

Summary
Mitochondria play a central role in regulating apoptosis by releasing proapoptotic contents such as cytochrome c, and generating reactive oxygen species (ROS). Early in apoptosis, proteins translocate to mitochondria to promote the release of their contents. Here, we show that the actin- and cofilin-interacting protein CAP1 has a role in apoptosis. When we induced apoptosis, CAP1 rapidly translocated to the mitochondria independently of caspase activation. Translocation was proapoptotic because CAP1-knockdown cells were resistant to apoptosis inducers. Overexpression of wild-type CAP1 did not stimulate apoptosis on its own, but stimulated cofilin-induced apoptosis. Apoptosis induction required a mitochondrial-targeting domain, localized in the N-terminus and also the actin-binding domain in the C-terminus. Taken together, these studies suggest that CAP1 provides a direct link from the actin cytoskeleton to the mitochondria by functioning as an actin shuttle.
Keywords: Cyclase-associated protein, Srv2, ASP-56, Reactive oxygen species, Cytochrome c, Staurosporine, Etoposide
Introduction
Mitochondria play a central role in programmed cell death through both apoptotic and necrotic signals (Gross et al., 1999; Jiang and Wang, 2004; Vander Heiden and Thompson, 1999; Wang, 2001). Under survival conditions, the mitochondrial membranes present a barrier to contain proapoptotic contents such as cytochrome c, and apoptosis-inducing factor (AIF). Proapoptotic signals that disrupt the mitochondrial barrier cause the release of cytochrome c, which activates a cascade of caspases, whereas release of AIF promotes nuclear changes in a caspase-independent manner (Vander Heiden and Thompson, 1999). Mitochondria also generate reactive oxygen species (ROS), which when present in excess, can harm cells.
Proteins that regulate cell survival often shuttle to and from the mitochondria, modifying the mitochondrial barrier to either promote apoptosis or protect cells (Gross et al., 1999; Jiang and Wang, 2004; Vander Heiden and Thompson, 1999; Wang, 2001). The best characterized example of this is the regulation of the Bcl-2 proto-oncogene (official protein symbol BCL2), a protective apoptosis regulator protein concentrated in mitochondria. Bcl-2 is regulated by protein-protein complexes, some of which are protective complexes and others are inhibitory complexes. The balance between the protective complexes and the inhibitory complexes is thought to be critical in determining whether a cell will survive or undergo apoptosis. Proteins that form protective complexes include the protein kinase RAF1 and another Bcl-2 family member, Bcl-XL. By contrast, BAD (Bcl-associated death promoter) and BAX are Bcl-2 family members that translocate to mitochondria and form inhibitory complexes with Bcl-2. The complexes can be regulated by survival factors through the action of protein kinases. For example, PKA, RAF1 and AKT promote cell survival by phosphorylating BAD and preventing it from forming inhibitory complexes with Bcl-2 (Zha et al., 1996).
One emerging and poorly defined area of research in cell survival pathways is the role of the actin cytoskeleton in mitochondrial-dependent cell survival. In some systems, stabilization of actin filaments trigger apoptosis (Gourlay and Ayscough, 2005b; Gourlay and Ayscough, 2005c; Gourlay et al., 2004), whereas in other systems destabilization stimulates apoptosis (Martin and Leder, 2001; Martin and Vuori, 2004). Actin can be cleaved by caspases to create a 15 kDa fragment which then translocates to mitochondria and promotes apoptosis (Mashima et al., 1995; Utsumi et al., 2003). Actin is also required for tumor necrosis factor α (TNFα) to trigger mitochondrial clustering and cell death through a necrosis pathway (Li et al., 2004). Another cytoskeletal protein, cofilin, promotes apoptosis by translocating to mitochondria to deliver actin (Chua et al., 2003). Although different routes of actin-dependent cell survival have been found depending on the experimental systems, in all cases the actin cytoskeletal pathways are upstream of the mitochondria. Actin itself may be the primary sensor to reach the mitochondria because a common observation is that targeting actin to mitochondria, either directly, or indirectly through intermediate proteins, promotes apoptosis.
Cofilin, a member of the ADF (actin-depolymerizing factor) family, is a small cytoskeletal protein that depolymerizes actin filaments (Lappalainen and Drubin, 1997). Its activity is regulated by phosphorylation by LIM kinase (Edwards et al., 1999; Yang et al., 1998). In its mitochondrial role, cofilin rapidly translocates to mitochondria upon exposure to apoptosis-inducing agents. Mitochondrial translocation is regulated by phosphorylation because only dephosphorylated cofilin can associate with mitochondria (Chua et al., 2003). In the comparison with the Bcl-2/BAD pathway, cofilin occupies the position of BAD, because it is a proapoptotic protein that, when dephosphorylated, associates with the mitochondria. Because of its direct interactions with both actin and the mitochondrial pathway, cofilin may link the cytoskeleton to cell-survival pathways.
One protein that interacts with cofilin is CAP1, which was first identified as a component of the yeast adenylyl cyclase complex (Fedor-Chaiken et al., 1990; Field et al., 1988; Field et al., 1990). In yeast, CAP facilitates Ras activation of adenylyl cyclase (Shima et al., 2000). Although CAP does not regulate cAMP in animal cells, its cytoskeletal functions are widely conserved. CAP is conserved in all eukaryotic organisms, including Drosophila, plants and mammals (Hubberstey and Mottillo, 2002). The two mammalian isoforms CAP1 and CAP2 are about 64% identical (Swiston et al., 1995). CAP1 is expressed in most cells and tissues whereas CAP2 expression is more restricted; at least one of the two isoforms is expressed in nearly all cells (Peche et al., 2007; Swiston et al., 1995).
CAP proteins are all characterized by an N-terminal coiled-coil domain, a middle proline-rich region, and a highly conserved C-terminal domain. The N-terminal domain binds adenylyl cyclase (in yeast) and associates with cofilin (Mintzer and Field, 1994; Moriyama and Yahara, 2002; Nishida et al., 1998). The proline-rich region binds profilin and SH3 domains and is important for localization (in yeast) (Bertling et al., 2007; Freeman et al., 1996; Yu et al., 1999). Finally, the C-terminus binds actin monomers (Freeman et al., 1995; Gieselmann and Mann, 1992). Functional studies with microinjection and RNAi suggested a role in actin filament turnover (Bertling et al., 2004; Freeman and Field, 2000). The discovery of its interaction with cofilin led to the elucidation of its role in filament assembly (Moriyama and Yahara, 2002). Although CAP has little effect on actin turnover by itself in vitro, it accelerates cofilin-promoted disassembly of actin filaments (Lappalainen and Drubin, 1997; Moriyama and Yahara, 2002). The interactions between CAP and cofilin are conserved in the yeast homologs of the two proteins (Balcer et al., 2003).
In this study, we demonstrate a proapoptotic role for CAP1. CAP1 rapidly translocates to the mitochondria upon treatment with a number of agents that induce apoptosis. Translocation occurs independently of actin binding but requires the cofilin-binding domain. Translocation is a proapoptotic event because CAP1-knockdown cells are resistant to induction of apoptosis. Studies with cofilin suggest that cofilin translocates independently of CAP1 but that CAP1 assists cofilin to promote apoptosis.
Results
CAP1 translocates to mitochondria during early stages of apoptosis
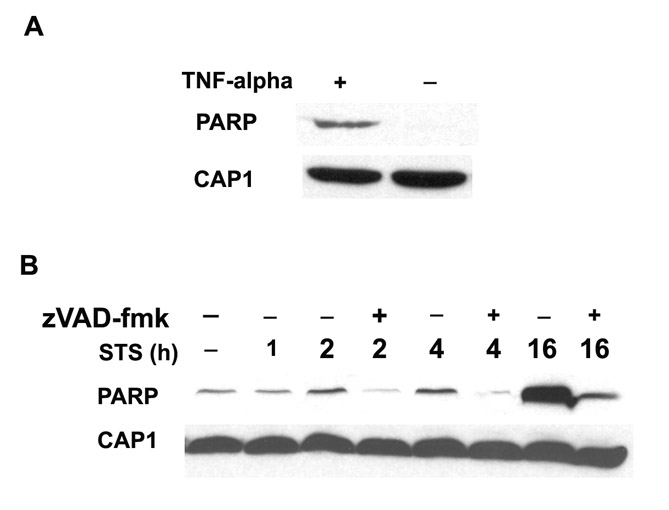
Because cofilin and actin are both proapoptotic and act via mitochondrial-dependent apoptosis pathways, we sought to determine whether CAP1 regulated cell survival. We first treated cells with staurosporine (STS), a broad acting kinase inhibitor, to induce apoptosis and isolated mitochondria by sucrose gradient centrifugation. Fractions were then analyzed on western blots to determine whether CAP1 translocates to mitochondria. The blots were also probed with the mitochondrial marker voltage-dependent anion channel (VDAC) or HSP60 to monitor mitochondrial fractions and normalize the blots. As expected, we observed that STS stimulated cofilin to translocate to the mitochondrial fraction (Fig. 1A, lanes 1 and 2) and that cytochrome c was not present in the cytosol of untreated cells. When blots were probed with CAP1, we found that CAP1 also translocated to the mitochondria (Fig. 1A, lanes 1 and 2). Significantly less translocation was caused by the receptor-dependent ligands, TNFα and TRAIL (tumor necrosis factor ligand superfamily member 10, also known as TNF10) suggesting that CAP1 responds primarily to endogenous apoptosis inducers (Fig. 1B, data not shown for TRAIL). PARP cleavage confirmed the activity of TNFα under similar experimental conditions (supplemental material Fig. S1A). Translocation was also stimulated by a DNA-damaging agent, etoposide (Fig. 1C). Because CAP1 is an actin-sequestering protein, it is possible that destabilization of the actin cytoskeleton was driving its translocation to mitochondria. To address this, we treated cells with latrunculin A (LA), a compound that binds to actin monomers and disrupts the cytoskeleton, which can cause apoptosis in some cells (Coue et al., 1987). LA caused the cells to round up (supplementary material Fig. S2), which is consistent with loss of the cytoskeleton. However, it did not cause CAP1 to translocate (Fig. 1B), suggesting that the translocation was not driven by actin depolymerization. We also tested whether translocation was directed by caspases by treating cells with zVAD-fmk, which is a broad-range caspase inhibitor (Fig. 1D). This compound failed to prevent CAP1 from translocating, suggesting that translocation was caspase independent. As a control, we showed that zVAD-fmk inhibits STS-induced PARP cleavage (supplementary material Fig. S1B); additionally, we found that the bulk of PARP cleavage occurs after 16 hours of STS treatment. Thus, caspase activation occurs long after we begin detecting CAP1 translocation, which is as soon as 5 minutes (data not shown) after STS treatment and steadily increased throughout the course of the experiment (Fig. 1D). We also detected translocation by immunofluorescence using a monoclonal antibody to label CAP1 and Mitotracker dye to stain the mitochondria (Fig. 2A). The specificity of the antibody was confirmed by western blot (Fig. 2B) and immunostaining of CAP1-knockdown cells (supplementary material Fig. S3). The micrographs show that the predominant staining for CAP1 is a diffuse cytoplasmic staining, as previously reported, although some cells showed CAP at the peripheral ruffles. In treated cells, CAP1 translocated to the mitochondria. The localization was more prominent when the mitochondria were clustered on one side of the nucleus, as is often observed in apoptotic cells. In confocal imaging, we found that the CAP1 staining did not precisely align with the Mitotracker dye (Fig. 2C). CAP1 stained as punctuate structures on the outer surface of the mitochondria with one to three spots per mitochondrion. The localization resembled that of BAX and BAK, two Bcl-2 family members that translocate to the mitochondria in clusters and do not circumscribe the membrane (Nechushtan et al., 2001), as shown in supplementary material Fig. S4. This suggested that CAP1 was not translocating within the mitochondria, but was perhaps associating with the outer membrane. We next treated the mitochondria collected from HEK293T cells transfected with GFP-CAP1, with proteinase K prior to running western blots with CAP1 antibody (Fig. 2D). GFP-CAP1 was completely digested by the proteinase, whereas VDAC, which spans the mitochondrial outer membrane, was partially digested and COX4, an inner mitochondrial membrane protein, was resistant. Translocation was detected using either western blots of purified mitochondria or immunofluorescence in numerous cell types, including HeLa (Fig. 1), NIH3T3 (Fig. 2A,C) and with transfected CAP1 (Fig. 3B), HEK293T (Fig. 2D) and COS-7 (Fig. 4C). We conclude that CAP translocates to the mitochondria upon induction of apoptosis and that translocation was not dependent on caspases or actin depolymerization.
Fig. 1.
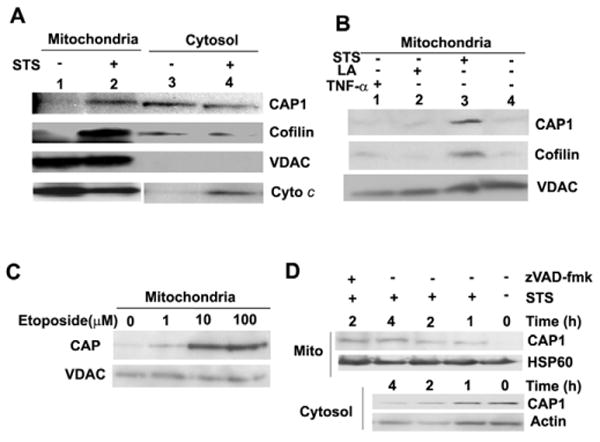
CAP1 translocates to mitochondria upon treatment to induce apoptosis. (A) STS stimulates CAP1 translocation to mitochondria. HeLa cells were treated with 1 μM STS for 4 hours, mitochondria were isolated by sucrose gradient centrifugation and mitochondria or cytosol fractions were subjected to western blot. Cytochrome c (Cyto c) images were from the same gel, although mitochondrial and cytosol results were scanned separately because a longer exposure was required to detect signals in the cytosol. (B) STS but not LA stimulated translocation of CAP1 to mitochondria. HeLa cells were treated with STS, LA and TNFα, mitochondria were isolated as above and CAP1 and cofilin were detected. (C) Treatment with etoposide also induces CAP1 translocation to mitochondria. HeLa cells were treated for 24 hours with etoposide concentrations ranging from 1 μM to 100 μM, mitochondria were purified and subjected to western blots with CAP1. (D) Time course of CAP1 translocation to the mitochondria. HeLa cells were treated with STS (1 μM) in the presence of 50 μM zVAD-fmk, where indicated. Gradient-purified mitochondrial (top) and cytosolic (bottom) samples were subjected to western blots to detect CAP1. HSP60 and actin were used as loading standards.
Fig. 2.
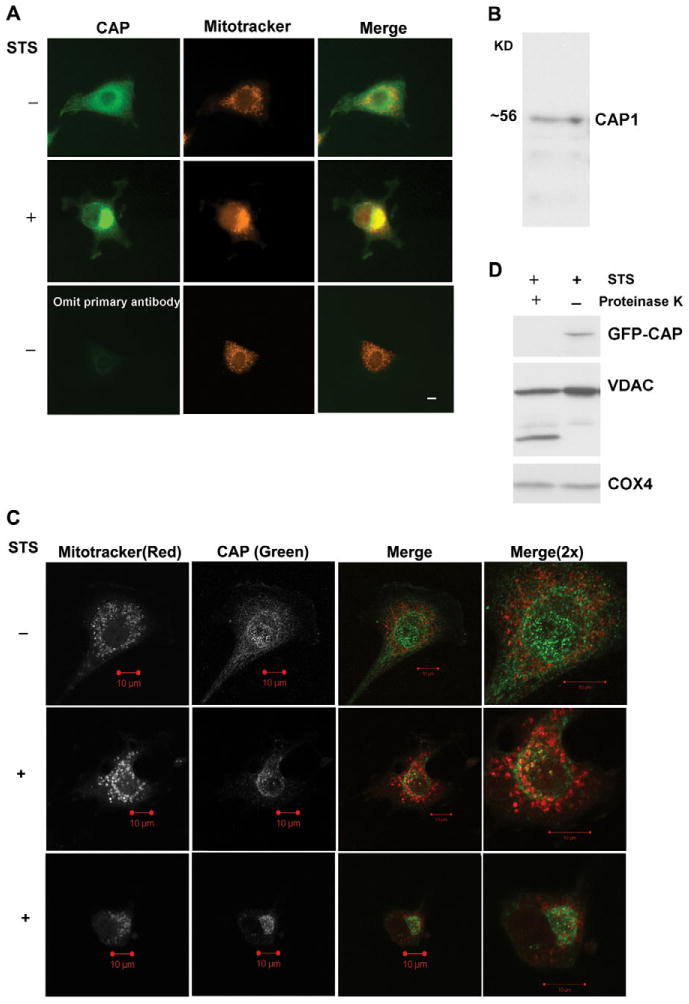
CAP1 translocates to the outer mitochondrial membrane. (A) CAP1 translocates to mitochondria upon apoptosis induction with STS in NIH3T3 cells. Cells were plated on coverslips overnight and stained with 50 nM MitoTracker Red CMXROS for 1 hour followed by treatment for 1 hour with (+) or without (−) 1 μM STS. Fixed and permeabilized cells were incubated with mouse anti-CAP1 antibody for 2 hours and then incubated for 1 hour with anti-mouse FITC conjugate. The slides were analyzed using a 40× objective lens by fluorescence microscopy. Scale bar: 10 μm. (B) Western blots of NIH 3T3 cell lysate with anti-CAP1 antibody demonstrate specificity for the antibody. (C) Confocal imaging of CAP1 reveals punctate staining at the mitochondria in treated cells. NIH3T3 cells were treated and processed as described above. Fluorescence images were collected at similar focal planes by confocal microscopy with a 63× objective lens; two independent STS-treated cells are shown. Scale bars: 10 μm. (D) Mitochondrial CAP1 is proteinase sensitive. HEK293T cells were transfected with GFP-CAP1 plasmid and treated with STS for 2 hours. Gradient-purified mitochondria were digested with proteinase K as indicated and then subjected to western blot analysis.
Fig. 3.
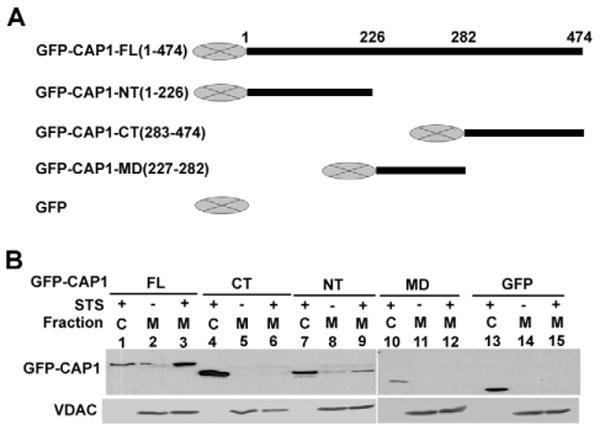
CAP1 is directed to mitochondria through the N-terminal domain. (A) GFP fusion constructs. (B) Translocation of GFP fusion CAP1 functional domains to mitochondria after apoptosis induction. HEK293T cells were transfected with the indicated CAP1constructs. 24 hours after transfection, cells were treated with 1 μM STS for 2 hours. Gradient-purified mitochondria were subjected to western blotting with anti-GFP. VDAC blotting was used to normalize total proteins from mitochondrial fractions.
Fig. 4.
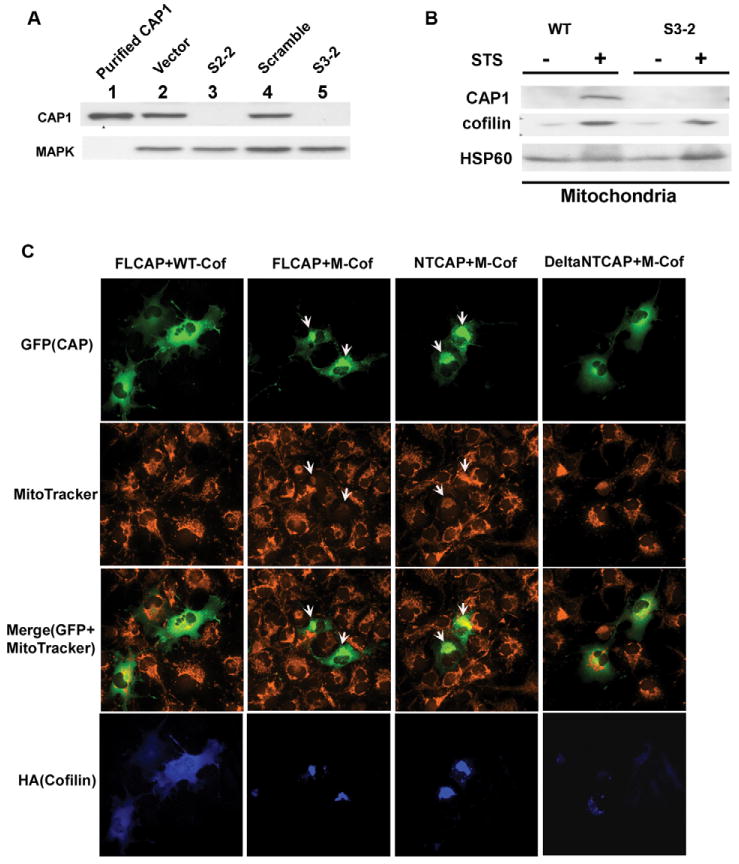
Knockdown of CAP1 and its interaction with cofilin. (A) CAP1 knockdown in HeLa cells. Stable cell lines S2-2 and S3-2 are from two independent shRNA constructs, S2 and S3, which target different sequences within CAP1 (see Materials and Methods). Control cell lines include HeLa cells harboring empty vector or a scrambled shRNA S2. Purified CAP1 from pig platelet was used as a positive control and MAPK was blotted to normalize total protein. CAP1 levels in the cell lysates were detected by western blotting. (B) CAP1 knockdown does not affect cofilin translocation to mitochondria during apoptosis induction. HeLa wild-type or CAP1-knockdown cells (S3-2) were treated with 1 μM STS for 2 hours. Sucrose-gradient-purified mitochondria were isolated and analyzed by western blot. HSP60 was monitored to normalize total mitochondrial protein. (C) Mitochondrial cofilin stimulates CAP1 translocation to mitochondria. GFP-tagged CAP1 constructs, FL-CAP, NT-CAP and ΔNTCAP1 were cotransfected with mitochondria-targeted cofilin (M-cof) into COS-7 cells and stained with MitoTracker. Cofilin was stained with anti-HA monoclonal antibody 12CA5. Images were viewed on a fluorescence microscope with a 40× objective lens. Arrows indicate cells with localization of GFP fusion CAP1 (domains) to mitochondria.
CAP1 translocation to mitochondria is independent of actin binding
To determine the domain of CAP1 responsible for mitochondrial shuttling, we constructed several deletion mutants to express GFP fusion CAP1 domains and then tested them in a transfection paradigm. HEK293T cells were transfected with the constructs and the mitochondrial and cytosolic fractions were analyzed on western blots (Fig. 3B) to evaluate CAP translocation to mitochondria. As expected, STS treatment stimulated mitochondrial translocation of the full-length protein (Fig. 3B, lanes 1-3) but not GFP alone (lanes 13-15). Translocation was observed with two N-terminal constructs, 1-226 (lanes 7-9) and 1-282 (which has the C-terminus deleted, not shown); however, no translocalization was observed with the construct that expressed the C-terminus alone (lanes 4-6). The middle region construct 227-282 did not translocate either (lanes 10-12). Similar results were obtained by immunofluorescence (data not shown). As the actin-binding domain localizes to the C-terminus, we concluded that actin binding was not essential for localization. Instead, localization was dependent on the N-terminal regions.
Knockdown of CAP1 in HeLa cells
To determine whether CAP1 plays a role in apoptosis, we first employed a knockdown strategy and assessed the effects on apoptosis. We found that two (of the three tested) independently targeted shRNA constructs, designated S2 and S3, resulted in stable cell lines that had a reduction in CAP1 levels by over 90% as determined using western blots (Fig. 4A), compared with wild-type cells (not shown), cells harboring vector controls or scrambled controls. The reduced CAP1 level in knockdown cells was also verified by immunofluorescence (supplementary material Fig. S3). These shRNA constructs target distinct regions of mRNA encoding the CAP protein. S2 targets the N-terminal half (residues 173-179) whereas S3 targets the C-terminal half (residues 358-364). To control for the specificity of the shRNA, we tested their ability to prevent expression of the GFP-CAP1 mutants described in Fig. 3. Both shRNAs efficiently reduced the expression of full-length CAP1, and when tested with GFP fusion NT-CAP1 and CT-CAP1 fragments, S2 reduced expression of only NT-CAP1 and S3 reduced expression of only CT-CAP1 (data not shown), thus confirming the specificity for the shRNA constructs. We recovered two stable cell lines S2-2 and S3-2, with efficient CAP1 knockdown (Fig. 4A). When compared with the control cells, the CAP1-knockdown cells were larger, had more ruffles, more prominent stress fibers and were often multinucleated (data not shown).
CAP1 interaction with cofilin and actin
To determine whether CAP1 was functionally interacting with cofilin, we performed cell survival and localization experiments. We first tested whether knockdown of CAP1 affected cofilin translocation. Although we found cofilin localized in aggregates in CAP1-knockdown HeLa cells (not shown), as previously reported in a transient knockdown paradigm (Bertling et al., 2004), cofilin still translocated to mitochondria upon treatment with STS (Fig. 4B). Knockdown of cofilin did not affect CAP1 translocation (supplementary material Fig. S5). However, we note that the knockdown of cofilin was considerably less efficient than CAP1 knockdown. Since the cofilin-binding domain was required for mitochondrial translocation of CAP1 (Fig. 3), we tested whether CAP1 may be targeted by cofilin. To test this, we transfected a mitochondria-targeted cofilin (Chua et al., 2003) into cells, and determined the localization of various CAP1 constructs. The mitochondria-targeted cofilin stimulated the localization of full-length GFP-CAP1 to the mitochondria (Fig. 4C). When we tested deletion mutants, we found that cofilin stimulated the translocation of the N-terminal construct and the full-length CAP1 (see arrow in merged images) but not the ΔNTCAP construct (a mutant including the middle domain and the C-terminus) or GFP alone (GFP alone largely localized to nucleus, data not shown). As expected, wild-type cofilin showed localization across the whole cell, whereas mitochondria-targeted cofilin staining is confined to the mitochondria (Fig. 4C lower panels). In addition, cells expressing mitochondrial cofilin showed signs of apoptosis, such as cell shrinking, mitochondrial clustering and reduced staining with Mitotracker (Fig. 4C, with arrow). Thus, the N-terminus of CAP1 is required for localization induced by both cofilin and STS. This suggests that cofilin associates with the mitochondria independently of CAP1, but that cofilin may bind CAP1 to facilitate CAP1 translocation.
Role of CAP1 in cofilin-induced apoptosis
We next tested the effects of altering CAP1 levels on apoptosis. To determine whether knocking down CAP1 affected apoptosis we cotransfected HeLa cells with CAP1 shRNA constructs together with GFP (we verified that the shRNA constructs also effectively knocked down CAP1 in a transient transfection paradigm, data not shown), and then treated the cells with 1 μM STS for 2 hours to induce apoptosis. The cells were then stained with Hoechst 33342 and GFP-positive cells were scored for apoptosis by nuclear condensation. As shown in Fig. 5A, both shRNA constructs, S2 and S3, inhibited apoptosis, suggesting that CAP1 translocating to mitochondria is pro-apoptotic.
Fig. 5.
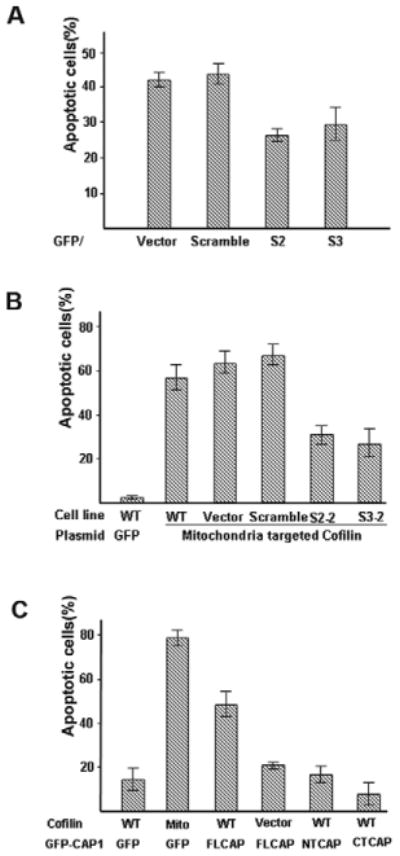
CAP1 interacts with cofilin to promote apoptosis. (A) Transient expression of CAP1 shRNA in HeLa cells reduces STS-induced apoptosis. HeLa control and CAP1-knockdown cell lines were cotransfected with GFP and ShRNA constructs (1:1). After 24 hours, cells were treated with 1 μM STS for 2 hours and labeled with Hoechst 33342. GFP-positive cells were counted. Cells with DNA fragmentation and nuclear collapse were scored as apoptotic cells. ∼200 cells were counted for each treatment and the experiment was done three times with similar results. Percentage of apoptotic cells are shown as mean ± s.e.m. (B) CAP1 knockdown reduces cofilin-induced apoptosis. CAP1-knockdown HeLa cells were transfected with mitochondrial cofilin along with GFP. 24 hours after transfection, cells were stained with Hoechst 33342 and scored for chromosome condensation. Approximately 100 transfected cells were scored for each of three fields and the experiment was repeated three times with similar results. Data are presented as mean ± s.e.m. (C) Overexpression of CAP1 stimulates cofilin-induced apoptosis. HEK293T cells were cotransfected with GFP-fused FL-CAP1, NT-CAP1 or CT-CAP1 along with either M-cofilin or wild-type cofilin (0.4 μg and 0.8 μg, respectively, per well). Cells were allowed to express proteins for 18 hours and then fixed cells were stained with 1 μg/ml Hoechst 33342 for 10 minutes. Apoptosis was identified by nuclear condensation and DNA fragmentation under fluorescence microscopy in GFP-positive cells. At least 300 GFP-positive cells were scored for each of three fields and experiment was repeated three times with similar results. Data are presented as mean ± s.e.m.
As previously reported, the mitochondria-targeted cofilin stimulates apoptosis (Chua et al., 2003). However, the rate of apoptosis was reduced by about 50% in the CAP1-knockdown cells (Fig. 5B) suggesting that CAP1 is required for efficient induction of apoptosis by cofilin. To explore this further, we transiently coexpressed GFP-CAP1 with cofilin in HEK293T cells. Neither CAP1 nor wild-type cofilin stimulated apoptosis (Fig. 5C) when expressed by themselves, but there was a substantial increase in the number of apoptotic cells when they were coexpressed. When we tested the deletion constructs, we found that neither the N-terminus nor the C-terminus of CAP1 stimulated cofilin-dependent apoptosis (Fig. 5C). These studies suggest that CAP1 and cofilin translocate to mitochondria to promote apoptosis. We also observed that in STS-treated HEK293T cells, CAP1 association with actin was not affected (supplementary material Fig. S6), suggesting that actin translocates with CAP1 to mitochondria. Together, these observations suggest that CAP1 and cofilin are part of an actin-mitochondria shuttling pathway. Since targeting of CAP1 requires the cofilin-binding site, it is probably directed by cofilin, but because the actin-binding site is also required for cell killing, it is likely that actin translocation stimulates cell death.
Cytochrome c translocation to cytosol is reduced in CAP1-knockdown cells
The mitochondria of apoptotic cells release cytochrome c and generate ROS. To measure cytochrome c, we performed western blots in isolated mitochondria and found that the cytochrome c release was reduced in the CAP1 knockdowns (Fig. 6A). Immunofluorescence of CAP1-knockdown cells (S3-2) revealed that the cytochrome c translocation to the cytosol was reduced compared with that in wild-type cells (supplementary material Fig. S7). Since the yeast apoptosis pathways are activated by ROS release, we also measured ROS with the ROS-sensitive dye 2′,7′ -dichlorofluorescein (DCF). STS stimulated an increase in the production of ROS in wild-type cells but not in S2-2 and S3-2 cells (Fig. 6B). Together, these data suggest that loss of CAP1 attenuates the response to STS by maintaining the integrity of the mitochondrial barrier.
Fig. 6.
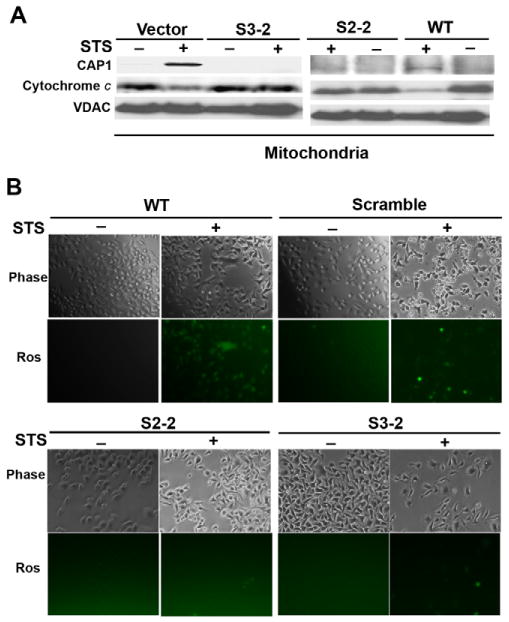
CAP1 knockdown reduces cytochrome c release and ROS accumulation. (A) CAP1-knockdown cells have reduced cytochrome c release when apoptosis is induced. HeLa stable cell lines harboring vector (vector) and CAP1 shRNA (S3-2) treated with STS (1 μM). After 4 hours of treatment, mitochondria were isolated by sucrose gradient centrifugation and subjected to western blots for CAP1 and cytochrome c. VDAC was monitored to normalize total protein. (B) Effect of CAP1 knockdown on ROS accumulation. HeLa wild-type cells, stable cell lines with scrambled S2 or CAP1 shRNA were cultured overnight and treated with 1 μM STS for 1 hour, cells were washed and stained with fresh 10 μM H2DCF-DA (Ros; Invitrogen) for 15 minutes and then examined under an inverted fluorescence microscope. All images were taken with the same exposure time.
Discussion
The actin cytoskeleton can regulate apoptosis through a number of mechanisms. For example, detaching cells from their substrates will often cause apoptosis; this mechanism of induction is called anoikis (Frisch and Screaton, 2001). However, anoikis is caused by inefficient growth-factor signaling in the absence of integrin-mediated cell-adhesion signals, and not by disrupting actin. More recently, several studies used cytoskeletal mutants and actin disrupting drugs to demonstrate that the actin cytoskeleton can regulate apoptosis in both mammalian cells and yeast (Gourlay and Ayscough, 2005a; Martin and Leder, 2001; Martin and Vuori, 2004). We propose that CAP1 provides a direct connection from the actin cytoskeleton to the mitochondria by shuttling actin in response to signals that cause apoptosis. Shuttling is stimulated by inducers including staurosporine and etoposide, but not by direct disruption of the cytoskeleton with latrunculin A. Translocation of CAP1 is proapoptotic because its loss protects cells, and when expressed with cofilin, CAP1 stimulates apoptosis. Cell death requires the N-terminus of CAP1 for targeting, but targeting is unable to promote apoptosis without the C-terminal actin-binding domain.
The proapoptotic role of CAP1 described here resembles that of cofilin, because both are cytoskeletal proteins that translocate to mitochondria upon stimulation of apoptosis. Translocation of both CAP1 and cofilin occurs independently of caspase activation and stimulates apoptosis. Additionally, both proteins require a functional actin-binding domain to promote apoptosis, suggesting that actin translocation is required for cell killing (Chua et al., 2003). Some observations we describe here suggest that CAP1 is downstream of cofilin: (1) mitochondrial targeting of cofilin can stimulate CAP1 translocation; (2) CAP1 knockdowns are resistant to cofilin-induced apoptosis; and (3) cofilin-induced apoptosis is stimulated by CAP1. However, other observations suggest that CAP1 may act independently of cofilin through an independent mechanism of actin delivery. We also did not see a loss of translocation of cofilin in CAP1 knockdowns or loss of CAP1 translocation with cofilin knockdowns. We are cautious about interpreting these knockdown data because our cofilin knockdowns were not efficient (with around 50-75% reduction of protein) and cofilin still translocated in the knockdown cells. In addition, homologs such as ADF may be functionally redundant for translocation. Finally, in yeast, localization is not driven by either actin or cofilin, but instead by SH3 proteins that bind to the central proline-rich region of CAP (Freeman et al., 1996; Lila and Drubin, 1997). Therefore, although cofilin remains an attractive candidate as the mitochondrial partner in CAP1 translocation and apoptosis, we do not rule out its participation in a parallel pathway of actin shuttling.
Although cofilin and CAP1 can serve as mitochondrial shuttles for actin, at least one other mechanism can translocate actin and promote apoptosis. Actin can be cleaved by a caspase-3-like activity to generate two fragments, 32 kDa and 15 kDa in size. The 15 kDa fragment is then modified by N-myristoylation to target it to mitochondria and stimulate apoptosis (Utsumi et al., 2003). The common observation in all three cases is that mitochondrial actin itself is promoting apoptosis.
Actin participates in extrinsic mechanisms of apoptosis as well as necrotic pathways. Although receptor-mediated apoptosis is generally reported as mitochondrial independent, agents such as TNFα also affect the mitochondria. In a retrovirial insertion mutagenesis screen for mutants that prevent TNFα killing, insertions were found in β-actin. The mutant had reduced rates of necrosis, reduced mitochondrial clustering and ROS generation (Li et al., 2004). Our observation that TNFα and TRAIL stimulate translocation poorly, suggest that cofilin and CAP1 are primarily responding to endogenous inducers of apoptosis. However, responses to ligands may occur later.
Regulation of ROS and apoptosis by CAP is conserved in yeast, although the pathway appears to be somewhat distinct from that in mammalian cells (Gourlay and Ayscough, 2006). In yeast, actin mutations that reduce actin cycling promote activation of Ras-cAMP and accumulation of ROS, which then stimulates ageing and apoptosis. (Hlavata et al., 2003). The ROS pathway has a PKA-dependent component as well as a PKA-independent component. Interestingly, actin appears to play a direct role in ROS generation, but not necessarily through actin delivery (Gourlay and Ayscough, 2005b; Gourlay and Ayscough, 2005c; Gourlay et al., 2004). Instead, actin dynamics itself drives ROS production because mutants that have reduced actin dynamics cause accumulation of large actin aggregates to stimulate ROS. Yeast CAP appears to provide the link between the actin aggregates and Ras-cAMP by transducing signals from the aggregates to activate Ras. Although the actin-binding domain is essential for CAP to activate Ras, the mechanism may not occur by mitochondrial translocation, but rather by sensing the aggregates (Gourlay and Ayscough, 2006). In yeast, neither CAP nor cofilin has been reported to translocate to mitochondria. It is interesting to note that we find that CAP1 translocates to mitochondria as discrete structures (Fig. 2C), suggesting that mammalian proteins may form aggregates similar to those in yeast.
In conclusion, CAP1, similarly to cofilin, BAD and BAX, translocates to mitochondria to promote apoptosis. It provides a direct link between the actin cytoskeleton and apoptosis-signaling pathways. Although distinct mechanisms of linking the cytoskeleton to apoptotic pathways have been documented, remarkably, all of the known examples find the mitochondria downstream of actin and in most cases, the mechanism of cell death can be directly attributed to the proapoptotic properties of actin localized to mitochondria.
Materials and Methods
Plasmids
The cDNA fragments of mouse CAP1 were amplified by PCR and cloned into the XhoI and BamHI sites of pEGFP vector. The primers are as follows: Full-length (FL, 1-474): 5′-TCTCGAGTTATGGCTGACATGCAAAATCTTGTA-3′ (F) and 5′-TGGATCCTTATCCAGCGATTTCTGTCACTGT-3′ (R); N terminus (NT, 1-226): 5′-TCTCGAGTTATGGCTGACATGCAAAATCTTGTA-3′ (F) and 5′-TGGATCCTTATCCCACAGAGGGTCCAGATGG-3′ (R); Middle domain (MD, 227-282): 5′-TCTCGAGGATCAGGCCCACCTCCTCCCCCA-3′ (F) and 5′-TGGATCCTTAAGGGTTCTTGTGAGTCTT-3′ (R); C terminus (CT, 283-474): 5′-TCTCGAGCTGCCCTGAAAGCTCAGAGCGGT-3′ (F) and 5′-TGGATCCT-TATCCAGCGATTTCTGTCACTGT-3′ (R). Appropriate combinations of primers were also used to amplify CAP1 mutants with NT deleted [ΔNT CAP1(227-474)] or CT deleted [ΔCT CAP1(1-282)]. Cofilin plasmids, expressing non-muscle cofilin, were previously described elsewhere (Chua et al., 2003).
shRNA constructs and CAP1 knockdown
shRNA constructs based on a pRNA-U6.1/Neo vector were from GenScript Corporation (Piscataway, NJ). Three constructs targeting independent sequences of human CAP1 mRNA were made by cloning the fragments into BamHI and HindIII sites. The target sequences for two shRNA constructs that efficiently knocked down CAP1 are S2, 5′-AGATGTGGATAAGAAGCAT-3′ (nucleotides 519-537; residues 173-179) and S3, 5′-CACGACATTGCAAATCAAG-3′ (nucleotides 1074-1092; residues 358-364). Stable cell lines were established in HeLa cells by transfection with FuGENE 6 (Roche) and selection with 1000 μg/ml G418 for 2 weeks. Multiple clones were recovered from cells transfected with both constructs with efficient knockdown of CAP1. Two clones, designated S2-2 and S3-2 decreased CAP1 expression to levels undetectable by western blot compared with wild-type cells. Cells harboring the empty vector or a scrambled S2 shRNA (GGAGAGT -AACGATTAAGA), made with a construct using the same composition of S2 but without any good match to any human mRNA, were used as controls.
Cell culture and treatment
Cell lines including HeLa, NIH3T3, HEK 293T and COS-7 were grown in DMEM (Dulbecco's modified Eagle's medium, Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), penicillin (100 U/ml) and streptomycin (100 μg/ml) (Invitrogen) at 37°C in 5% CO2. Cells were treated, where indicated, with one of following reagents: 1 μM staurosporine (STS), the indicated concentrations of etoposide for 24 hours, 5 μM latrunculin A (LA) (Molecular Probes) for 1 hour or 50 ng/ml TNFα for 6 hours. Caspase inhibitors (50 μM zVAD-fmk or DEVD-fmk, Calbiochem) were added to culture medium 30 minutes before drug treatment.
Mitochondria preparation
HeLa cells or HEK293T cells were gently washed three times with cold PBS and then harvested from plates by scraping with a rubber policeman to a final concentration of 1×107 cells/ml. Cells were centrifuged at 600 g for 5 minutes and the cell pellets were resuspended in 2 ml MS buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HCl at pH 7.5 and 1 mM EDTA) containing protease inhibitor cocktail (Roche). Cells were homogenized by gentle douncing (50 strokes for HeLa cells; 25 strokes for 293T cells). The homogenate was spun at 1300 g for 5 minutes at 4°C to pellet nuclei and unbroken cells. The supernatant was centrifuged again for 5 minutes and the pellet was removed. The heavy-membrane (HM) fraction, which is enriched in mitochondria, was isolated by spinning the supernatants at 17,000 g for 30 minutes at 4°C and collecting the pellets. Mitochondria were purified from the HM fraction by sucrose step gradient centrifugation. Briefly, the HM pellet was suspended in 1 ml MS buffer and layered on a sucrose gradient, consisting of 1 M and 1.5 M sucrose buffer (10 mM Tris-HCl at pH 7.5 and 1 mM EDTA) following centrifugation at 60,000 g for 20 minutes at 4°C. Mitochondria were collected at the interface of the sucrose gradients and dissolved in dilution buffer (5 mM Tris-HCl pH 7.5 and 1 mM EDTA). The diluted mitochondria were spun at 17,000 g for 30 minutes and the pellets were used for protein analysis. For proteinase K digestion assays, mitochondria were resuspended in MS buffer (without proteinase inhibitors) and incubated with proteinase K (10 ng/ml) on ice for 10 minutes. The reaction mixture was spun at 17,000 g for 10 minutes and the pellet dissolved in SDS lysis buffer for western blots.
Western blotting
Samples were mixed with same volume of 2× SDS buffer, boiled for 5 minutes at 95°C and resolved on 10-12% SDS-PAGE gels. Samples were transferred to PVDF membranes and western blots were performed. The antibodies and their dilutions used for western blots were: monoclonal mouse anti-CAP1 antibodies (NLF5, NLF8, 12 B1) (Freeman and Field, 2000), anti-VDAC (1:1000; PharMingen), anti-COX4 (1:1000; BD Biosciences Clontech), polyclonal rabbit anti-cofilin which recognizes both non-muscle and muscle cofilin (1:1000; Cell Signaling Technology), anti-cytochrome c (1:500; Cell Signaling Technology), anti-cleaved PARP (Asp214) (1:1000, Cell Signaling Technology), anti-GFP (1:500; Santa Cruz) and polyclonal goat anti-actin (1:500; Santa Cruz).
Immunofluorescence
Cells were seeded in 12-well plates on 12 mm coverslips or Nunc chambers and grown overnight. They were stained with 50 nM MitoTracker Red CMXRos (Molecular Probes) for 1 hour and washed with DMEM before treatment, where appropriate. After treatment with the indicated drugs, cells were washed with PBS twice and fixed in 3.3% paraformaldehyde for 15 minutes. Cells were permeabilized with 0.5% Triton X-100 in PBS for 20 minutes and then incubated with a solution of PBS containing 3% BSA and normal goat IgG for 1 hour to block nonspecific binding. Cells were stained with mouse monoclonal anti CAP1 12B1 (Freeman and Field, 2000), anti-HA(12CA5) (Field et al., 1988) or rabbit anti-Bak (Sigma) for 2 hours followed by staining with appropriate secondary antibodies conjugated with Alexa Fluor 488 (Molecular Probes) or Alexa Fluor 350 for 1 hour. After three washes with PBS containing 0.1% Triton X-100, the samples were mounted with mounting medium with or without DAPI (Vector Laboratories). Fluorescence images were collected and analyzed with a Leica Polyvar-2 fluorescence microscope using a Hamamatsu ORCA charge-coupled device camera operated with Open Lab software (Improvision, Lexington, MA). Confocal imaging was performed on a Zeiss Axiovert 200 M inverted fluorescence microscope using the LSM510 META confocal system and Zeiss LSM510META Version 3.2 software. The images were processed using Adobe Photoshop.
ROS formation assay
Cells were plated on 12-well culture plates, incubated overnight, treated with STS for 1 hour then washed three times with DMEM without Phenol Red. The cells were maintained in the same medium and treated with freshly prepared H2-DCFDA (Invitrogen) using a final concentration of 10 μM and then incubated for 15 minutes. The cells were then washed with pre-warmed DMEM (free of Phenol Red and serum) three times and examined under an inverted fluorescence microscope. Images were taken on a Nikon Eclipse TE200-a microscope using the same exposure times.
Apoptosis assay
Apoptosis was assessed by Hoechst 33342 staining. Cells were stained with 1 μg/ml Hoechst 33342 (Molecular Probes, Eugene, OR) for 10 minutes after fixing the cells with 3.3% paraformaldehyde for 15 minutes. Apoptotic cells were identified by nuclear condensation and DNA fragmentation visualized by fluorescence microscopy. In cotransfection experiments with GFP-CAP1 constructs, only GFP-positive cells were scored.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Wafik S. El-Deiry for providing TRAIL. This work was supported by a National Scientist Development Grant from American Heart Association to G.L.Z. (0630394N), a grant from the Hong Kong Research Grant Council to P.L. (HKUST6242/04M) and a grant from the NIH to J.F. (GM48241).
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/17/2913/DC1
References
- Balcer HI, Goodman AL, Rodal AA, Smith E, Kugler J, Heuser JE, Goode BL. Coordinated regulation of actin filament turnover by a high-molecular-weight Srv2/CAP complex, cofilin, profilin, and Aip1. Curr Biol. 2003;13:2159–2169. doi: 10.1016/j.cub.2003.11.051. [DOI] [PubMed] [Google Scholar]
- Bertling E, Hotulainen P, Mattila PK, Matilainen T, Salminen M, Lappalainen P. Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol Biol Cell. 2004;15:2324–2334. doi: 10.1091/mbc.E04-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertling E, Quintero-Monzon O, Mattila PK, Goode BL, Lappalainen P. Mechanism and biological role of profilin-Srv2/CAP interaction. J Cell Sci. 2007;120:1225–1234. doi: 10.1242/jcs.000158. [DOI] [PubMed] [Google Scholar]
- Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P. Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol. 2003;5:1083–1089. doi: 10.1038/ncb1070. [DOI] [PubMed] [Google Scholar]
- Coue M, Brenner SL, Spector I, Korn ED. Inhibition of Actin Polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nature Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Fedor-Chaiken M, Deschenes RJ, Broach JR. SRV2, a gene required for RAS activation of adenylate cyclase in yeast. Cell. 1990;61:329–340. doi: 10.1016/0092-8674(90)90813-t. [DOI] [PubMed] [Google Scholar]
- Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J, Vojtek A, Ballester R, Bolger G, Colicelli J, Ferguson K, Gerst J, Kataoka T, Michaeli T, Powers S, et al. Cloning and characterization of CAP, the S. cerevisiae gene encoding the 70 kd adenylyl cyclase-associated protein. Cell. 1990;61:319–327. doi: 10.1016/0092-8674(90)90812-s. [DOI] [PubMed] [Google Scholar]
- Freeman NL, Field J. Mammalian homolog of the yeast adenylyl cyclase associated protein, CAP/Srv2p, regulates actin filament assembly. Cell Motil Cytoskel. 2000;45:106–120. doi: 10.1002/(SICI)1097-0169(200002)45:2<106::AID-CM3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Freeman NL, Chen Z, Horenstein J, Weber A, Field J. An actin monomer binding activity localizes to the carboxyl half of the Saccharomyces cerevisiae cyclase associated protein. J Biol Chem. 1995;270:5680–5695. doi: 10.1074/jbc.270.10.5680. [DOI] [PubMed] [Google Scholar]
- Freeman NL, Lila T, Mintzer KA, Chen Z, Pahk AJ, Ren R, Drubin DG, Field J. A conserved proline-rich region of the Saccharomyces cerevisiae cyclase-associated protein binds SH3 domains and modulates cytoskeletal localization. Mol Cell Biol. 1996;16:548–556. doi: 10.1128/mcb.16.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- Gieselmann R, Mann K. ASP-56, a new actin sequestering protein from pig platelets with homology to CAP, an adenylylate cyclase-associated protein from yeast. FEBS Lett. 1992;298:149–153. doi: 10.1016/0014-5793(92)80043-g. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. The actin cytoskeleton in ageing and apoptosis. FEMS Yeast Res. 2005a;12:1193–1198. doi: 10.1016/j.femsyr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol. 2005b;6:583–589. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. Identification of an upstream regulatory pathway controlling actin-mediated apoptosis in yeast. J Cell Sci. 2005c;118:2119–2132. doi: 10.1242/jcs.02337. [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR. Actin-induced hyperactivation of the Ras signaling pathway leads to apoptosis in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:6487–6501. doi: 10.1128/MCB.00117-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay CW, Carpp LN, Timpson P, Winder SJ, Ayscough KR. A role for the actin cytoskeleton in cell death and aging in yeast. J Cell Biol. 2004;164:803–809. doi: 10.1083/jcb.200310148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Hlavata L, Aguilaniu H, Pichova A, Nystrom T. The oncogenic RAS2(val19) mutation locks respiration, independently of PKA, in a mode prone to generate ROS. EMBO J. 2003;22:3337–3345. doi: 10.1093/emboj/cdg314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubberstey AV, Mottillo EP. Cyclase-associated proteins: CAPacity for linking signal transduction and actin polymerization. FASEB J. 2002;16:487–499. doi: 10.1096/fj.01-0659rev. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Drubin D. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- Li J, Li Q, Xie C, Zhou H, Wang Y, Zhang N, Shao H, Chan SC, Peng X, Lin SC, et al. Beta-actin is required for mitochondria clustering and ROS generation in TNF-induced, caspase-independent cell death. J Cell Sci. 2004;117:4673–4680. doi: 10.1242/jcs.01339. [DOI] [PubMed] [Google Scholar]
- Lila T, Drubin D. Evidence for physical and functional interactions among two Saccharomyces cerevisiae SH3 domain proteins, an adenylyl cyclase-associated protein and the actin cytoskeleton. Mol Biol Cell. 1997;8:367–385. doi: 10.1091/mbc.8.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS, Leder P. Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol Cell Biol. 2001;21:6529–6536. doi: 10.1128/MCB.21.19.6529-6536.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS, Vuori K. Regulation of Bcl-2 proteins during anoikis and amorphosis. Biochim Biophys Acta. 2004;1692:145–157. doi: 10.1016/j.bbamcr.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Mashima T, Naito M, Fujita N, Noguchi K, Tsuruo T. Identification of actin as a substrate of ICE and an ICE-like protease and involvement of an ICE-like protease but not ICE in VP-16-induced U937 apoptosis. Biochem Biophys Res Commun. 1995;217:1185–1192. doi: 10.1006/bbrc.1995.2894. [DOI] [PubMed] [Google Scholar]
- Mintzer KA, Field J. Interactions between adenylyl cyclase, CAP and RAS from Saccharomyces cerevisiae. Cell Signal. 1994;6:681–694. doi: 10.1016/0898-6568(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Moriyama K, Yahara I. Human CAP1 is a key factor in the recycling of cofilin and actin for rapid actin turnover. J Cell Sci. 2002;115:1591–1601. doi: 10.1242/jcs.115.8.1591. [DOI] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Lamensdorf I, Yoon SH, Youle RJ. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol. 2001;153:1265–1276. doi: 10.1083/jcb.153.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Shima F, Sen H, Tanaka Y, Yanagihara C, Yamawaki-Kataoka Y, Kariya K, Kataoka T. Coiled-coil interaction of N-terminal 36 residues of cyclase-associated protein with adenylyl cyclase is sufficient for its function in Saccharomyces cerevisiae Ras pathway. J Biol Chem. 1998;273:28019–28024. doi: 10.1074/jbc.273.43.28019. [DOI] [PubMed] [Google Scholar]
- Peche V, Shekar S, Leichter M, Korte H, Schroder R, Schleicher M, Holak TA, Clemen CS, Ramanath YB, Pfitzer G, et al. CAP2, cyclase-associated protein 2, is a dual compartment protein. Cell Mol Life Sci. 2007;64:2702–2715. doi: 10.1007/s00018-007-7316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima F, Okada T, Kido M, Sen H, Tanaka Y, Tamada M, Hu CD, Yamawaki-Kataoka Y, Kariya KI, Kataoka T. Association of yeast adenylyl cyclase with cyclase-associated protein CAP forms a second Ras-binding site which mediates its Ras-dependent activation. Mol Cell Biol. 2000;20:26–33. doi: 10.1128/mcb.20.1.26-33.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiston J, Hubberstey A, Yu G, Young D. Differential expression of CAP and CAP2 in adult-rat tissues. Gene. 1995;165:273–277. doi: 10.1016/0378-1119(95)00522-8. [DOI] [PubMed] [Google Scholar]
- Utsumi T, Sakurai N, Nakano K, Ishisaka R. C-terminal 15 kDa fragment of cytoskeletal actin is posttranslationally N-myristoylated upon caspase-mediated cleavage and targeted to mitochondria. FEBS Lett. 2003;539:37–44. doi: 10.1016/s0014-5793(03)00180-7. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Thompson CB. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-Kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- Yu J, Wang C, Palmieri SJ, Haarer BK, Field J. A cytoskeletal localizing domain in the cyclase-associated protein, CAP/Srv2p, regulates access to a distant SH3-binding site. J Biol Chem. 1999;274:19985–19991. doi: 10.1074/jbc.274.28.19985. [DOI] [PubMed] [Google Scholar]
- Zha JH, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-xL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.