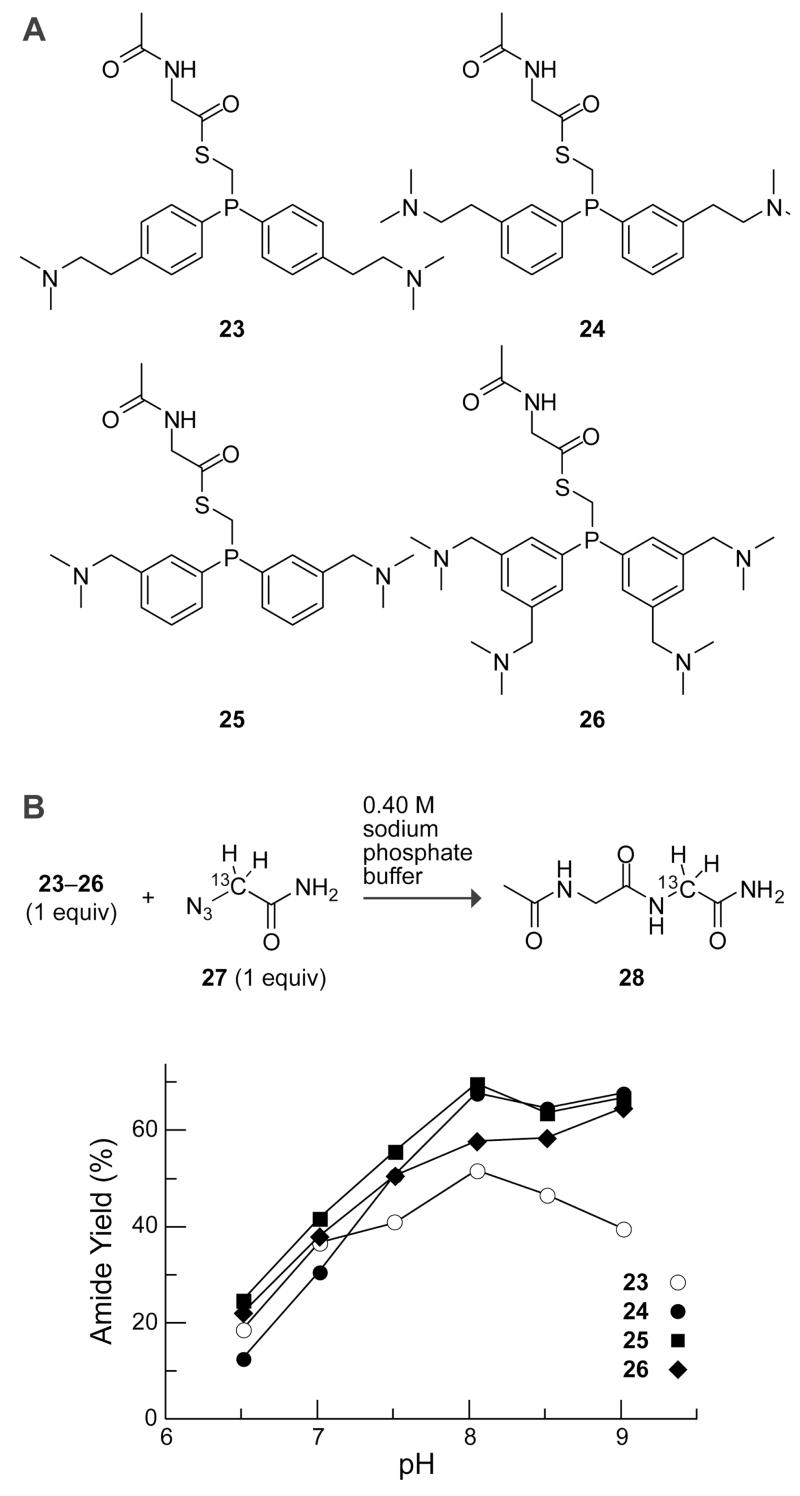
(A) Phosphinothioesters 23–26 were synthesized by reaction of the respective phosphinothiol with AcGlyOPfP. (B) Dependence of amide yield on the solution pH. Reactions were performed with equimolar amounts of phosphinothioesters and azide 27 (60 mM) in 0.40 M sodium phosphate buffers of varying pH. Data are mean values (SE = ±2%) from two experiments, and yields were determined by 13C NMR spectroscopy.10 Data for phosphinothioester 23 are from ref. 10.
Interestingly, the additional cationic charges present in the molecule, as in 26, did not improve yields further. We reason there are competing through-bond effects that promote amine formation, which is the only byproduct of this reaction.10 Unprotonated N,N-dimethylaminomethyl groups are slightly electron-donating substituents and would increase electron density on the phosphorus of the iminophosphorane, rendering the nitrogen more susceptible to protonation. Such an inductive effect is apparent in the phosphorous pKa values of (p-OMe-C6H4)3P (4.57), Ph3P (2.73), and (p-F-C6H4)3P (1.97) in water.14
We sought to determine the pKa values of phosphinothiols 2–4 so as to quantitate the impact of Coulombic effects. Using 31P NMR spectroscopy, we found that the phosphorus atoms of all positively-charged phosphinothiols remained largely unprotonated at pH ≥1.0 in water (data not shown). Thus, the phosphorous pKa value of our cationic phosphinothiols appears to be below the limit of experimental detection. We note, however, that EtPPh2, which is a neutral mimic of HSCH2PPh2, has a phosphorous pKa of 4.90.14 That value is much larger than the phosphorous pKa of phosphinothiols 2–4, consistent with our hypothesis that through-space Coulombic effects lowers the pKa of neighboring atoms, and in particular discourages protonation of the key iminophosphorane intermediate.
3. Conclusions
Careful consideration of the proximity of cationic groups to the key iminophosphorane intermediate permitted the development of improved phosphinothiol reagents for mediating the traceless Staudinger ligation in water. With its positively-charged N,N-dimethylamino groups closer to the iminophosphorane nitrogen, phosphinothiol 3 proved to be superior to our previous reagent (1), approaching yields of 70% near pH 8.0. The synthesis of 3 is efficient and high-yielding, facilitating its widespread use in peptide and protein chemistry. An attempt to amplify the Coulombic effects with additional positive charges proved to be unsuccessful, presumably because of deleterious inductive effects. These results show that an effective use of Coulombic interactions can be used to tune pKa values, which are an important consideration in aqueous traceless Staudinger ligations. Studies are ongoing in our laboratory to use such water-soluble phosphinothiols in the semisynthesis of proteins.
4. Experimental
4.1 General
Reagent chemicals were obtained from commercial suppliers, and reagent grade solvents were used without further purification. Procedures were performed at room temperature (<23 °C) unless indicated otherwise. Reactions were monitored by thin-layer chromatography with visualization by ultraviolet light or staining with KMnO4, ninhydrin, PMA, or I2. Compound purification was carried out with flash chromatography on silica gel, which had a mesh of 230–400 (ASTM) and a pore size of 60 Å. The removal of solvents and other volatile materials “under reduced pressure” refers to the use of a rotary evaporator at water-aspirator pressure (<20 torr) and a water bath of <40 °C.
4.1.1 Instrumentation
NMR spectra were acquired at ambient temperature with a Bruker AC-300 spectrometer (1H, 300 MHz; 13C, 75 MHz; 31P, 121 MHz) at the University of Wisconsin Chemistry Department Nuclear Magnetic Resonance Facility or a Bruker DMX-400 Avance spectrometer (1H, 400 MHz; 13C, 100.6 MHz; 31P, 161 MHz) or Bruker Avance DMX-500 spectrometer (1H, 500 MHz; 13C, 125.7 MHz; 31P, 202 MHz) at the National Magnetic Resonance Facility at Madison (NMRFAM) or a Varian Inova 500 (1H, 500 MHz; 13C, 125.7 MHz; 31P, 202 MHz) spectrometer at the University of Wisconsin Nuclear Magnetic Resonance Facility. Carbon-13 and phosphorus-31 spectra were proton-decoupled, and phosphorus-31 spectra were referenced against an external standard of deuterated phosphoric acid (0 ppm).
Mass spectrometry was performed with a Micromass LCT (electrospray ionization, ESI) in the Mass Spectrometry Facility in the Department of Chemistry.
4.2 Synthesis
4.2.1 m-Br-C6H4CH2CH2OMs (5)
Triethylamine (5.2 mL, 37.3 mmol) was added to a solution of 4-bromophenethyl alcohol (5.0 g, 24.9 mmol) in CH2Cl2 (200 mL), and the resulting solution was cooled to 0 °C with an ice bath. Methanesulfonyl chloride (2.7 mL, 34.8 mmol) was added dropwise to the reaction mixture, and the resulting solution was allowed to warm slowly to room temperature overnight. The solution was washed with 0.1 N HCl and brine, and the combined organic extracts were dried over anhydrous MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The crude yellow solid was purified by flash chromatography (silica gel, 70% v/v hexanes in CH2Cl2) to give mesylate 5 as a white solid in 98% yield. 1H NMR (CDCl3, 400 MHz) δ 7.42–7.40 (m, 2H), 7.21–7.16 (m, 2H), 4.41 (t, J = 6.9 Hz, 2H), 3.03 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H) ppm; observed 13C NMR (CDCl3, 100.6 MHz) δ 138.87, 132.25, 130.51, 127.87, 123.92, 69.79, 37.67, 35.46 ppm; MS (ESI) m/z 300.9519 (MNa+ [C9H11BrO3SNa+] = 300.9595).
4.2.2 m-Br-C6H4CH2CH2NMe2 (6)
Mesylate 5 (6.68 g, 23.9 mmol) was dissolved in anhydrous THF (60 mL) under Ar(g), and dimethylamine (2 M in THF, 47 mL, 94 mmol) was added to this solution. The resulting solution was heated at 45 °C for 30 h. The white solid that formed was removed by filtration, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel, 10% v/v MeOH in CH2Cl2) to give dimethylamine 6 as a yellow oil in 63% yield. 1H NMR (CDCl3, 400 MHz) δ 7.30–7.26 (m, 2H), 7.21–7.17 (m, 2H), 2.80–2.75 (m, 2H), 2.55–2.51 (m, 2H), 2.30 (s, 6H) ppm; observed 13C NMR (CDCl3, 100.6 MHz) δ 140.64, 128.87, 128.87, 128.61, 126.28, 61.86, 45.73, 34.65 ppm; MS (ESI) m/z 228.0387 (MNa+ [C10H14BrNNa+] = 228.0388).
4.2.3 1-Br-3,5-bis(CH2Br)C6H3 (7)
Bromide 7 was synthesized according to reports published previously.12 Spectral data were as reported previously.12
4.2.4 1-Br-3,5-bis(CH2NMe2)C6H3 (8)
Bromide 8 was synthesized according to reports published previously.12 Spectral data were as reported previously.12
4.2.5 HP(O)(C6H4-m-CH2NMe2)2 (10)
Dimethylamine 9 (1.31 g, 6.13 mmol) was dissolved in anhydrous THF (15 mL) under Ar(g) in a flame-dried roundbottom flask equipped with a reflux condenser. To facilitate generation of the Grignard reagent, a catalytic amount of I2 was added to the solution. Crushed magnesium turnings (223 mg, 9.2 mmol) were then added to this solution, and the resulting solution was heated at reflux for 2 h to generate the Grignard reagent. In a separate flamed-dried flask, diethyl phosphite (237 μL, 1.84 mmol) was dissolved in anhydrous THF (1.0 mL), and cooled to 0 °C with an ice bath. The solution of Grignard reagent was added dropwise to this solution, and the resulting solution was allowed to warm to room temperature and stirred overnight. The reaction mixture was then quenched with water (1 mL), and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2, and the resulting solution was washed with water and brine. The combined organic extracts were dried over anhydrous MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2) to give phosphine oxide 10 as a colorless oil in 66% yield. 1H NMR (CDCl3, 400 MHz) δ 8.06 (d, J = 1.2 Hz, 1H), 7.69 (s, 1H), 7.66 (s, 1H), 7.58 (t, J = 7.5 Hz, 2H), 7.54 (dd, J = 8.9, 1.2 Hz 2H), 7.45 (dd, J = 7.6, 3.1 Hz 2H), 3.45 (s, 4H), 2.22 (s, 12H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 140.22 (d, J = 10.7 Hz), 133.45, 132.15, 131.31 (d, J = 12.2 Hz), 129.67 (d, J = 11.2 Hz), 129.14 (d, J = 12.2 Hz), 64.06, 45.58 ppm; 31P NMR (CDCl3, 161 MHz) δ 22.02 ppm; MS (ESI) m/z 288.0393 (MH+ [C18H25N2OPH+] = 288.0383).
4.2.6 HP(O)(C6H4-m-CH2CH2NMe2)2 (11)
Dimethylamine 6 (1.70 g, 7.47 mmol) was dissolved in anhydrous THF (18 mL) under Ar(g) in a flame-dried round-bottom flask equipped with a reflux condenser. To facilitate generation of the Grignard reagent, a catalytic amount of I2(s) was added to the solution. Crushed magnesium turnings (272 mg, 11.2 mmol) were then added, and the resulting solution was heated at reflux for 2 h to generate the Grignard reagent. In a separate flamed-dried flask, diethylphosphite (289 μL, 2.24 mmol) was dissolved in anhydrous THF (1.5 mL), and cooled to 0 °C with an ice bath. The solution of Grignard reagent was added dropwise to this solution, and the resulting solution was allowed to warm to room temperature and stirred overnight. The reaction mixture was then quenched with water (1 mL), and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2, and the resulting solution was washed with water and brine. The combined organic extracts were dried over anhydrous MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2) to give phosphine oxide 11 as a colorless oil in 69% yield. 1H NMR (CDCl3, 400 MHz) δ 8.03 (d, J = 1.2 ppm, 1H), 7.62 (s, 1H), 7.59 (s, 1H), 7.44–7.41 (m, 6H), 2.84-2.80 (m, 4H), 2.55-2.51 (m, 4H), 2.28 (s, 12H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 141.62 (d, J = 14.0 Hz), 133.21, 132.09, 131.10 (d, J = 11.4 Hz), 129.13 (J = 15.4 Hz), 128.53 (d, J = 14.0 Hz), 61.26, 45.62, 34.33 ppm; 31P NMR (CDCl3, 161 MHz) δ 22.14 ppm; MS (ESI) m/z 345.2079 (MNa+ [C20H29N2OPNa+] = 345.2096).
4.2.7 HP(O)(C6H3-bis-m,m-CH2NMe2)2 (12)
Bromide 8 (2.88 g, 10.6 mmol) was dissolved in anhydrous THF (30 mL) under Ar(g) in a flame-dried round-bottom flask equipped with a reflux condenser. To facilitate generation of the Grignard reagent, two drops of 1,2-dibromoethane was added to the solution. Crushed magnesium turnings (387 mg, 16.0 mmol) were then added to this solution, and the resulting solution was heated at reflux for 2 h to generate the Grignard reagent. In a separate flamed-dried flask, diethyl phosphite (256 μL, 3.19 mmol) was dissolved in anhydrous THF (15 mL), and cooled to 0 °C with an ice bath. The solution of Grignard reagent was added dropwise to this solution, and the resulting solution was allowed to warm to room temperature and stirred overnight. The reaction mixture was then quenched with water (1 mL), and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2, and the resulting solution was washed with water and brine. The combined organic extracts were dried over anhydrous MgSO4(s) and filtered, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2 with 1% v/v NEt3) to give phosphine oxide 12 as a yellow oil in 50% yield. 1H NMR (CDCl3, 400 MHz) δ 8.09 (d, J = 1.2 ppm, 1H), 7.58 (s, 2H), 7.54 (s, 2H), 7.51 (s, 2H), 3.44 (s, 8H), 2.21 (s, 24H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 140.00 (d, J = 13.4 Hz), 133.91, 131.36, 129.98 (d, J = 11.6 Hz), 63.75, 45.33 ppm; 31P NMR (CDCl3, 161 MHz) δ 22.48 ppm; MS (ESI) m/z 431.2946 (MH+ [C24H39N4OPH+] = 431.2935).
4.2.8 BH3·PH(C6H4-m-CH2NMe2)2 (13)
A solution of phosphine oxide 10 (383 mg, 1.21 mmol) in anhydrous CH2Cl2 (2.6 mL) was added dropwise slowly to a solution of DIBAl-H (1 M in CH2Cl2, 6.05 mL, 6.05 mmol) under Ar(g) in a flame-dried three-neck round-bottom flask. The resulting solution was stirred for 20 min, then cooled to 0 °C with an ice bath. The solution was then diluted with CH2Cl2 (10 mL), and a sparge needle of Ar(g) was allowed to blow through the solution for 5 min. A solution of 2 N NaOH (5 mL) was added dropwise slowly to the reaction mixture (Caution! Gas evolution!) followed by a saturated solution of Rochelle’s salt (5 mL) to dissipate the emulsion that forms. The resulting biphasic solution was transferred to a separatory funnel, and the organic layer was separated, dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure to ~12 mL. The resulting solution was cooled to 0 °C with an ice bath under Ar(g), and borane·dimethyl sulfide complex (10 M, 387 μL, 3.87 mmol) was added dropwise. The resulting reaction mixture was allowed to warm slowly to room temperature overnight. The solvent was removed under reduced pressure, and the crude oil was purified by flash chromatography (silica gel, CH2Cl2) to give phosphine–borane complex 13 as a white solid in 87% yield. 1H NMR (CDCl3, 400 MHz) δ 7.73–7.70 (m, 4H), 7.55–7.49 (m, 4H), 6.38 (dq, J = 381.3 Hz, 7.1 Hz, 1H), 3.95 (s, 4H), 2.53 (s, 6H), 2.52 (s, 6H), 2.15–0.80 (m, 9H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 137.01 (d, J = 8.4 Hz), 135.91, 133.80 (d, J = 8.5 Hz), 132.85 (d, J = 10.2 Hz), 129.41 (d, J = 56.7 Hz), 126.67 (d, J = 55.9 Hz), 67.39, 50.76 (d, J = 21.6 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ 0.98 ppm; MS (ESI) m/z 360.3095 (MNH4+ [C18H34B3N2PNH4+] = 360.3078).
4.2.9 BH3·PH(C6H4-m-CH2CH2NMe2)2 (14)
A solution of phosphine oxide 11 (288 mg, 0.84 mmol) in anhydrous CH2Cl2 (1.8 mL) was added dropwise slowly to a solution of DIBAl-H (1 M in CH2Cl2, 4.18 mL, 4.18 mmol) under Ar(g) in a flame-dried three-neck round-bottom flask. The resulting solution was stirred for 20 min, then cooled to 0 °C with an ice bath. The solution was then diluted with CH2Cl2 (10 mL), and a sparge needle of Ar(g) was allowed to blow through the solution for 5 min. A solution of 2 N NaOH (5 mL) was added dropwise slowly to the reaction mixture (Caution! Gas evolution!) followed by a saturated solution of Rochelle’s salt (5 mL) to dissipate the emulsion that forms. The resulting biphasic solution was transferred to a separatory funnel, and the organic layer was separated, dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure to ~8 mL. The resulting solution was cooled to 0 °C with an ice bath under Ar(g), and borane·dimethyl sulfide complex (10 M, 268 μL, 2.68 mmol) was added dropwise. The resulting reaction mixture was allowed to warm slowly to room temperature overnight. The solvent was removed under reduced pressure, and the crude oil was purified by flash chromatography (silica gel, CH2Cl2) to give phosphine–borane complex 14 as a white solid in 69% yield. 1H NMR (CDCl3, 400 MHz) δ 7.54–7.50 (m, 4H), 7.44–7.37 (m, 4H), 6.30 (dq, J = 380.2 Hz, 1H), 3.14–3.09 (m, 4H), 2.97–2.93 (m, 4H), 2.66 (s, 12H), 2.15–0.70 (m, 9H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 139.50 (d, J = 9.8 Hz), 133.20 (d, J = 10.4 Hz), 132.48, 131.59 (d, J = 8.8 Hz), 129.77 (d, J = 56.7 Hz), 126.57 (d, J = 58.7 Hz), 65.89, 52.12 (d, J = 9.4 Hz), 30.92 ppm; 31P NMR (CDCl3, 161 MHz) δ 1.64 ppm; MS (ESI) m/z 388.3372 (MNH4+ [C20H36B3N2PNH4+] = 388.3391).
4.2.10 BH3·PH(C6H3-bis-m,m-CH2NMe2)2 (15)
A solution of phosphine oxide 12 (200 mg, 0.46 mmol) in anhydrous CH2Cl2 (1.5 mL) was added dropwise slowly to a solution of DIBAl-H (1 M in CH2Cl2, 2.32 mL, 2.32 mmol) under Ar(g) in a flame-dried three-neck round-bottom flask. The resulting solution was stirred for 20 min, then cooled to 0 °C with an ice bath. The solution was then diluted with v (10 mL), and a sparge needle of Ar(g) was allowed to blow through the solution for 5 min. A solution of 2 N NaOH (3 mL) was added dropwise slowly to the reaction mixture (Caution! Gas evolution!) followed by a saturated solution of Rochelle’s salt (5 mL) to dissipate the emulsion that forms. The resulting biphasic solution was transferred to a separatory funnel, and the organic layer was separated, dried over anhydrous MgSO4(s), filtered, and concentrated under reduced pressure to ~5 mL. The resulting solution was cooled to 0 °C with an ice bath under Ar(g), and borane·dimethyl sulfide complex (10 M, 241 μL, 2.40 mmol) was added dropwise. The resulting reaction mixture was allowed to warm slowly to room temperature overnight. The solvent was removed under reduced pressure, and the crude oil was purified by flash chromatography (silica gel, 5% v/v EtOAc and 25% v/v hexanes in CH2Cl2) to give phosphine–borane complex 15 as a white solid in 50% yield. 1H NMR (CDCl3, 400 MHz) δ 7.85 (s, 2H), 7.82 (s, 2H), 7.70 (s, 2H), 6.47 (dq, J = 0.963 ppm, 7.1 Hz, 1H), 3.98-3.91 (m, 8H), 2.56 (s, 12H), 2.53 (s, 12H), 1.80–0.80 (m, 15H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 140.27, 137.53 (d, J = 9.8 Hz), 133.05 (d, J = 10.0 Hz), 125.69 (d, J = 56 Hz), 67.12, 51.38 (d, J = 50.9 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ −1.12 ppm; MS (ESI) m/z 507.4447 (MNa+ [C24H54B5N4PNa+] = 507.4444).
4.2.11 BH3·AcSCH2P(C6H4-m-CH2NMe2)2 (17)
Phosphine-borane complex 13 (81 mg, 0.24 mmol) was dissolved in dry DMF (2.5 mL) under Ar(g) and cooled to 0 °C. NaH (5.7 mg, 0.24 mmol) was added slowly, and the mixture was stirred at 0 °C until bubbling ceased. Bromide 16 (40 mg, 0.24 mmol) was then added, and the resulting mixture was allowed to warm to room temperature and stirred for 12 h. Solvent was removed under reduced pressure, and the residue was purified by flash chromatography (silica gel, 2% v/v ethyl acetate and 28% hexanes in CH2Cl2) to give phosphine–borane complex 17 as a white solid in 78% yield. 1H NMR (CDCl3, 400 MHz) δ 7.78–7.73 (m, 4H), 7.57–7.49 (m, 4H), 3.95 (d, J = 2.4 Hz, 4H), 3.73 (d, J = 6.3 Hz, 2H), 2.53 (d, J = 6.3 Hz, 12H), 2.25 (s, 3H), 2.20–0.60 (m, 9H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 192.88, 136.42 (d, J = 9.5 Hz), 135.93, 133.41 (d, J = 9.4 Hz), 132.53 (d, J = 9.9 Hz), 129.03 (d, J = 10.0 Hz), 127.73 (d, J = 54.6 Hz), 67.45, 53.65, 30.18, 23.70 (d, J = 35.7 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ 19.97 ppm; MS (ESI) m/z 453.2595 (MNa+ [C21H38B3N2OPSNa+] = 453.2614).
4.2.12 BH3·AcSCH2P(C6H4-m-CH2CH2NMe2)2 (18)
Phosphine-borane complex 14 (100 mg, 0.27 mmol) was dissolved in dry DMF (3 mL) under Ar(g) and cooled to 0 °C. NaH (6.5 mg, 0.27 mmol) was added slowly, and the mixture was stirred at 0 °C until bubbling ceased. Bromide 16 (46 mg, 0.27 mmol) was then added, and the resulting mixture was allowed to warm to room temperature and stirred for 12 h. Solvent was removed under reduced pressure, and the residue was purified by flash chromatography (silica gel, 2% v/v ethyl acetate and 28% hexanes in CH2Cl2) to give phosphine–borane complex 18 as a white solid in 75% yield. 1H NMR (CDCl3, 400 MHz) δ 7.56–7.52 (m, 4H), 7.45–7.38 (m, 4H), 3.71 (d, J = 6.8 Hz, 2H), 3.13–3.09 (m, 4H), 2.97–2.92 (m, 4H), 2.67 (d, J = 2.7 Hz, 12H), 2.28 (s, 3H), 2.40–0.60 (m, 9H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 193.34, 139.26 (d, J = 9.9 Hz), 132.77 (d, J = 11.7 Hz), 132.58, 130.96 (d, J = 8.3 Hz), 129.54 (d, J = 10.0 Hz), 128.23 (d, J = 55.9 Hz), 65.83, 52.05, 30.95, 30.32, 23.81 (d, J = 35.3 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ 19.65 ppm; MS (ESI) m/z 481.2918 (MNa+ [C23H42B3N2OPSNa+] = 481.2927).
4.2.13 BH3·AcSCH2P(C6H3-bis-m,m-CH2NMe2)2 (19)
Phosphine-borane complex 15 (86 mg, 0.18 mmol) was dissolved in dry DMF (2 mL) under Ar(g) and cooled to 0 °C. NaH (4.3 mg, 0.18 mmol) was added slowly, and the mixture was stirred at 0 °C until bubbling ceased. Bromide 16 (30 mg, 0.18 mmol) was then added, and the resulting mixture was allowed to warm to room temperature and stirred for 12 h. Solvent was removed under reduced pressure, and the residue was purified by flash chromatography (silica gel, 10% v/v ethyl acetate and 20% v/v hexanes in CH2Cl2) to give phosphine–borane complex 19 as a white solid in 56% yield. 1H NMR (CDCl3, 400 MHz) δ 7.91 (d, J = 1.1 Hz, 2H), 7.88 (d, J = 1.1 Hz, 2H), 7.74 (s, 2H), 3.93 (q, J = 13.7 Hz, 8H), 3.73 (d, J = 6.3 Hz, 2H), 2.58 (s, 12H), 2.57 (s, 12H), 2.23 (s, 3H), 2.00–1.50 (m, 15H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 192.70, 140.46, 137.24 (d, J = 9.5 Hz), 132.84 (d, J = 10.2 Hz), 126.98 (d, J = 54.9 Hz), 67.94, 52.04, 51.09, 30.28 24.21 (d, J = 34.9 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ 18.74 ppm; MS (ESI) m/z 590.4409 (MNa+ [C27H58B5N4OPSNa+] = 590.4427).
4.2.14 AcSCH2P(C6H4-m-CH2NMe2)2 (20)
Phosphine–borane complex 17 (100 mg, 0.23 mmol) was dissolved in toluene (2 mL) under Ar(g). DABCO (84 mg, 0.74 mmol) was added, and the resulting solution was heated to 40 °C for 4 h. The solvent was removed under reduced pressure, and the residue was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2) to give phosphinothioester 20 as a colorless oil in 95% yield. 1H NMR (CDCl3, 400 MHz) δ 7.38–7.29 (m, 8H), 3.53 (d, J = 3.7 Hz, 2H), 3.41 (s, 4H), 2.28 (s, 3H), 2.22 (s, 12H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 194.74, 138.88 (d, J = 4.7 Hz), 136.91 (d, J = 13.4 Hz), 133.83 (t, J = 19.2 Hz), 131.56, 130.17 (d, J = 29.9 Hz), 128.63 (d, J = 32.2 Hz), 64.15, 45.63, 30.76, 20.96 (d, J = 23.5 Hz) ppm; 31P NMR (CDCl3, 161 MHz) –15.23 ppm.
4.2.15 AcSCH2P(C6H4-m-CH2CH2NMe2)2 (21)
Phosphine–borane complex 18 (38 mg, 0.096 mmol) was dissolved in toluene (1 mL) under Ar(g). DABCO (35 mg, 0.30 mmol) was added, and the resulting solution was heated to 40 °C for 4 h. The solvent was removed under reduced pressure, and the residue was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2) to give phosphinothioester 21 as a colorless oil in 95% yield. 1H NMR (CDCl3, 400 MHz) δ 7.28–7.20 (m, 8H), 3.50 (d, J = 3.4 Hz, 2H), 2.80–2.76 (m, 4H), 2.57–2.53 (m, 4H), 2.32 (s, 12H), 2.30 (s, 3H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 194.87, 140.54 (d, J = 7.7 Hz), 136.96 (d, J = 14.2 Hz), 133.25 (d, J = 22.8 Hz), 130.48 (d, J = 16 Hz), 129.70, 128.78 (d, J = 4.9 Hz), 64.26, 46.94, 45.47, 34.25, 25.99 (d, J = 23.4 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ −14.99 ppm.
4.2.16 AcSCH2P(C6H3-bis-m,m-CH2NMe2)2 (22)
Phosphine–borane complex 19 (59 mg, 0.10 mmol) was dissolved in toluene (1.5 mL) under Ar(g). DABCO (60 mg, 0.54 mmol) was added, and the resulting solution was heated to 40 °C for 4 h. The solvent was removed under reduced pressure, and the residue was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2 with 1% v/v NEt3) to give phosphinothioester 22 as a colorless oil in 91% yield. 1H NMR (CDCl3, 400 MHz) δ 7.28–7.21 (m, 6H), 3.53 (d, J = 3.7 Hz, 2H), 3.39 (m, 8H), 2.27 (s, 3H), 2.21 (s, 24H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 194.82, 139.07 (d, J = 7.4 Hz), 136.88 (d, J = 14.6 Hz), 132.48 (d, J = 18.8 Hz), 131.01, 64.12, 45.39, 30.43, 26.05 ppm; 31P NMR (CDCl3, 161 MHz) δ −15.32 ppm.
4.2.17 HSCH2P(C6H4-m-CH2NMe2)2·2 HCl (3)
Phosphinothioester 20 (35 mg, 0.09 mmol) was dissolved in degassed MeOH (1 mL), and NaOH (3.7 mg) was added to this solution under Ar(g). The resulting solution was stirred at room temperature for 1.5 h. The solvent was removed under reduced pressure, and the residue was acidified with 4 N HCl in dioxane and filtered to give hydrochloride salt of phosphosphinothiol 3 as a white solid in quantitative yield. 1H NMR (CD3OD, 400 Hz) δ 7.81 (d, J = 5.9 Hz, 2H), 7.61–7.56 (m, 4H), 7.49 (d, J = 7.4 Hz, 2H), 4.37 (s, 4H), 3.27 (d, J = 2.6 Hz, 2H), 2.84 (s, 12H) ppm; 13C NMR (CD3OD, 100.6 MHz) δ 140.34 (d, J = 17.4 Hz), 136.25 (d, J = 17.2 Hz), 135.45 (d, J = 23.1 Hz), 133.02, 132.18, 130.65 (d, J = 7.0 Hz), 61.99, 54.95, 43.16 ppm; 31P NMR (CDCl3, 161 MHz) δ −8.59 ppm.
4.2.18 HSCH2P(C6H3-bis-m,m-CH2NMe2)2·4 HCl (4)
Phosphinothioester 22 (47 mg, 0.09 mmol) was dissolved in degassed MeOH (1 mL), and NaOH (3.7 mg) was added to this solution under Ar(g). The resulting solution was stirred at room temperature for 1.5 h. The solvent was removed under reduced pressure, and the residue was acidified with 4 N HCl in dioxane and filtered to give hydrochloride salt of phophosphinothiol 4 as a white solid in quantitative yield. 1H NMR (CDCl3, 400 Hz) δ 7.26–7.24 (m, 6H), 4.05 (d, J = 5.2 Hz, 2H), 3.38, (s, 8H), 3.07 (m, 2H), 2.20 (s, 24H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 139.20 (d, J = 6.3 Hz), 137.12 (d, J = 14.4 Hz), 132.48 (d, J = 17.0 Hz), 130.99, 64.20, 45.48, 21.14 (d, J = 23.3 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ −7.71 ppm.
4.2.19 AcGlySCH2P(C6H4-m-CH2CH2NMe2)2 (24)
Phosphinothioester 21 (40 mg, 0.10 mmol) was dissolved in degassed MeOH (1 mL), and NaOH (3.8 mg) was added to this solution under Ar(g). The resulting solution was stirred at room temperature for 1.5 h. The solvent was removed under reduced pressure, and the residue was acidified with 4 N HCl in dioxane and filtered to give hydrochloride salt of phophosphinothiol 2 as a white solid in quantitative yield. The residue was used without further purification. N-Acetyl glycine pentafluorophenol ester (28.3 mg, 0.1 mmol) was dissolved in anhydrous DMF (1 mL) under Ar(g). The hydrochloride salt of phosphinothiol 2 from above was added, followed by DIEA (67 μL, 0.38 mmol). The resulting solution was stirred for 4 h at room temperature under Ar(g). The solvent was removed under reduced pressure, and the resulting crude oil was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2) to give phosphinothioester 24 as a colorless oil in 75% yield. 1H NMR (CDCl3, 400 Hz) δ 7.30–7.22 (m, 8H), 4.19 (d, J = 6.2 Hz, 2H), 3.54 (d, J = 3.9 Hz, 2H), 2.80–2.76 (m, 4H), 2.55–2.51 (m, 4H), 2.30 (s, 12H), 2.05 (s, 3H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 196.48, 170.50, 140.66 (d, J = 6.9 Hz), 136.78 (d, J = 13.4 Hz), 133.37 (d, J = 20.0 Hz), 130.66 (d, J = 18.0 Hz), 129.92, 128.93, 61.38, 49.35, 45.62, 34.43, 25.60 (d, J = 24.8 Hz), 23.25 ppm; 31P NMR (CDCl3, 161 MHz) δ −14.48 ppm.
4.2.20 AcGlySCH2P(C6H4-m-CH2NMe2)2 (25)
Phosphinothioester 20 (72 mg, 0.21 mmol) was dissolved in degassed MeOH (2 mL), and NaOH (8.3 mg) was added to this solution under Ar(g). The resulting solution was stirred at room temperature for 1.5 h. The solvent was removed under reduced pressure, and the residue was acidified with 4 N HCl in dioxane and filtered to give the hydrochloride salt of phophosphinothiol 3 as a white solid in quantitative yield. The residue was used without further purification. N-Acetyl glycine pentafluorophenol ester (62 mg, 0.22 mmol) was dissolved in anhydrous DMF (2 mL) under Ar(g). A solution of the above phosphinothiol in DMF (1 mL) was added to the mixture. DIEA (144 μL, 0.83 mmol) was added to the resulting solution, and the mixture was stirred for 4 h at room temperature under Ar(g). The solvent was removed under reduced pressure, and the resulting crude oil was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2) to give phosphinothioester 25 as a colorless oil in 95% yield. 1H NMR (CDCl3, 400 Hz) δ 7.40–7.28 (m, 8H), 6.68 (bs, 1H), 4.11 (d, J = 6.0 Hz, 2H), 3.53 (d, J = 4.4 Hz, 2H), 3.48 (s, 4H), 2.27 (s, 12H), 2.04 (s, 3H) ppm; 13C NMR (CDCl3, 100.6 MHz) δ 196.63, 170.75, 137.01 (d, J = 14.2 Hz), 136.38, 134.18 (d, J = 18.8 Hz), 132.65 (d, J = 19.8 Hz), 130.93, 128.96 (d, J = 6.2 Hz), 63.37, 49.15, 44.65, 25.32, 23.11 ppm; 31P NMR (CDCl3, 161 MHz) δ −14.33 ppm.
4.2.21 AcGlySCH2P(C6H3-bis-m,m-CH2NMe2)2 (26)
N-Acetyl glycine pentafluorophenol ester (27.7 mg, 0.1 mmol) was dissolved in anhydrous DMF (1 mL) under Ar(g). The hydrochloride salt of phosphinothiol 4 (41.4 mg, 0.09 mmol) was added, followed by DIEA (129 μL, 0.74 mmol). The resulting solution was stirred for 4 h at room temperature under Ar(g). The solvent was removed under reduced pressure, and the resulting crude oil was purified by flash chromatography (silica gel, 20% v/v MeOH in CH2Cl2 with 1% v/v NEt3) to give phosphinothioester 26 as a mixture with unreacted 4. This mixture was dissolved in CH2Cl2 (2 mL) and 2-chlorotrityl chloride resin (200 mesh) (3 equiv) and TBD-methyl polystyrene resin (5 equiv) was added and gently stirred at RT for 30 min. The resin was filtered and washed with CH2Cl2. The filtrate was concentrated under reduced pressure to give 26 as a colorless oil in 72% yield. 1H NMR (CDCl3, 400 Hz) δ 7.27–7.22 (m, 6H), 6.50 (bs, 1H), 4.09 (d, J = 5.7 Hz, 2H), 3.54 (d, J = 4.6 Hz, 2H), 3.37 (s, 8H), 2.20 (s, 24H), 2.02 (s, 3H) ppm; observed 13C NMR (CDCl3, 100.6 MHz) δ 196.41, 170.35, 139.19 (d, J = 7.5 Hz), 136.51 (d, J = 13.6 Hz), 132.51 (d, J = 18.2 Hz), 64.22, 53.61, 49.20, 45.51, 25.29 (d, J = 26.5 Hz) ppm; 31P NMR (CDCl3, 161 MHz) δ −13.68 ppm.
4.3 Staudinger ligations of 23 + 27, 24 + 27, 25 + 27, and 26 + 27
The respective phosphinothioester (36 μmol) and azide 27 (3.6 mg, 36 μmol) were dissolved in 0.40 M sodium phosphate buffers (0.6 mL) of various pH. The resulting reaction mixtures were stirred for 16 h and the reaction was monitored with quantitative 13C NMR spectroscopy. Amide yields were obtained by integration of all normalized product 13C NMR signals.
4.4 pKa Determination of phosphinothiols
Phosphinothioesters 20–22 (10 mg) were each dissolved in sodium phosphate buffer containing 20% v/v D2O. The solution was adjusted with a solution of either 6 N HCl or 2 N NaOH, and chemical shifts at each pH were determined by 31P NMR spectroscopy. An external standard (D3PO4) was used to calibrate the chemical shifts. A control experiment using tris(2-carboxyethyl)phosphine (TCEP) gave a pKa value of 7.7, which is similar to the reported value of 7.66.15
Acknowledgments
This paper is dedicated to Professor Jonathan A. Ellman on the occasion of his winning the 2006 Tetrahedron Young Investigator Award. We acknowledge E. L. Myers for comments on this manuscript. This work was supported by grant GM044783 (NIH).
Footnotes
NMR spectra of the novel compounds reported herein.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dawson PE, Kent SB. Annu Rev Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]
- 2.Muir TW. Annu Rev Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson BL, Soellner MB, Raines RT. Annu Rev Biophys Biomol Struct. 2005;34:91–118. doi: 10.1146/annurev.biophys.34.040204.144700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]; (b) Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2001;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]; (c) Köhn M, Breinbauer R. Angew Chem Int Ed. 2004;43:3106–3116. doi: 10.1002/anie.200401744. [DOI] [PubMed] [Google Scholar]
- 5.Soellner MB, Tam A, Raines RT. J Org Chem. 2006;71:9824–9830. doi: 10.1021/jo0620056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Soellner MB, Nilsson BL, Raines RT. J Am Chem Soc. 2006;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]; (b) Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]; (c) Bräse S, Gil C, Knepper K, Zimmermann V. Angew Chem Int Ed. 2005;44:5188–5240. doi: 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]
- 7.Soellner MB, Nilsson BL, Raines RT. J Org Chem. 2002;67:4993–4996. doi: 10.1021/jo025631l. [DOI] [PubMed] [Google Scholar]
- 8.Nilsson BL, Hondal RJ, Soellner MB, Raines RT. J Am Chem Soc. 2003;125:5268–5269. doi: 10.1021/ja029752e. [DOI] [PubMed] [Google Scholar]
- 9.(a) Soellner MB, Dickson KA, Nilsson BL, Raines RT. J Am Chem Soc. 2003;125:11790–11791. doi: 10.1021/ja036712h. [DOI] [PubMed] [Google Scholar]; (b) Watzke A, Kohn M, Gutierrez-Rodriguez M, Wacker R, Schroder H, Breinbauer R, Kuhlmann J, Alexandrov K, Niemeyer CM, Goody RS, Waldmann H. Angew Chem Int Ed. 2006;45:1408–1412. doi: 10.1002/anie.200502057. [DOI] [PubMed] [Google Scholar]
- 10.Tam A, Soellner MB, Raines RT. J Am Chem Soc. 2007;129:11421–11430. doi: 10.1021/ja073204p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown HC, McDaniel DH, Häfliger O. In: Determination of Organic Structures by Physical Methods. Braude EA, Hachod FC, editors. Academic Press; New York: 1955. pp. 567–662. [Google Scholar]
- 12.Vandekuil LA, Luitjes H, Grove DM, Zwikker JW, Vanderlinden JGM, Roelofsen AM, Jenneskens LW, Drenth W, Vankoten G. Organometallics. 1994;13:468–477. [Google Scholar]
- 13.Farrington GK, Kumar A, Wedler FC. Org Prep Proced Int. 1989;21:390–392. [Google Scholar]
- 14.Moore SJ, Marzilli LG. Inorg Chem. 1998;37:5329–5335. doi: 10.1021/ic971299y. [DOI] [PubMed] [Google Scholar]
- 15.Podlaha J, Podlahova J. Collect Czech Chem Commun. 1973;38:1730–1736. [Google Scholar]