

Abstract
A facile synthetic route to substituted trans-2-arylcyclopropylamines was developed to provide access to mechanism-based inhibitors of the human flavoenzyme oxidase lysine-specific histone demethylase LSD1 and related enzyme family members such as monoamine oxidases A and B.
The functional capacity of genetically-encoded proteins is powerfully expanded by reversible posttranslational modification. Within eukaryotic cells, the regulation of gene expression is intimately linked with posttranslational modification of histone proteins. Reversible histone post-translational modifications include acetylation of lysine, phosphorylation of serine and threonine, and methylation of lysine and arginine.1 Additionally, lysine residues may be mono-, di-, or trimethylated, while arginine residues may be mono- or dimethylated. The resulting complexity of modifications has been postulated to act as a "histone code", by which these patterns of modifications are "read" by cellular machinery to produce a specific gene regulatory outcome.1
Two classes of human enzymes capable of demethylating methylated lysine residues within histones have been recently identified. JmjC domain-containing demethylases including JHDM3A/JMJD2A, GASC1/JMJD2C, JHDM2A, and JHDM1 comprise one class;2–6 these enzymes appear to utilize the cofactors Fe(II) and α-ketoglutarate to demethylate via hydroxylation of the methyl group with subsequent elimination of formaldehyde. The second class of histone demethylases includes the amine oxidase domain-containing enzyme lysine-specific demethylase 1 (LSD1).7 Catalysis by this enzyme is a flavin-dependent process in which formaldehyde and peroxide are produced as by-products of histone demethylation (Scheme 1).7, 8 The requirement for formation of positive charge on the ε-nitrogen atom of lysine in the imine intermediate limits LSD1 to demethylation of dimethylated and monomethylated lysine. The amine oxidase domain of LSD1 is homologous to equivalent domains found in polyamine oxidase (PAO, 22.4% identity) and monoamine oxidases A and B (MAO A, 17.6% identity; MAO B, 17.6% identity).
Scheme 1.

The reaction catalyzed by the histone demethylase LSD1
Monoamine oxidase inhibitors (MAOIs) are well-known drugs that have been used clinically for the treatment of depression, anxiety, and Parkinson's disease. Because of the similarities between the catalytic mechanisms and architecture of the enzyme active sites of MAO A and B and LSD1, we hypothesized that flavin-targeted irreversible inhibitors of monoamine oxidases might simultaneously inhibit the function of LSD1, perhaps leading to pleiotropic effects beneficial to the biological action of these therapeutics.
Indeed, upon evaluation of this hypothesis, we discovered that clinically-relevant concentrations of trans-2-phenylcyclopropylamine9 (2-PCPA, brand name Parnate), an irreversible inhibitor of monoamine and polyamine oxidases, proved effective at inhibiting LSD1-mediated histone demethylation in vivo and in vitro. Our group subsequently reported the first mechanistic studies of LSD1 inactivation by 2-PCPA.10 As depicted in Scheme 2, LSD1 inhibition by 2-PCPA occurs via formation of a covalent adduct joined at the N5 and C4a positions of the flavin ring (Scheme 2).11 To pave the way for ongoing LSD1-selective inhibitor discovery, we report here a facile route to substituted trans-2-arylcyclopropylamines and the kinetic evaluation of a representative subset of 2-PCPA derivatives as mechanism-based inactivators of the human flavoenzyme oxidase lysine-specific histone demethylase LSD1 and related monoamine oxidases A and B.
Scheme 2.

The proposed mechanism of inhibition of LSD1 by 2-PCPA via flavin adduct formation
Results
The general synthetic route to trans-2-arylcyclopropylamines commenced with the synthesis of esters 2a–k either by esterification of the commercially available carboxylic acids or by palladium catalyzed cross-coupling between the appropriate aryl iodide and methyl acrylate (Scheme 3). Two cyclopropanation methods were employed for the preparation of compounds 3a–k (Table 1). Reaction of α,β-unsaturated esters 2a–j with the Corey-Chaykovsky reagent in DMSO (Method A) generally gave poor yields of the desired cyclopropanated products.12–14 Detosylation of the indole nitrogen in 2k was anticipated under the strongly basic conditions of Method A, thus cyclopropanation by this method was not attempted. On the other hand, use of diazomethane with palladium (II) acetate15 (Method B) afforded the corresponding cyclopropanated products in excellent yields with only two exceptions (entries 9 and 10, Table 1).
Scheme 3.

Reagents and conditions: (a) TMSCHN2, C6H6, MeOH; (b) Pd(OAc)2, K2CO3, NBu4Cl, DMF; (c) 1.6 mol% Pd(OAc)2, CH2N2, THF or (d) Me3S(O)I/NaH, DMSO; (e) (i) aq. NaOH, MeOH, (ii) aq. HCl; (f) (i) diphenylphosphoryl azide, Et3N, t-BuOH, (ii) aq. HCl, THF; or (g) (i) ethyl chloroformate, Et3N, acetone, (ii) aq. NaN3, (iii) 2-(trimethylsilyl)ethanol, (iv) NBu4F, THF.
Table 1.
Cyclopropanation methods for the preparation of esters 3a–k
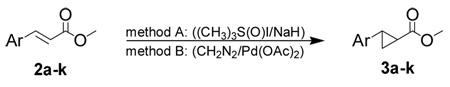 | ||||
|---|---|---|---|---|
| entry | compound | Ar | % yielda, (method A)4 | % yielda, (method B)5 |
| 1 | 3a | 4-CF3C6H4 | 27 | 96 |
| 2 | 3b | 4-BrC6H4 | 50 | 94 |
| 3 | 3c | 4-CH3OC6H4 | 50 | 98 |
| 4 | 3d | 4-NO2C6H4 | 5 | 65 |
| 5 | 3e | 2-thienyl | 40 | 97 |
| 6 | 3f | 3-thienyl | 59 | 99 |
| 7 | 3g | 2-furyl | 27 | 94 |
| 8 | 3h | 3-furyl | 21 | 99 |
| 9b | 3i | 3-pyridyl | 34 | - |
| 10b | 3j | 1-napthyl | 65 | - |
| 11c | 3k | N(Ts)-5-indoyl | - | 97 |
Yields are for isolated product using 1.4 – 1.8 mmol of alkene substrate.
Cyclopropanation by Method B gave inseparable and/or complex mixtures.
Cyclopropanation by Method A not attempted on this substrate.
For these two substrates, cyclopropanation by Method B gave complex reaction mixtures whereas all other substrates reacted under these conditions yielded the cyclopropyl adduct as the sole reaction product. However, cyclopropanation with Method A provided 3i and 3j in acceptable yield as the sole reaction products without the need for further purification.
The trans stereochemistry of the 2-arylcyclopropylcarboxylate esters, obtained by either method, was confirmed by the vicinal 1H-1H coupling constants of the cyclopropyl moiety (Jab = 4.2 – 4.5 Hz). Alkaline hydrolysis gave the desired trans-arylcyclopropanecarboxylic acids in high yields. These acids were transformed to the corresponding primary amines by Curtius rearrangements in yields ranging from 41–56% over 4 steps (Scheme 3).16, 17
Additionally, as shown in Scheme 4, the synthesis of 2,2-diphenylcyclopropylamine (7) was accomplished in four steps.18 Cyclopropanation of 5 was achieved via copper(II) sulfate catalyzed addition of ethyl diazoacetate to 1,1-diphenylethene. The resulting ester was hydrolyzed to give 6. Curtius rearrangement afforded the desired amine 7.
Scheme 4.

Reagents and conditions: (a) N2CHCO2Et, 3 mol% CuSO4, C6H6 reflux ; (b) NaOH, EtOH (87% after 2 steps); (c) diphenylphosphoryl azide, Et3N, t-BuOH; (d) 1M HCl, MeOH (52% overall)
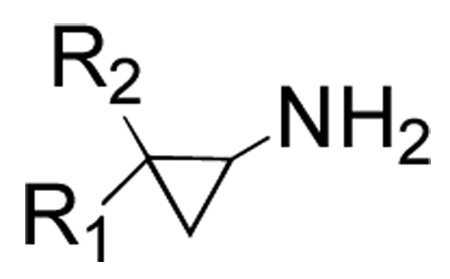
We next examined inhibition kinetics for a representative subset of the 2-PCPA derivatives against human LSD1 and monoamine oxidases A and B enzymes (Table 2). Intriguingly, biphenyl (7) and naphthyl (8) 2-PCPA analogues showed only minor inhibitory potential with respect to LSD1 at concentrations up to 1 mM (Table 3). However, the remaining derivatives displayed approximately four- to five-fold increased values of kinact/KI for inactivation of LSD1. Each of the inhibitors was tested for their ability to irreversibly inhibit LSD1 by incubating 50 µM LSD1 with 250 µM inhibitor overnight, then diluting 100-fold into assay solution. Only LSD1 incubated with the biphenyl derivative (7) showed any residual activity, approximately 2% (data not shown). This demonstrates that both compounds 7 and 8 are able to inhibit LSD1 over a longer period of time. Absorbance scans of LSD1 inactivated by the compounds showed the formation of the reduced flavin species previously reported with 2-PCPA (data not shown).10
Table 2.
Representative 2-PCPA derivatives synthesized and kinetically evaluated for LSD1, MAOA, and MAOB inhibition
 | ||
|---|---|---|
| Compound | R1 | R2 |
| 7 | Ph | Ph |
| 8 | 1-naphthyl | H |
| 9 | 4-CF3C6H4 | H |
| 10 | 4-BrC6H4 | H |
| 11 | 4-CH3OC6H4 | H |
| 12 | 3-thienyl | H |
Table 3.
Kinetic parameters for inactivation of LSD1, MAO B, and MAO A by trans-2-phenylcyclopropylamine derivatives.
| LSD1 | MAO B | MAO A | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibitor | kinact (s−1) | KI (µM) | kinact/KI (M −1s−1) | kinact (s−1) | KI(µM) | kinact/KI (M−1s −1) | kinact (s−1) | KI (µM) | kinact/KI (M−1s−1) |
| 2-PCPA | 0.026 ± 0.001 | 477 ± 79 | 55 | 0.024 ± 0.002 | 13.6 ± 3.0 | 1779 | 0.018 ± 0.001 | 37.3 ± 8.4 | 469 |
| 7 | N.D.a | N.D. | < 1 | N.D. | N.D. | < 1.5 | N.D. | N.D. | < 14 |
| 8 | N.D. | N.D. | < 28 | 0.047 ± 0.001 | 9.8 ± 0.6 | 4837 | 0.014 ± 0.001 | 38.7 ± 8.3 | 362 |
| 9 | 0.067 ± 0.008 | 352 ± 95 | 190 | 0.058 ± 0.002 | 7.7 ± 1.4 | 7507 | 0.085 ± 0.004 | 29.8 ± 3.3 | 2859 |
| 10 | 0.104 ± 0.007 | 566 ± 73 | 184 | 0.078 ± 0.005 | 32.9 ± 9.1 | 2356 | 0.070 ± 0.005 | 36.5 ± 6.2 | 1918 |
| 11 | 0.049 ± 0.004 | 188 ± 52 | 262 | 0.070 ± 0.002 | 86.6 ± 7.8 | 802 | 0.023 ± 0.001 | 20.5 ± 3.0 | 1112 |
| 12 | N.D. | N.D. | < 10 | 0.035 ± 0.005 | 1548 ± 312 | 22.3 | N.D. | N.D. | < 18 |
N.D., not determined.
The structure of the 2-PCPA-flavin adduct was recently determined11 and was found to differ from that seen in the crystal structure of MAO B inactivated by 2-PCPA19 (Figure 1 and Figure 2). In LSD1, cyclopropyl ring opening appears to lead to formation of a benzylic radical which attacks the flavin at C4(a) to form an initial adduct, which then undergoes cyclization to form a five-membered ring joining the N5 and C4(a) atoms of the isoalloxazine ring.
Figure 1.

Structures of the 2-PCPA-flavin adducts identified in the crystal structures of MAO B (left) and LSD1 (right)
Figure 2.

Active sites of (a) LSD1, (b) MAOB, and (c) MAOA.
Formation of the adduct structure seen in MAO B is apparently prohibited in LSD1 by a steric clash between the phenyl ring of 2-PCPA and F538 and Y761.11 There appears to be ample room at the para position of the phenyl ring of the 2-PCPA-flavin adduct in LSD1 to accommodate substituents of varying sizes at this position. However, formation of an adduct with naphthyl derivative 8 is predicted to be limited in either possible orientation by Val333 on one side of the active site and Ala809 on the other. The active site cavity in the vicinity of the adduct carbon adjacent to flavin C4(a) is not large enough to efficiently accommodate an additional phenyl moiety in the biphenyl derivative 7.
Three of the five tested 2-PCPA derivatives showed increased inhibitory potential towards MAO B relative to the starting compound; these included the naphthyl (8), para-trifluoromethyl (9), and para-bromo (10) derivatives (Table 2 and Table 3). It is somewhat unclear how the active site of MAO B is able to accept the naphthyl derivative, but rotation of the carbon-carbon bond between the naphthyl group and the rest of the adduct likely prevents a steric clash with Gln206 (Figure 2). As with LSD1, the biphenyl derivative showed little inhibitory potential at concentrations up to 1 mM. Interestingly, the para-methoxy substituted derivative showed a decrease in kinact/KI of approximately 50% relative to 2-PCPA, with a nearly seven-fold increase in KI. In the active site of MAO B, Leu171, Tyr326, and Gln206 form a "cap" above the para position of the phenyl ring of 2-PCPA, likely limiting the length of substituents permissible at that position, and suggesting a means to develop inhibitors selective for LSD1 over MAO B. We are currently conducting enantioselective syntheses of the most potent and selective inhibitors of LSD1 to determine the degree to which absolute stereochemistry affects inhibitory potency.
With the exception of the biphenyl derivative 7, all of the tested 2-PCPA derivatives were equal to or greater than 2-PCPA in their ability to inhibit MAO A (Table 3). The naphthyl derivative was equal in potency to 2-PCPA and less potent than the p-bromo and p-methoxy derivatives, opposite of the trend in MAO B. For both monoamine oxidases compound 9 was the most potent. Although the exact structure of the 2-PCPA-flavin adduct formed in MAO A is unknown, superposition of the LSD1 and MAO B adducts in the active site of MAO A suggests that the MAO B adduct will be formed (data not shown). Ile335 in MAO A replaces Tyr326 in MAO B, potentially creating additional inhibitor access. Cys323 serves as an upper limit on the space above the para carbon of the phenyl group of 2-PCPA; the sulfur is located 10.2 Å away.
Presently we are investigating whether analysis of cells exposed to 2-PCPA or other MAOIs reveal some of the contributing factors responsible for the poorly understood therapeutic effects and side effects of these antidepressants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang Y, Reinberg D. Genes Dev. 2001;15:2343. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 2.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. Nature. 2006;442:307. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 3.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. Nature. 2006;442:312. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 4.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Nature. 2006;439:811. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 5.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Cell. 2006;125:467. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 6.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. Cell. 2006;125:483. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 7.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Cell. 2004;119:941. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Forneris F, Binda C, Vanoni MA, Mattevi A, Battaglioli E. FEBS Lett. 2005;579:2203. doi: 10.1016/j.febslet.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Burger A, Yost L, Yost J. J. Amer. Chem. Soc. 1948;70:2198. [Google Scholar]
- 10.Schmidt DM, McCafferty DG. Biochemistry. 2007;46:4408. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 11.Yang M, Culhane JC, Szewczuk LM, Jalili P, Ball HL, Machius M, Cole PA, Yu H. Biochemistry. 2007;46:8058. doi: 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- 12.Ciaccio JA, Aman CE. Synth. Comm. 2006;36:1333. [Google Scholar]
- 13.Corey EJ, Chaykovsky M. J. Am. Chem. Soc. 1962:867. [Google Scholar]
- 14.Corey EJ, Chaykovsky M. J. Am. Chem. Soc. 1965;87:1353. [Google Scholar]
- 15.Arvidsson LE, Johansson AM, Hacksell U, Nilsson JL, Svensson K, Hjorth S, Magnusson T, Carlsson A, Lindberg P, Andersson B, et al. J. Med. Chem. 1988;31:92. doi: 10.1021/jm00396a014. [DOI] [PubMed] [Google Scholar]
- 16.Vallgarda J, Appelberg U, Arvidsson L, Hjorth S, Svensson BE, Hacksell U. J. Med. Chem. 1996;39:1485. doi: 10.1021/jm9507136. [DOI] [PubMed] [Google Scholar]
- 17.Rosen TC, Yoshida S, Frolich R, Kirk KL, Haufe GJ. J. Med. Chem. 2004;47:5860. doi: 10.1021/jm049957t. [DOI] [PubMed] [Google Scholar]
- 18.Tichilibon S, Kim SK, Bao ZG, Harris BA, Blaustein JB, Gross AS, Duong HT, Melman N, Jacobson KA. Bio. Org. Med. Chem. 2004;12:2021. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Proc. Nat. Acad. Sci. U.S.A. 2003;100:9750. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]