

Abstract
Purpose
Tumor necrosis factor-α has a significant role in renal tubular cell apoptosis during obstruction induced renal injury. While we have previously reported the role of tumor necrosis factor-α in extrinsic pathway apoptotic signaling during renal obstruction, to our knowledge its effect on intrinsic pathway signaling and mitochondrial release of cytochrome C has not previously been evaluated.
Materials and Methods
Male Sprague-Dawley rats were anesthetized and underwent unilateral ureteral obstruction vs sham operation. At 24 hours before surgery and every 84 hours thereafter the animals received vehicle or a pegylated form of soluble tumor necrosis factor receptor type 1. The kidneys were harvested 1 week postoperatively. The renal cortex was analyzed for tumor necrosis factor-α production (enzyme-linked immunosorbent assay), apoptosis (TUNEL and enzyme-linked immunosorbent assay), Bcl-2, Bcl-xL, Bax, caspase 8 and truncated Bid expression (Western blot), and mitochondrial cytochrome C release (immunohistochemistry).
Results
Renal obstruction induced increased tumor necrosis factor-α production, apoptotic renal tubular death, the expression of Bax, caspase 8 and truncated BID, and mitochondrial release of cytochrome C, while simultaneously stimulating decreased Bcl-2 and Bcl-xL expression. Treatment with the pegylated form of soluble tumor necrosis factor receptor type 1 significantly decreased obstruction induced tumor necrosis factor-α production, apoptosis, Bax, caspase 8, truncated Bid expression and mitochondrial cytochrome C release, and increased Bcl-2 and Bcl-xL expression.
Conclusions
These results demonstrate that tumor necrosis factor-α stimulates Bid and subsequent intrinsic apoptotic signaling pathway activation during unilateral ureteral obstruction, resulting in mitochondrial cytochrome C release and apoptotic cell death. We identified tumor necrosis factor-α neutralization as a potential therapeutic option for ameliorating obstruction induced renal injury.
Keywords: cytokines, apoptosis, mitochondria
Upper urinary tract obstruction is a common cause of renal dysfunction in children and adults. Histologically the condition is characterized by significant collecting system dilatation, increasing tubulointerstitial fibrosis and a loss of renal parenchymal mass secondary to progressive apoptotic tubular cell death.1 Renal tubular cells are highly susceptible to obstruction induced apoptosis with peak apoptotic cell death occurring after 7 to 14 days of renal obstruction.2,3
As a gene directed process, apoptosis can be triggered by an extrinsic death receptor signaling pathway or an intrinsic pathway involving mitochondrial membrane disturbances and cytochrome C release.4 Bcl family members are important mediators of the mitochondrial pathway of apoptotic cell death because they control the permeability of the mitochondrial membrane and, therefore, influence the mitochondrial release of cytochrome C.4 This family of proteins is divided into 1 group with pro-apoptotic properties (Bax, Bak, Bad, etc) and 1 group with anti-apoptotic properties (Bcl-2 and Bcl-XL).4,5 Bax is located in the cytosol in its inactive state but upon activation it translocates into the mitochondrial membrane and initiates its pro-apoptotic effect.6 On the other hand, Bcl-2 is located on the outer surface of the mitochondrial membrane as well as the membranes of the endoplasmic reticulum and nuclear envelope.6 The relative expression of these 2 groups of proteins determines the ability of Bcl proteins to form pore-like complexes in the mitochondrial membrane, which facilitate the release of cytochrome C and subsequently commitment of the cell to apoptosis. Chevalier et al examined the interaction of pro-apoptotic Bax and anti-apoptotic Bcl-2 during renal obstruction and found that chronic renal obstruction inhibits Bcl-2 expression, thereby promoting obstruction induced renal tubular cell apoptosis.2
TNF-α is a cytotoxic cytokine that has emerged as an important mediator of inflammatory renal injury.7–9 Recently TNF-α was shown to stimulate obstruction induced renal cell apoptosis by increasing the activation of caspase 8 and 3 in the extrinsic death receptor signaling pathway.9 A link between the intrinsic and extrinsic pathways of apoptosis appears to exist with the cytoplasmic protein Bid.10 Bid is activated by caspase 8, an initiator caspase in the death receptor signaling pathway, and its truncated form t-Bid subsequently becomes incorporated into the mitochondrial cell wall. Through an unclear mechanism t-Bid activates the intrinsic pathway of apoptosis following its incorporation into mitochondria.11 While the intrinsic and extrinsic pathways of apoptosis are activated during renal obstruction,2,9 to our knowledge the effect of obstruction induced TNF-α production on Bid activation, intrinsic apoptotic signaling and mitochondrial cytochrome C release has not previously been evaluated. Therefore, we determined 1) TNF-α production, 2) renal tubular cell apoptosis, 3) Bax, Bcl-2 and Bcl-xL expression, 4) active caspase 8 and t-Bid expression, 4) mitochondrial release of cytochrome C and 4) the impact of TNF-α neutralization on these parameters in a rat model of UUO.
MATERIALS AND METHODS
Animals, Experimental Groups and Operative Techniques
The animal protocol was reviewed and accepted by the animal care and research committee at Indiana University School of Medicine. Male Sprague-Dawley rats weighing 200 to 250 gm were acclimated and maintained on a standard pellet diet for 1 week before experiment initiation. Rats were anesthetized using isoflurane inhalation. Following the induction of anesthesia the left ureter was isolated and completely ligated via a midline laparotomy. Sham treated animals underwent an identical surgical procedure without ureteral ligation. At the completion of the experiment the animals were re-anesthetized, the left kidneys were removed and snap frozen in liquid nitrogen, and the animals were subsequently sacrificed. The animals were divided into group 1—1-week sham operation plus vehicle, group 2—1-week UUO plus vehicle and group 3—1-week UUO plus PEG-sTNFR1, with 6 in each group.
PEG-sTNFR1
TNF-α neutralization was achieved using a soluble, long acting form of TNFR1. Recombinant sTNFR1 is an Escherichia coli derived, 2 domain, monomeric form of the 4 domain sTNFR1. For prolonged half-life a high molecular mass PEG molecule has been attached at the NH2-terminal position. Preclinical studies to date have demonstrated that subcutaneous administration of PEG-sTNFR1 is effective for limiting the inflammatory reaction of rheumatoid arthritis in rat models at a dose as low as 0.3 mg/kg.12
Tissue Homogenization
A portion of the renal cortex from each kidney was homogenized for testing in a TNF-α ELISA. Homogenization was performed after the tissue samples were diluted in 5 volumes of homogenate buffer composed of 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM egtazic acid, 1 mM dithiothreitol and 0.5 mM phenylmethylsulfonyl fluoride, using a Virtis-hear tissue homogenizer (Virtis, Gardiner, New York). Renal homogenates were centrifuged at 3,000 × gravity for 15 minutes at 4C. Supernatants were subsequently stored at −80C until TNF-α ELISA could be performed.
TNF-α Protein Expression
Renal cortical homogenate TNF-α protein content was determined using ELISA. ELISA was performed by adding 100 μl of each sample to wells in a 96-well plate of a rat ELISA kit (BD™ Biosciences). Samples were tested in duplicate. TNF-α ELISA was performed according to manufacturer instructions. Final results are expressed in pg TNF-α/mg protein.
Apoptosis ELISA
A portion of the renal cortex from each kidney was homogenized in liquid nitrogen and resuspended in 4 volumes of ice-cold 50 mM tris-HCl (pH 7.4). Samples were sheared by passing them through a 19 gauge needle and agitated on ice for 30 minutes. Four volumes of 0.1% Triton X-100 were added to each sample and the samples were agitated on ice for 60 minutes. The samples were then centrifuged at 13,000 rpm for 20 minutes at 4C. Cell Death Detection ELISA Plus photometric enzyme immunoassay (Roche Diagnostics, Mannheim, Germany) was performed to quantitatively determine cytoplasmic histone associated DNA fragments representative of apoptosis using 20 μl supernatant from each sample, as described by the manufacturer. Samples were tested in duplicate and results are expressed as optical density per mg protein.
Western Blot Analysis
Protein extracts (50 μg per lane) from homogenized samples were electrophoresed onto tris-glycine gel (Invitrogen™) and transferred to nitrocellulose or polyvinylidene fluoride membrane. Immunoblotting was performed by incubating each membrane in 5% dry milk overnight at 4C, followed by incubation with anti-Bcl-2, anti-Bax and anti-Bcl-xL (each 1:500) each overnight at 4C (Cell Signaling Technology®), anti-caspase 8 (Santa Cruz Biotechnology, Santa Cruz, California) (1:200) for 2 hours at RT or anti-t-Bid antibody (1:500) overnight at 4C. After being washed 3 times in tris-PBS each membrane was incubated for 1 hour with secondary antibody. The membranes were then developed using enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, New Jersey). Equivalent protein loading for each lane was confirmed by stripping and reblotting each membrane for β-actin. Analysis was repeated in triplicate to ensure the reproducibility of results. The membranes were developed using enhanced chemiluminescence. The density of each band was determined using National Institutes of Health image analysis software and expressed as a percent of β-actin density.
Mitochondrial Cytochrome C Release
Renal cortical tissue sections (6 μm) were prepared from each sample with a cryostat and fixed in 4% methanol-free formaldehyde for 15 minutes. The slides were washed in PBS 3 × 5 minutes. Normal goat serum was applied as a blocking agent for 1 hour at RT in a dark humidifying chamber and the slides were washed in PBS 3 × 5 minutes. Sections were incubated with anti-cytochrome C antibody (Calbiochem®) (1:100) for 1 hour at RT. After washing in PBS 3 × 5 minutes the slides were incubated with rabbit anti-sheep secondary antibody (Invitrogen) (1:200) for 1 hour at RT, washed and exposed to Texas red labeled donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania) (1:200) for 1 hour at RT. Nuclei were counterstained with bisbenzimide (10 mg/ml in PBS) for 30 seconds. The slides were washed with PBS 3 × 2 minutes and mounted. To test for nonspecific fluorescence adjacent sections from each experimental group were incubated with nonimmune purified sheep IgG instead of primary antibody. Nonspecific fluorescence was digitally subtracted and sections were photographed.
Statistical Analysis
Data are presented as the mean ± SD. Differences at the 95% CI were considered significant. Experimental groups were compared using ANOVA with the post hoc Bonferroni-Dunn test using JMP®, version 5.0.
RESULTS
TNF-α Protein Production
Renal cortical tissue obtained from sham operated animals revealed minimal TNF-α production (70 ± 3.1 pg/mg protein) (fig. 1, A). TNF-α levels increased significantly in renal cortical tissue in response to 1 week of obstruction vs sham operation (116.0 ± 5.3 vs 70 ± 3.1 pg/mg protein, p <0.0001). However, animals exposed to PEG-sTNFR1 during the time course of renal obstruction showed a significant decrease in TNF-α production compared to levels for sham treatment with vehicle (71 ± 2.8 vs 116 ± 5.3 pg/mg protein, p <0.0001).
Fig. 1.

ELISA in renal cortical tissue after 1-week obstruction in presence of vehicle (OB) or PEG-sTNFR1 (OB + PEG-sTNFR1). A, TNF-α production in pg/mg protein. B, histone associated DNA fragment accumulation. Sham, sham operation.
Apoptosis
Renal cortical samples from each treatment group were homogenized and analyzed quantitatively for cytoplasmic histone associated DNA fragments, a marker of apoptosis. The quantity of cytoplasmic histone associated DNA fragments increased significantly in response to 1 week of obstruction vs sham operation (0.19 ± 0.02 vs 0.04 ± 0.007, p <0.0001, fig. 1, B). However, animals exposed to PEG-sTNFR1 during the time course of renal obstruction showed a significant decrease in the quantity of cytoplasmic histone associated DNA fragments, which approached levels of sham treatment with vehicle (0.06 ± 0.009 vs 0.19 ± 0.02, p <0.0001).
Bcl Family Expression
Protein extracts from homogenized renal samples from each treatment group were analyzed for the activity of Bcl-XL, Bcl-2 and Bax activity, which are mediators of the intrinsic apoptotic signaling pathway. As anti-apoptotic factors, Bcl-XL and Bcl-2 showed increased expression in sham treated samples (1.2% ± 0.2% and 0.93% ± 0.2% β-actin density, respectively, fig. 2). Bcl-XL and Bcl-2 expression decreased significantly in response to 1 week of renal obstruction vs sham treatment (0.28% ± 0.08% β-actin density vs 1.2% ± 0.2%, p <0.005, and 0.03% ± 0.008% vs 0.93% ± 0.2%, p <0.005). Exposure to TNF-α neutralization during the time course of renal obstruction preserved Bcl-XL and Bcl-2 expression vs vehicle (0.88% ± 0.07% β-actin density vs 0.28% ± 0.08%, p <0.005, and 0.62% ± 0.01% vs 0.03% ± 0.008%, p <0.0001).
Fig. 2.

Western blots and graphs show renal cortical Bcl-2, Bcl-xL and Bax expression after 1-week obstruction in presence of vehicle (OB) or PEG-sTNFR1 (OB + PEG-sTNFR1). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Sham, sham operation.
The pro-apoptotic factor Bax showed minimal expression in sham treated samples. However, expression of this mediator was significantly increased in response to 1 week of renal obstruction vs sham operation (0.87% ± 0.13% vs 0.14% ± 0.03% β-actin density, p <0.05, fig. 2). Animal exposure to TNF-α neutralization during the period of renal obstruction significantly decreased Bax expression vs vehicle (0.27% ± 0.03% vs 0.87% ± 0.13% β-actin density, p <0.05).
Caspase 8 and Bid Activation
To evaluate the mechanisms by which TNF-α activates the intrinsic mitochondrial signaling pathway of apoptosis caspase 8 and Bid activation were evaluated. Cleaved caspase 8 and t-Bid expression were low in sham treated animals, while the expression of each mediator increased significantly in response to 1 week of obstruction vs sham treatment (0.82% ± 0.08% β-actin density vs 0.15% ± 0.06%, p <0.05, and 0.43% ± 0.03% vs 0.07% ± 0.06%, p <0.0005, fig. 3). However, TNF-α neutralization during the time course of renal obstruction markedly decreased cleaved caspase 8 and t-Bid expression vs vehicle (0.18% ± 0.04% β-actin density vs 0.82% ± 0.08%, p <0.005, and 0.03% ± 0.015% vs 0.43% ± 0.03%, p <0.0005), elucidating t-Bid as an important intermediary in TNF-α mediated intrinsic apoptotic signaling pathway activation.
Fig. 3.

Western blots and graphs demonstrate renal cortical cleaved 20 kDa (kD) active caspase 8 and t-Bid expression after 1-week obstruction in presence of vehicle (OB) or PEG-sTNFR1 (OB + PEG-sTNFR1). Sham, sham operation.
Mitochondrial Cytochrome C Release
Renal cortical tissue sections from each treatment group were evaluated for mitochondrial cytochrome C release, which represents the final common pathway of intrinsic apoptotic signal activation. Sham treated samples demonstrated low levels of cytochrome C staining in discrete bundles, representing the mitochondria of tubular epithelial cells (fig. 4, A). In contrast, tissue samples exposed to 1 week of obstruction showed a marked increase in cytochrome C staining, which was diffuse throughout the cytoplasm of tubular epithelial cells, representing mitochondrial release of cytochrome C (fig. 4, B). Samples exposed to PEG-sTNFR1 during the time course of renal obstruction demonstrated low levels of cytochrome C staining in discrete mitochondrial bundles, suggesting that TNF-α neutralization prevents the obstruction induced release of mitochondrial cytochrome C (fig. 4, C).
Fig. 4.
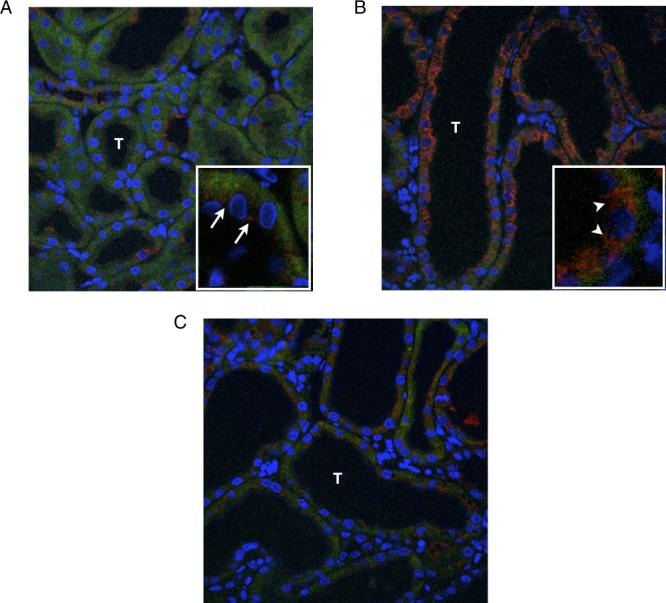
Photographs reveal mitochondrial cytochrome C release in renal tubule (T) cells in response to 1-week UUO and effects of PEG-sTNFR1. Blue areas represent nuclei stained blue. Red areas represent cytochrome C. A, sham operated kidney. Arrows indicate packets of intramitochondrial cytochrome C. B, obstructed kidney in presence of vehicle. C, obstructed kidney in presence of PEG-sTNFR1. Arrowheads indicate cytochrome C release into cytoplasm. Reduced from ×400.
DISCUSSION
Obstruction induced renal injury is a significant clinical problem that culminates in renal functional deterioration due to progressive tubulointerstitial fibrosis and a loss of renal tubular epithelial cells via apoptotic cell death. TNF-α is a well established mediator of inflammatory renal injury7–9 that has been shown to contribute to obstruction induced renal cell apoptosis by increasing caspase activation in the extrinsic death receptor signaling pathway.9 While the intrinsic and extrinsic pathways of apoptosis are activated during renal obstruction,2,9 to our knowledge we report the initial demonstration that TNF-α directly stimulates the intrinsic pathway of apoptotic signaling during renal obstruction, resulting in the mitochondrial release of cytochrome C. Our observation that TNF-α stimulates the activation of caspase 8 and Bid provides a mechanism by which TNF-α binding to its receptor can activate intrinsic apoptotic signaling cascades.
TNF-α is a cytotoxic cytokine that induces apoptotic cell death by interacting with its membrane bound receptor TNFR1. It has been previously demonstrated that TNF-α binding to TNFR1 allows TRADD (TNF receptor 1-associated death domain) to interact specifically with the death domain of TNFR1 and serve as a common platform for activating various signaling molecules. The TNFR1-TRADD complex can couple with Fas associated death domain, thereby committing the cell to apoptosis by stimulating caspase 8 and subsequently caspase 3 activation.13 We have previously reported that TNF-α induces caspase 8 and 3 activation, and renal tubular cell apoptosis via death receptor signaling pathways during renal obstruction.9 To examine the TNF-α effect on obstruction induced intrinsic apoptotic signaling animals underwent a 1-week course of UUO in the presence and absence of TNF-α neutralization. We found that renal cortical TNF-α levels and apoptotic cell death were significantly increased in response to obstruction, consistent with prior observations in the literature.7,9 However, administering PEG-sTNFR1 during the time course of renal obstruction decreased renal TNF-α production and apoptotic cell death to sham treatment levels. This corroborates our prior observations and reinforces the critical role of TNF-α in the pathophysiology of obstruction induced renal injury.9
Bcl-2 family members are important mediators of the mitochondrial pathway of apoptotic cell death because they control the permeability of the mitochondrial membrane and the release of cytochrome C.4 Each Bcl-2 family member has 1 to 4 Bcl-2 homology domains, referred to as BH domains. In each BH domain is a conserved structural motif that is essential for the activity of the proteins in the family. The anti-apoptotic family members Bcl-2 and Bcl-xL each contain 4 BH domains, while pro-apoptotic family members contain 3 BH domains, ie Bax, Bak and Bok, or 1 BH domain, ie Bid, Bad, Bik and Blk.5 Previously groups have examined interactions among the Bcl-2 family of proteins and determined that interactions among the 3 separate groups are crucial to cell survival.14,15 For example, Bcl-2 and Bcl-xL are capable of inhibiting the activation and incorporation of BAX into the mitochondrial membrane.16 After BAX is activated and incorporates into the mitochondrial membrane, the cell is committed to releasing cytochrome C, thereby propagating the machinery of apoptosis.17
In experimental models of obstructive nephropathy decreasing levels of Bcl-2 and increasing levels of Bax have correlated with progressive apoptotic cell death.2 Our results confirm these observations, revealing a significant decrease in anti-apoptotic Bcl-2 and Bcl-xL expression, and a significant increase in pro-apoptotic Bax expression in response to renal obstruction. Interestingly we further observed that the expression of these Bcl-2 family members is mediated by TNF-α. Neutralization of TNF-α activity during the time course of renal obstruction effectively preserved Bcl-2 and Bcl-xL expression, decreased Bax expression to almost sham treatment levels and importantly prevented the mitochondrial release of cytochrome C, which heralds imminent cell death. While TNF-α has a well established role in apoptotic renal tubular cell death during obstruction and ischemia-reperfusion injury, to our knowledge this is the first study to demonstrate the significant influence of TNF-α on the intrinsic apoptotic signaling pathway during obstructive nephropathy.
In the BH 3 single domain group of proteins Bid has been shown to have a significant role in connecting the extrinsic and intrinsic pathways of apoptosis.18 Upon cleavage by caspase 8 into its truncated form, t-Bid becomes incorporated into the mitochondrial membrane, triggering Bax and/or Bak oligomerization, and initiating the intrinsic pathway of apoptosis.19,20 Bid and its family members also appear to bind and inhibit the anti-apoptotic function of BH 3 multidomain proteins.14,15 To evaluate the mechanism of the stimulatory effect of TNF-α on the intrinsic apoptotic signaling pathway we examined active caspase 8 and t-Bid expression during UUO in the presence and absence of TNF-α neutralization. Our results demonstrate that TNF neutralization dramatically decreases caspase 8 and Bid activation during renal obstruction, implicating Bid activation as the mechanism by which TNF-α stimulates the intrinsic apoptotic signaling pathway. However, it remains unclear whether the pro-apoptotic effect of t-Bid during UUO is primarily related to the direct activation of Bax or is an indirect effect resulting from its binding and inhibition of Bcl-2. Further studies are required to elucidate the specific pro-apoptotic mechanism of action of Bid during obstructive nephropathy.
CONCLUSIONS
Apoptotic cell death causes a significant decrease in renal parenchymal mass and it is an important contributing factor to obstructive nephropathy. The intrinsic and extrinsic apoptotic signaling pathways are involved in stimulating cell execution and TNF-α has previously been shown to be an important mediator of this process via death receptor signaling. To our knowledge this study is the initial demonstration that TNF-α also directly stimulates the intrinsic mitochondrial pathway of apoptosis during renal obstruction by stimulating Bax expression, and the simultaneous suppression of Bcl-2 and Bcl-xL expression. Our results reveal that TNF-α stimulation of caspase 8 and Bid activation is the likely mechanism by which TNF-α, binding to its receptor, can activate the intrinsic apoptotic pathway, resulting in the mitochondrial release of cytochrome C. The modulation of TNF-α activity during renal obstruction provides a potential therapeutic strategy for ameliorating obstruction induced renal injury.
ACKNOWLEDGMENTS
Anti-t-Bid antibody was provided by Dr. Xiao-Ming Yin, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania.
Abbreviations and Acronyms
- ELISA
enzyme-linked immunosorbent assay
- PBS
phosphate buffered saline
- PEG
pegylated
- RT
room temperature
- sTNFR1
soluble TNFR1
- t-BID
truncated BID
- TNF
tumor necrosis factor
- TNFR1
TNF receptor type 1
- UUO
unilateral ureteral obstruction
Footnotes
Study received approval from the Indiana University School of Medicine animal care and research committee.
REFERENCES
- 1.Troung L, Sheikh-Hamad D, Chakraborty S, Suki W. Cell apoptosis and proliferation in obstructive uropathy. Semin Nephrol. 1998;18:641. [PubMed] [Google Scholar]
- 2.Chevalier R, Smith C, Wolstenhome J, Krajewski S, Reed J. Chronic ureteral obstruction in the rat suppresses renal tubular Bcl-2 and stimulates apoptosis. Exp Nephrol. 2000;8:115. doi: 10.1159/000020657. [DOI] [PubMed] [Google Scholar]
- 3.Choi Y, Baranowska-Daca E, Nguyen V, Koji T, Ballantyne C, Sheikh-Hamad D, et al. Mechanism of chronic obstructive uropathy: increased expression of apoptosis-promoting molecules. Kidney Int. 2000;58:1481. doi: 10.1046/j.1523-1755.2000.00310.x. [DOI] [PubMed] [Google Scholar]
- 4.Joza N, Kroemer G, Penninger J. Genetic analysis of the mammalian cell death machinery. Trends Genet. 2002;18:142. doi: 10.1016/s0168-9525(01)02618-x. [DOI] [PubMed] [Google Scholar]
- 5.Reed J. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Diff. 2006;13:1378. doi: 10.1038/sj.cdd.4401975. [DOI] [PubMed] [Google Scholar]
- 6.Guo G, Morrissey J, McCracken R, Tolley T, Klahr S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am J Physiol. 1999;277:F766. doi: 10.1152/ajprenal.1999.277.5.F766. [DOI] [PubMed] [Google Scholar]
- 7.Meldrum K, Metcalfe P, Leslie J, Misseri R, Hile K, Meldrum D. TNF-a neutralization decreases nuclear factor-kB activation and apoptosis during renal obstruction. J Surg Res. 2006;131:182. doi: 10.1016/j.jss.2005.11.581. [DOI] [PubMed] [Google Scholar]
- 8.Misseri R, Meldrum D, Dinarello C, Dagher P, Hile K, Rink R, et al. TNF-a mediates obstruction-induced renal tubular cell apoptosis and proapoptotic signaling. Am J Physiol Renal Physiol. 2005;288:F406. doi: 10.1152/ajprenal.00099.2004. [DOI] [PubMed] [Google Scholar]
- 9.Yin X. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10:161. doi: 10.1038/sj.cr.7290045. [DOI] [PubMed] [Google Scholar]
- 10.Werner AB, de Vries E, Tait SW, Bontjer I, Borst J. Bcl-2 family member Bfl-1/A1 sequesters truncated bid to inhibit is collaboration with pro-apoptotic Bak or Bax. J Biol Chem. 2002;277:22781. doi: 10.1074/jbc.M201469200. [DOI] [PubMed] [Google Scholar]
- 11.Bendele A, McComb J, Gould T, Frazier J, Chilipala E, Seely J, et al. Combination benefit of PEGylated soluble tumor necrosis factor receptor type 1 (PEG sTNF-R1) and dexamethasone or indomethacin in adjuvant arthritic rats. Inflam Res. 1999;48:453. doi: 10.1007/s000110050486. [DOI] [PubMed] [Google Scholar]
- 12.Hsu H, Xiong J, Goeddel D. The TNF receptor 1-associated protein TRADD signals death and NF-kappa B activation. Cell. 1995;81:495. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 13.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 14.Uren R, Dewson G, Chen L, Coyne S, Huang D, Adams J, et al. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J Cell Biol. 2007;177:277. doi: 10.1083/jcb.200606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Willis S, Wei A, Smith B, Fletcher J, Hinds M, et al. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Liu X, Bhalla K, Kim C, Ibrado A, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 17.Lalier L, Cartron PF, Juin P, Nedelkina S, Manon S, Bechinger B, Vallette FM. Bax activation and mitochondrial insertion during apoptosis. Apoptosis. 2007;12:887. doi: 10.1007/s10495-007-0749-1. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Ding W, Ni H, Gao W, Shi Y, Gambotto A, et al. Bid-independent mitochondrial activation in tumor necrosis factor alpha-induced apoptosis and liver injury. Mol Cell Biol. 2007;27:541. doi: 10.1128/MCB.01166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-Induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gross A, Yin X, Wang K, Wei M, Jockel J, Milliman C, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while Bcl-Xl prevents this release but not tumor necrosis- R1/FAS death. J Biol Chem. 1999;274:1156. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]