

Abstract
Moderate heat stress (40 °C for 30 min) on spinach thylakoid membranes induced cleavage of the reaction center-binding D1 protein of photosystem II, aggregation of the D1 protein with the neighboring polypeptides D2 and CP43, and release of three extrinsic proteins, PsbO, -P, and -Q. These heat-induced events were suppressed under anaerobic conditions or by the addition of sodium ascorbate, a general scavenger of reactive oxygen species. In accordance with this, singlet oxygen and hydroxyl radicals were detected in spinach photosystem II membranes incubated at 40 °C for 30 min with electron paramagnetic resonance spin-trapping spectroscopy. The moderate heat stress also induced significant lipid peroxidation under aerobic conditions. We suggest that the reactive oxygen species are generated by heat-induced inactivation of a water-oxidizing manganese complex and through lipid peroxidation. Although occurring in the dark, the damages caused by the moderate heat stress to photosystem II are quite similar to those induced by excessive illumination where reactive oxygen species are involved.
Photosystem II (PS II)3 in higher plants is a multisubunit complex composed of more than 25 proteins and the associated cofactors. Excitation energy captured by the chlorophylls and carotenoids in the light-harvesting chlorophyll protein complexes of PS II is finally transferred to P680, the reaction center chlorophyll of PS II, where charge separation takes place. In particular, PS II performs oxidation of water and reduction of plastoquinone molecules via chlorophyll-mediated photochemical reactions. Although it plays such an important role in the primary photochemical reaction of photosynthesis, PS II is vulnerable to various environmental stresses such as excessive visible light and high temperature.
When irradiated with excessive visible light, the D1 protein is oxidatively
damaged, and electron transport is inhibited. This process is referred to as
photoinhibition of PS II
(1–4).
The photo-damaged D1 protein is subsequently degraded by specific proteases
(5), and the repair of PS II is
accomplished by the integration of a newly synthesized D1 protein to the PS II
complex (6). Photoinhibition of
PS II is caused by either the so-called acceptor-side or donor-side mechanism
or both (4,
7). The acceptor-side
photoinhibition takes place when the acceptor side of PS II is over-reduced by
excessive illumination and the double-reduced QA molecule is
released from its binding site. Reversed electron flow from the primary
electron acceptor pheophytin to P680 in the absence of QA generates
the triplet state P680, which reacts with molecular oxygen to form singlet
oxygen (1O2). The 1O2 eventually
damages the nearby polypeptide, the D1 protein. Alternatively, oxygen
molecules may be reduced at the acceptor side of PS II to produce superoxide
anion radicals
(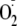 ),
which are turned into hydrogen peroxide (H2O2) and
finally hydroxyl radical (HO·) through the Fenton reaction
(8). It is claimed, however,
that the generation of
),
which are turned into hydrogen peroxide (H2O2) and
finally hydroxyl radical (HO·) through the Fenton reaction
(8). It is claimed, however,
that the generation of
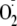 is more prominent at the acceptor side of PS I than at PS II under excessive
illumination (9).
is more prominent at the acceptor side of PS I than at PS II under excessive
illumination (9).
In the donor-side photoinhibition of PS II, the extrinsic proteins in PS II, such as PsbO as well as manganese, should be destabilized before illumination of PS II. This can be done in vitro by washing the thylakoid membranes or PS II membranes with a solution of high concentration salts such as 1 m CaCl2 or alkaline solutions such as Tris-HCl (pH 8.0) (10, 11). Illumination of the donor side-impaired PS II gives rise to cationic radicals including P680+, which causes oxidative damage to the D1 protein.
Moderate heat stress is another environmental factor that affects the D1 protein significantly. It was shown that the treatment of spinach thylakoids at elevated temperatures induced inhibition of oxygen evolution, dephosphorylation of the phosphoproteins (12, 13), cleavage and aggregation of the D1 protein (13, 14), and release of three extrinsic proteins PsbO, -P, and -Q from PS II (13, 15–17). The cleavage products of the D1 protein induced by heat treatment at 40 °C for 30 min have a mass of 23 and 9 kDa, which are similar in size to the fragments detected in the acceptor-side photoinhibition of PS II (13, 14). These results suggest that the site of cleavage in the D1 protein during heat stress is the stroma-exposed DE-loop of the D1 protein, being identical to that of the acceptor-side photoinhibition of PS II. In PS II, the moderate heat stress was suggested to destabilize the secondary quinone electron acceptor QB in the DE-loop of the D1 protein (14), and such a conformational change may be a prerequisite condition for the subsequent cleavage of the D1 protein at the DE-loop.
As the profile of cleavage of the D1 protein is similar between the acceptor-side photoinhibition and heat inhibition of PS II, we presumed that it was the reactive oxygen species (ROS) that were responsible for the damage to PS II during the heat inhibition. To examine this possibility, we first studied heat-induced damage to the D1 protein and then release of PsbO, -P, and -Q proteins from PS II and other related processes under aerobic and anaerobic conditions using spinach thylakoid membranes. We also carried out EPR spin-trapping spectroscopy to detect free oxygen radicals in the PS II membranes. Our results demonstrated that the damage to the D1 protein and the release of extrinsic proteins under moderate heat stress were prevented under the anaerobic conditions. As 1O2 and HO· were detected in PS II by EPR spin-trapping spectroscopy under the same heat stress conditions, we suggest that the ROS are responsible for the heat-induced damage to PS II.
EXPERIMENTAL PROCEDURES
Preparation of the Thylakoid and PS II Membranes from Spinach—Intact thylakoids were prepared from spinach as described previously (18). PS II membranes were prepared according to Berthold et al. (19) with modifications described by Ford and Evans (20) and suspended in a medium containing 0.4 m sucrose, 15 mm NaCl, 5 mm MgCl2, and 40 mm MES (pH 6.5). Both preparations were stored at -80 °C. PS II membranes lacking the water-oxidizing manganese complex were prepared by incubation of PS II membranes (1 mg of chlorophyll ml-1) in a buffer containing 0.8 m Tris-HCl (pH 8.0). The PS II membranes were incubated for 30 min on ice, in the darkness with a continuous gentle stirring. After treatment, the PS II membranes were washed several times in 0.4 m sucrose, 10 mm NaCl, 5 mm CaCl2, and 40 mm MES-NaOH (pH 6.5).
Heat Treatment of Thylakoid and PS II Membranes—For heat treatment, the thylakoid membranes or the PS II membranes were put in plastic microtubes (volume, 1.0 ml) and incubated at 40 °C for the indicated periods in a thermostated water bath in the dark. The incubation was immediately terminated by transferring the sample tubes to an ice bath.
Treatment of Thylakoids and PS II Membranes with Chelex—To remove metals from the samples, 1 ml each of the samples containing 1 mg of chlorophyll was mixed with 2 ml of 10% Chelex with a vortex mixer for 1–2 min. After standing for 15 min at 20 °C, the mixture was centrifuged, and the supernatant was collected and used as the Chelex-treated sample.
Measurements of PS II Activity and Manganese Content—The PS II activity was measured either by monitoring the chlorophyll fluorescence, Fv/Fm (where Fv/Fm indicates the optimum quantum yield of PS II measured by the ratio of the variable fluorescence (Fv) and the maximum fluorescence (Fm) from chlorophyll excited by saturating flashes), with a minimal pulse amplitude modulation (Walz, Germany), or by monitoring oxygen evolution with an oxygen electrode (Hansatech, UK). The reaction mixture for the chlorophyll fluorescence measurement contained 0.4 m sucrose, 5 mm MgCl2, and 50 mm MES-NaOH (pH 6.5). For measurement under anaerobic conditions, 10 mm glucose, 0.1 mm glucose oxidase, and 0.1 mm catalase were added to the reaction mixture. Mineral oil was layered on top of the cuvette containing the reaction mixture, which was kept for 40 min in the dark at 25 °C to remove molecular oxygen. The reaction mixture for the measurement of oxygen evolution was the same as that for the fluorescence measurements, except that 0.5 mm phenyl p-benzoquinone and 1 mm potassium ferricyanide were added as electron acceptors, and 10 mm NH4Cl was used as an uncoupler for the thylakoids. The temperature of the samples was regulated by a thermostated water bath. After heat treatment of the samples at 40 °C for the indicated periods, the PS II activity was measured at 25 °C.
For determination of manganese, the PS II membranes (0.1 mg of chlorophyll) were incubated with 1 ml of 0.1 n HNO3 at 90 °C. After centrifugation at 5,000 × g for 5 min, the content of manganese in the supernatants was measured with an Hitachi Z-200 atomic absorption spectrophotometer. The content of manganese in PS II was determined on the basis of the antenna size of PS II in the PS II membranes reported previously (21).
Determination of Protein Oxidation—The carbonyl content of protein was measured according to the manufacturer's protocol (Oxi-Blot protein oxidation detection kit, Chemicon International). Briefly, protein samples (5 μg/lane) were derivatized with dinitrophenyl hydrazine, fractionated by 12.5% SDS-PAGE, and electroblotted to PVDF membranes (Millipore). The derivatized proteins were sequentially reacted with rabbit anti-dinitrophenyl and horseradish peroxidase-conjugated goat anti-rabbit IgG antibodies and visualized by enhanced chemiluminescence (ECL) (Amersham Biosciences).
Assay of Cleavage, Degradation, and Aggregation of the D1 Protein—The membrane samples were treated by moderate heat as described above under either aerobic or anaerobic conditions. Anaerobic conditions were obtained using 10 mm glucose, 0.1 mm glucose oxidase, and 0.1 mm catalase as described above. Depletion of molecular oxygen in the reaction mixture was checked by an oxygen electrode (Hansatech, UK). SDS-urea/PAGE and Western blot analysis with specific antibodies were carried out as described (11, 18). Immuno-decorated bands were detected by fluorography with ECL. The density of the proteins in the fluorogram was measured by the use of Scion Image software.
Measurement of Lipid Peroxidation—Lipid peroxidation of spinach thylakoids under heat stress was measured using a thiobarbituric acid (TBA) test (22) according to Mishra and Singhal (23), where the amount of malondialdehyde (MDA) was estimated from the absorbance at 532 nm of the TBA·MDA complex in the sample using an Hitachi U-2000 spectrophotometer, with absorbance at 600 nm as a reference wavelength to monitor the nonspecific absorption.
EPR Spin-trapping Spectroscopy—EPR spin-trapping spectroscopy was used to measure 1O2 and HO· production. Spin trapping of 1O2 was accomplished using 2,2,6,6-tetramethylpiperidine (TEMP) (Sigma), whereas an EMPO spin trap compound (5-(ethoxycarbonyl)-5-methyl-1-pyrroline N-oxide) (Alexis Biochemicals, Lausen, Switzerland) was used for HO· detection. To avoid precipitation of the TEMP spin trap, 5% ethanol was used. PS II membranes (500 μg of chlorophyll ml-1) in 40 mm MES (pH 6.5) in the presence of 50 mm TEMP or 75 mm EMPO were heated in Eppendorf tubes immersed in a water bath. Heating was performed at 40 °C with a digitally controlled heater in complete darkness for the times indicated in the figures. Following heat treatment, PS II membranes were put into a glass capillary tube (Blaubrand® intraMARK, Brand, Germany), and EPR spectra were recorded using an EPR spectrometer MiniScope MS200 (Magnettech GmbH, Germany). Signal intensity was evaluated from the relative height of the central peak (TEMPO) and the central doublet (EMPO-OH). The EPR spectrum was obtained as a first derivate of the EPR absorption signal. EPR conditions were as follows: microwave power (10 milliwatts), modulation amplitude (1 G), modulation frequency (100 kHz), sweep width (100 G), and scan rate (1.62 G s-1).
Detection of PsbO Released from PS II upon Heat Treatment—PS II membranes were treated with heat at 40 °C for the indicated periods under the aerobic and anaerobic conditions, and then the membranes were centrifuged at 35,000 × g for 30 min. After separation and collection, the supernatants and the precipitates were subjected to SDS/urea-PAGE and Western blot analysis with the antibody against the PsbO protein.
Artificial Production of ROS—1O2 was produced by the addition of 1 mm H2O2 and 1 mm NaClO. Hydroxyl radical was generated by the addition of 1 mm H2O2 and 1 mm FeSO4. The PS II membranes were exposed to the ROS at either 20 or 40 °C for 30 min.
RESULTS
Heat-induced Cleavage and Aggregation of the D1 Protein in Spinach Thylakoid Membranes under Aerobic and Anaerobic Conditions—Spinach thylakoid membranes were treated at 40 °C for 30 min under aerobic and anaerobic conditions. After SDS/urea-PAGE and Coomassie Blue staining to check the profile of the proteins (Fig. 1A), cleavage of the D1 protein was monitored by Western blot analysis with specific antibodies against the D1-DE loop and the C-terminal part of the D1 protein. Two fragments of the D1 protein, a 23-kDa N-terminal fragment and a 9-kDa C-terminal fragment, were detected under aerobic conditions, as reported previously (13) (Fig. 1B). In addition, aggregates of the D1 protein, having a mass of 65 kDa and higher and being identified previously as D1/D2 and D1/CP43 adducts (13), were also observed under the aerobic conditions (Fig. 1B). As the fate of the D1 protein seems to be very similar to that observed under illumination with excessive light, we assumed that ROS, which are related to the photo-damage to PS II, are responsible for the heat-induced processes. To examine this possibility, we compared the profile of the D1 protein exposed to moderate heat stress under aerobic conditions with the profile of the D1 protein exposed to anaerobic conditions. Interestingly, neither the D1 fragments nor the D1 aggregates were detected when the spinach thylakoid membranes were treated by heat under the anaerobic conditions (Fig. 1, B and C). These results strongly suggest that the heat-induced cleavage and aggregation of the D1 protein are caused by oxidative processes.
FIGURE 1.

Cleavage and aggregation of the D1 protein from spinach thylakoid membranes induced by moderate heat stress under aerobic and anaerobic conditions. A, profile of the thylakoid proteins in SDS/urea-PAGE with Coomassie staining after heat treatment of the thylakoids at 40 °C for the indicated periods under the aerobic (+O2) and anaerobic (-O2) conditions. The control temperature is 20 °C. Molecular markers are shown on the left-hand side of the gel. The positions of several proteins of PS II are indicated on the right-hand side of the gel. B, Western blot analysis with the antibody against the DE-loop and the C-terminal part of the D1 protein, showing the profile of the D1 protein from the thylakoid membranes treated at 40 °C for the indicated periods. The positions of the molecular mass markers are on the left side, and those of the degradation fragments and the aggregates of the D1 protein are on the right-hand side of the gel. C, quantification of the 23-kDa degradation fragments of the D1 protein detected after heat treatment of the thylakoid membranes at 40 °C for the indicated periods under the aerobic (+O2) and anaerobic (-O2) conditions. The data are the average of three independent measurements ± S.D. D, effects of sodium ascorbate on the heat-induced cleavage and aggregation of the D1 protein. Sodium ascorbate (2 mm) was added before the heat treatment of the thylakoid membranes at 40 °C under both the aerobic and anaerobic conditions, and after SDS/urea-PAGE, Western blot analysis was carried out with the antibody against the DE-loop of the D1 protein.
Effect of a Radical Scavenger, Sodium Ascorbate, on the Heat-induced Damage to the D1 Protein—Sodium ascorbate is a general scavenger of ROS (24). The effects of sodium ascorbate on the heat-induced cleavage and aggregation of the D1 protein were examined. Apparently the addition of 1 mm sodium ascorbate to the thylakoid membranes significantly suppressed the D1 cleavage as well as the D1 aggregation induced by the heat stress under the aerobic conditions, suggesting that the ROS indeed damage the D1 protein (Fig. 1D).
Oxidation of the Proteins in the Thylakoid Membranes under Moderate Heat Stress—We next examined the oxidation of the proteins in the thylakoid membranes and PS II membranes by moderate heat stress (40 °C for 30 min) under aerobic conditions (Fig. 2). The Oxi-Blot protein oxidation detection kit (Chemicon International) was used to monitor the protein oxidation. To be able to detect small changes in the protein oxidation in the thylakoid membranes, and also to identify the proteins that were most oxidized under the heat-stressed aerobic conditions, we changed the contact time of the x-ray films and the PVDF membranes where the thylakoid proteins were electroblotted. After heat treatment at 40 °C for 30 min under the aerobic conditions, several proteins in PS II were oxidized. They included the D2, PsbO, and PsbQ proteins, as revealed by Western blot analysis with specific antibodies. The amount of D1 protein likely decreased because of its degradation and aggregation.
FIGURE 2.

Detection of oxidation of the proteins in the thylakoid membranes induced by moderate heat stress. Thylakoid membranes were heat-treated at 40 °C for 30 min under aerobic (+O2) and anaerobic (-O2) conditions, and the content of protein carbonyl was measured by an Oxi-Blot protein oxidation detection kit. The control temperature is 20 °C. A, fluorograms of Oxi-Blot (left) and of Western blot analysis with the antibody against the DE-loop of the D1 protein (right). B, fluorograms of Oxi-Blot (left) and of Western blot analysis with the antibody against the D2 and PsbO proteins (right). C, fluorograms of Oxi-Blot (left) and of Western blot analysis with the antibody against PsbQ. A–C, the same PVDF membranes with the thylakoid proteins blotted were exposed to x-ray films for different periods to clarify the differences in the degree of protein oxidation between the aerobic and anaerobic conditions, as well as between the stress temperature and the control conditions.
Detection of 1O2 and HO· upon Heat Treatment of Spinach PS II Membranes by EPR—Production of 1O2 and HO· in PS II membranes was monitored using EPR spin-trapping spectroscopy. Generation of 1O2 upon heat treatment was detected in the presence of a TEMP spin trap compound. Exposure of PS II membranes to moderate temperature (40 °C) resulted in the generation of TEMPO EPR spectra (Fig. 3, A and B). The TEMPO EPR signal observed in nonheated PS II membranes was due to an impurity of the spin trap compound. Production of HO· in the PS II membranes was monitored in the presence of the EMPO spin trap compound. In nonheated PS II membranes, no EPR signal was observed in the presence of EMPO, whereas the addition of spin trap compound to PS II membranes followed by exposure of the membranes to moderate heating resulted in the appearance of the characteristic EMPO-OH adduct EPR signal (Fig. 3, C and D). It has been recently reported that the appearance of the EMPO-OH EPR signal observed in heated PS II membranes is solely linked to the generation of HO· (25).
FIGURE 3.

Detection of reactive oxygen species in the heat-treated PS II membranes. A, TEMPO EPR spectra measured in the PS II membranes heated at 40 °C for the periods indicated in the figure. TEMPO spectra were measured in the presence of 50 mm TEMP, 5% ethanol, 500 μg of chlorophyll ml-1, and 40 mm MES (pH 6.5). B, time profile of a TEMPO EPR signal measured in the PS II membranes heated at 40 °C. TEMPO EPR signal was determined as the relative height of the central peak of the first derivative of the absorption spectra. C, EMPO-OH adduct EPR spectra measured in the PS II membranes heated at 40 °C for the periods indicated in the figure. EMPO-OH spectra were measured in the presence of 75 μm EMPO, 500 μg of chlorophyll ml-1, and 40 mm MES (pH 6.5). D, time profile of EMPO-OH adduct EPR signal measured in the PS II membranes heated at 40 °C. EMPO-OH adduct EPR signal was determined as the relative height of the central doublet of the first derivative of absorption spectra. B and D, for longer periods of heat treatment, the spin trap-radical adduct EPR signals decline because of the instability of spin trap-radical adduct.
To study the effect of a water-oxidizing manganese complex on the generation of 1O2 and HO·, EPR spin trapping was performed in Tris-treated PS II membranes. When the Tris-treated PS II membranes were exposed to 40 °C, a TEMPO EPR signal was generated (Fig. 4A). These observations indicate that generation of 1O2 is unlikely related to a water-splitting manganese complex. When Tris-treated PS II membranes were heated in the presence of EMPO, no EMPO-OH adduct EPR spin-trapping spectra were observed (Fig. 4B). These findings reveal that removal of the water-splitting manganese complex prevented production of HO·, and thus the water-splitting manganese complex is likely involved in HO· formation. The observations that the EMPO-OH adduct EPR signal is completely suppressed by H2O2 scavengers (catalase) and a strong iron chelator (desferal) (25) confirmed that HO· is formed by the reduction of H2O2, whereas direct interaction of a spin trap with tyrosine Z or D on the PS II electron donor side is unlikely.
FIGURE 4.

Effect of the removal of the water-splitting manganese complex on formation of reactive oxygen species in the heat-treated PS II membranes. A, TEMPO EPR spectra measured in the Tris-treated PS II membranes heated at 40 °C for the periods indicated in the figure. B, EMPO-OH adduct EPR spectra measured in the Tris-treated PS II membranes heated at 40 °C for the periods indicated in the figure. A and B, other experimental conditions were the same as those in Fig. 3, A and C, respectively.
Quantification of the free radical formation using spin counting is based on the counting of the spin that originated from the radical spin trap adduct. This approach is feasible in the system, where the interaction of the spin trap with free radicals is not limited. However, in the biological membranes, the accessibility of the spin trap to free radicals is determined by several factors (e.g. lipophilic properties of the spin trap). Thus, in the biological membrane, it is only a fraction of free radicals that are trapped and detected by EPR. Based on these considerations, use of an EPR spin-trapping technique for detection of free radicals in biological membranes is considered a qualitative method.
Heat-induced Lipid Peroxidation in Spinach Thylakoid and PS II Membranes—As described above, the ROS were detected in the PS II membranes under heat stress. However, we do not know how the ROS were generated under these conditions. It was shown that lipid peroxidation is a good candidate to produce 1O2 (24). Heat-induced lipid peroxidation was previously shown with wheat thylakoids (23). Therefore, we examined lipid peroxidation in spinach thylakoid and the PS II membranes after they were exposed to heat stress using the TBA method. Here, we detected a marked lipid peroxidation in both the thylakoid and PS II membranes by heat treatment under aerobic conditions, whereas the lipid peroxidation was completely prevented under the anaerobic conditions (Fig. 5). Interestingly, the lipid peroxidation was 10 times more prominent in the PS II membranes than the thylakoid membranes under the aerobic conditions.
FIGURE 5.

Heat-induced lipid peroxidation in the thylakoid and PS II membranes estimated by the TBA test. A, thylakoid membranes. B, PS II membranes. Absorbance at 532 nm of the TBA·MDA complex was measured under the aerobic (closed circles) and anaerobic conditions (open circles) after incubation of the samples at 40 °C for the periods indicated. The concentration of chlorophyll in the reaction mixture was 1 mg ml-1 chlorophyll in A, and 0.1 mg ml-1 chlorophyll in B, respectively. The data are the average of three independent measurements ± S.D.
Effects of Moderate Heat Stress on the PS II Activity and Manganese Content—We investigated the effects of moderate heat stress on the PS II activity of spinach thylakoid membranes and PS II membranes. Oxygen evolving activity was measured with phenyl p-benzoquinone and potassium ferricyanide as electron acceptors. During incubation at 40 °C for 30 min, the oxygen evolving activity of the thylakoids was almost lost completely, whereas the activity of the PS II membranes was more resistant to heat stress (Fig. 6A). The PS II activity was also monitored by chlorophyll fluorescence Fv/Fm using a minimal pulse amplitude modification under aerobic and anaerobic conditions (Fig. 6, B and C). Usually the fluorescence measurement is applied to leaves or cells to monitor the PS II activity in a noninvasive way. As well, when the Fv/Fm is measured at higher temperatures, it has been reported that the light-harvesting chlorophyll a/b complexes are detached from the PS II core, thereby decreasing the Fv level and therefore Fv/Fm, even when PS II is not very inactivated (26). Furthermore, it has been demonstrated that the redox potential of the QA-/QA redox couple is shifted to higher values when thylakoid membranes are exposed to heat treatment (27), which might affect Fv/Fm. Although limited by these problems, Fv/Fm is a good measurement to estimate the PS II activity of the thylakoid membranes under aerobic and anaerobic conditions. The profile of the heat inactivation of the PS II activity under aerobic conditions assayed by Fv/Fm was similar to that measured by oxygen evolution. We estimated that the decrease in the PS II activity under heat stress was much less than that under the aerobic conditions. These results suggested that the oxidative damage to PS II by ROS is one of the important causes of heat inactivation of PS II.
FIGURE 6.

The effects of heat stress on PS II activity in the thylakoids and PS II membranes and on release of manganese from PS II. The samples were incubated at 40 °C for the periods indicated either under aerobic or anaerobic conditions. A, oxygen evolving activity in the thylakoids (closed circles) and PS II membranes (open circles). The samples were incubated at 40 °C under the aerobic conditions. 100% of the oxygen evolving activity corresponds to 91 μmol of O2 evolved mg-1 chlorophyll h-1 with the thylakoids and 311 μmol of O2 evolved mg-1 chlorophyll h-1 with the PS II membranes. The antenna size of the PS II preparation was assumed to be 270 chlorophylls, based on Ref. 21. B, Fv/Fm of chlorophyll fluorescence in the thylakoids under the aerobic (closed circles) or anaerobic (open circles) conditions. The 100% values under the aerobic and anaerobic conditions are 0.68 and 0.64, respectively. C, Fv/Fm of chlorophyll fluorescence in the PS II membranes under aerobic (closed circles) and anaerobic (open circles) conditions. The 100% values under the aerobic and anaerobic conditions correspond to 0.75 and 0.67, respectively. D, the content of manganese in the PS II membranes under the aerobic (closed circles) or anaerobic (open circles) conditions. All the data are the average of three measurements ± S.D.
The content of manganese was measured with atomic absorption spectrometry in the PS II membranes subjected to the moderate heat stress. Three of four manganese in PS II were released by incubation of the PS II membranes at 40 °C for 30 min, under aerobic conditions (Fig. 6D). Interestingly, release of manganese was much less when the PS II membranes were stressed by moderate heat under anaerobic conditions, indicating that ROS affect the binding of manganese in PS II.
Heat-induced Structural Changes at the Donor Side of PS II—Structural changes at the donor side of PS II may also be the cause of the loss of PS II activity under heat stress. As reported previously, PsbO, -P, and -Q proteins were released from PS II upon heat treatment (13, 15–17) (Fig. 7). We confirmed the heat-induced protein release from the PS II membranes under the present conditions (Fig. 7, A and B). Importantly, under the anaerobic conditions, the release of the PsbO protein from PS II was significantly prevented. The addition of sodium ascorbate also suppressed the release of the PsbO protein (Fig. 7, C and D). Thus, ROS are presumably involved in the heat-induced release of the extrinsic protein at least in part. To determine which ROS are responsible for this process, the effects of artificially induced 1O2 and HO· were examined (Fig. 8). Apparently, both ROS were effective in the release of the PsbO protein from PS II. It should be noted here, however, that the concentrations of the chemicals used were relatively high, and therefore, the other PS II proteins such as D1 and D2 were significantly damaged under these conditions (data not shown). Thus, another approach is necessary to determine the reason why the extrinsic proteins are released from PS II under the moderate heat stress conditions.
FIGURE 7.

Heat-induced release of the PsbO protein from PS II under the aerobic and anaerobic conditions and the effects of addition of sodium ascorbate on the protein release. A, heat-induced release of the PsbO protein from the PS II membranes under the aerobic (left) and anaerobic (right) conditions. The samples were incubated at 40 °C for the periods indicated. T, P, and S represent the total PS II membranes, the precipitate, and the supernatant of centrifugation after heat treatment of the PS II membranes, respectively. The positions of PsbO and molecular markers are shown on the right and left side of the gel, respectively. B, estimation of the amounts of PsbO in each fraction shown in A. The data are the average of three independent measurements ± S.D. The blue, purple, and cyan bars represent the amount of PsbO protein in the total PS II membranes, the precipitate, and the supernatant of centrifugation after heat treatment of the PS II membranes. C, effects of sodium ascorbate (2 mm) on the release of the PsbO protein from the PS II membranes under heat stress at 40 °C for 30 min. Sodium ascorbate was added prior to the heat treatment where indicated. D, estimation of the amounts of PsbO in each fraction shown in C. The data are the average of three independent measurements ± S.D. The colors of the bars represent the same as those in B.
FIGURE 8.

Release of the PsbO from PS II by the addition of artificially produced reactive oxygen species. A, 1O2 was chemically produced by the addition of 1 mm NaClO and 1 mm H2O2. B, HO· was generated through the Fenton reaction by the addition of 1 mm H2O2 and 1 mm FeSO4. The effects of the ROS were assayed by incubation of the PS II membranes with the ROS generating solution for 30 min both at 20 and 40 °C. The positions of PsbO and the molecular markers are shown on the right and left side of the gel, respectively.
Effects of Heat Stress on the Thylakoids and PS II Membranes That Had Been Treated by Chelex to Remove Metals—The experiments with the addition of ROS-generating reagents to the samples described above indicate participation of a free radical mechanism in the PS II damage. The Fenton reaction, which generates HO· from H2O2, depends on the presence of transition metal ions, and therefore, it is necessary to remove the contaminated free metals in the thylakoids or PS II membrane to evaluate the production of ROS inherent in PS II. After removing free metals by treatment with Chelex, the thylakoids or PS II membranes were subjected to heat treatment at 40 °C for 30 min. The Chelex treatment induced no significant change in the profile of the proteins in the thylakoids (Fig. 9A). However, the heat-induced cleavage of the D1 protein was completely eliminated after the Chelex treatment as reported previously with another chelating agent, EDTA (14), and a strong band representing the D1 aggregation appeared instead (Fig. 9B). Thus, the cleavage and aggregation of the D1 protein seem to include the two alternative pathways that the D1 protein takes after being damaged by excessive light or moderate heat (13). From these results, it was concluded that heat-induced damage to the D1 protein is present even in the absence of free metals at the stroma-exposed surface of the thylakoids.
FIGURE 9.

Effects of moderate heat stress on the thylakoids and PS II membranes that had been pretreated with Chelex. A, profile of the proteins in the Chelex-treated thylakoids shown by Coomassie Blue staining of the SDS/urea-PAGE gel. The thylakoids with (+) or without (-) Chelex treatment were incubated at either 20 or 40 °C for 30 min. The positions of molecular mass markers are shown at the left-hand side of the gel, and several PS II proteins at the right-hand side of the gel, respectively. B, effects of heat stress on the D1 protein in the thylakoids with (+) or without (-) Chelex treatment. Heat stress was given at 40 °C for 30 min. The bands of the D1 fragments and aggregates are shown at the right-hand side of the gel. C, effects of heat stress on the lipid peroxidation in the thylakoids and the PS II membranes with and without Chelex treatment. Lipid peroxidation was measured as described in the legend to Fig. 5. Where indicated, the samples were treated with Chelex once or twice (shown as blue and red bars, respectively), and then subjected to heat stress at 40 °C for 30 min. D, effects of heat stress on the PsbO protein in the PS II membranes with and without Chelex treatment. T, P, and S at the top of the gel represent the total protein, the pellet, and the supernatant of the centrifugation after incubation at 20 or 40 °C for 30 min, respectively.
Lipid peroxidation induced by the moderate heat stress was suppressed to some extent, but not significantly after pretreatment of the thylakoids and the PS II membranes with Chelex (Fig. 9C). Addition of 1 mm sodium ascorbate or 1 mm H2O2 plus 1 mm FeSO4 had no significant effect on the level of lipid peroxidation (data not shown). These results suggest that the heat-induced lipid peroxidation is initiated not at the stroma-exposed surface of the thylakoids or the stromal or luminal side of the PS II membranes but in the interior of the membranes. Interestingly, the heat-induced release of the PsbO protein from PS II was not affected by the removal of free metals with Chelex (Fig. 9D), indicating that the protein release is caused by the disintegration of the D1 protein and is independent of the HO· generated in the presence of transition metal ions at the donor side of PS II.
DISCUSSION
Moderate heat stress (40 °C for 30 min) on spinach thylakoid membranes inhibited PS II activity, thereby inducing cleavage and aggregation of the D1 protein and release of the extrinsic proteins, PsbO, -P, and -Q, from PS II (Figs. 1, 6, and 7). Interestingly, such deteriorating effects of moderate heat stress were prevented under anaerobic conditions. These results strongly suggest that molecular oxygen is necessary for the heat-induced damage to PS II. Recently, dark production of 1O2 and HO· molecules in spinach PS II membranes exposed to high temperature (47 °C) was shown using an EPR spin-trapping technique (25). In this study, we showed that 1O2 and HO· molecules were produced in spinach PS II membranes incubated at 40 °C, a slightly lower temperature condition, using the same EPR spin-trapping spectroscopy (Fig. 3). Thus, these results confirmed the dark production of 1O2 and HO· under moderate heat stress. However, the details of the heat-induced production of the ROS, in particular the site and mechanism of their production, remain to be elucidated.
It has been proposed that there are two major heat-susceptible sites in PS II as follows: the QB plastoquinone binding pocket at the acceptor side of PS II, and the water-oxidizing manganese complex at the donor side of PS II. The perturbation of the QB site by heat stress was shown previously using a kinetic analysis of chlorophyll fluorescence (28, 29). Recently, we reported the heat-induced cleavage of the D1 protein, which was prevented by the addition of a QB-binding herbicide (14). These results suggested that thermal destabilization occurs at the QB site before the proteolysis. The mechanism of the heat-induced damage to the D1 protein and that of the light-induced damage observed in the acceptor-side photoinhibition shares similarities in that it is the QB site that is damaged in both cases. During the acceptor-side photoinhibition of PS II, a reverse electron transfer in the reaction center generates 1O2 molecules, which leads to damage to the D1 protein (30–32). Involvement of ROS in the strong light-induced degradation of the D1 protein was shown in the studies on the effects of scavengers of ROS and also on the effects of H2O2 treatment of PS II in the dark (33, 34). According to our present data, 1O2 is formed even in the dark by heat treatment of the PS II membranes. One possible cause for the dark production of 1O2 is lipid peroxidation in the thylakoid membranes. An earlier study showed that when thylakoid membranes from higher plants were exposed to high temperature, a thermoluminescence band was exhibited, which had a peak around 75 °C and was dependent on molecular oxygen (35). The thermoluminescence band was related to the formation of 1O2 detected using spin-trapping EPR measurements. In that study, the heat-induced generation of 1O2 was tentatively attributed to lipid peroxidation, although no evidence was presented to support the view at the time. Thylakoid membranes of higher plant chloroplasts contain a high proportion of glycolipids with highly unsaturated fatty acids. In particular, spinach thylakoid membranes contain glycolipids having considerable amounts of a C16 polyunsaturated fatty acid, 16:3Δ7,10,13 (36). In general, lipids peroxidation is known to be proportional to the lipid unsaturation level. Raising the temperature should increase the fluidity of the thylakoid membranes and may stimulate the lipid peroxidation. Indeed, heat treatment of wheat leaves was shown to induce an increase in the peroxidation of thylakoid lipids (23). We also detected lipid peroxidation in the thylakoid and the PS II membranes (Fig. 5). The question that remains unanswered is how 1O2 molecules are produced during the lipid peroxidation. A likely mechanism is that through the reaction of two peroxyl radicals a cyclic intermediate is formed, which then decomposes to produce 1O2 (24) (Fig. 10). As described above, the QB site is one of the sites that are primarily damaged by heat stress in the dark, and the 1O2 molecules are possibly generated at the lipid phase near the QB site. In a recent x-ray crystallographic data analysis of the PS II core complex from Thermosynechococcus elongatus, 14 bound lipids were identified, and lipid molecules in the vicinity of the QB binding pocket were indicated (37, 38). It is reasonable to assume that the membrane fluidity around the QB site is relatively high, because the swift diffusion of plastoquinone molecules from and to the QB pocket is essential for the electron transport of PS II, and for that to occur, the nearby lipids should be unsaturated. Under these conditions, moderate heat stress may cause lipid peroxidation rather easily around the QB site resulting in 1O2 production. Although lipid peroxidation was more significant in the PS II membranes than in the thylakoid membranes, this is not reflected in the oxygen evolving activity of both samples under heat stress; the oxygen evolving activity of the PS II membranes was more resistant to heat stress compared with that of the thylakoids (Fig. 6A). These results suggest that the lipid peroxidation does not explain all of the damaging processes occurring to PS II under the heat stress.
FIGURE 10.

A model for the effects of moderate heat stress on PS II. At the acceptor side of PS II, heat-induced lipid peroxidation produces 1O2, which damages the DE-loop of the D1 protein and causes cleavage as well as aggregation of the D1 protein. At the donor side of PS II, heat induces destabilization of the four manganese-calcium cluster and generates H2O2. The H2O2 then reacts with transition metal ions to produce OH·, which in turn damages proteins, including D1 and D2 and the extrinsic PsbO and Q proteins. The extrinsic proteins are probably released from PS II after damage takes place in the D1 protein.
The oxygen-evolving site at the donor side of PS II is another heat-susceptible location, and release of the extrinsic PsbO, -P, and -Q proteins and also the catalytic manganese from PS II has been reported at high temperatures (13, 15–17). The cause of the release of the extrinsic proteins and manganese has been thought to be through thermal destabilization of the proteins. However, the present data, which show that manganese and the PsbO protein are not released from PS II under anaerobic conditions (Figs. 6D and 7), strongly suggest that oxidative modification of the PsbO protein itself or of the D1 protein to which the PsbO protein binds is the direct cause of the liberation of manganese and the PsbO protein. It was shown previously that the oxidatively modified PsbO produced by strong illumination of PS II appeared as a diffused band in Coomassie staining after SDS/urea-PAGE (39). SDS/urea-PAGE of the heat-liberated PsbO protein showed a slightly diffused band (Fig. 7), and the results of the Oxi-Blot suggested oxidation of the PsbO protein takes place under moderate heat stress (Fig. 2). However, as discussed later, the ROS-mediated damage to the D1 protein rather than to the PsbO protein is likely the primary cause for the release of manganese and the PsbO protein.
Destabilization of the water-oxidizing manganese complex by heat stress may give rise to an anomalous environment for the interaction of water molecules with the manganese cluster. Earlier, production of H2O2 was reported during the illumination of the donor side impaired PS II (40). It was also suggested that H2O2 is formed by heat treatment of PS II through partial oxidation of water on the donor side of PS II (41). More recently, it was shown that HO· is produced in the dark at elevated temperatures, a phenomenon that is related to the thermal disassembly of the water-splitting complex (25). In this study, we detected the signal of HO· by EPR spin-trapping using the PS II membrane incubated at 40 °C in the dark. The HO· is probably produced by the Fenton reaction from H2O2 in the presence of reduced metal cations, typically Fe2+. It is possible that free Mn2+ released from the catalytic site also participates in the reaction. As HO· is a potent oxidant, it may damage the D1 protein as well as the PsbO protein easily (Fig. 10). It should be noted that the HO· can initiate lipid peroxidation (24). The data presented in Fig. 4, however, suggest that there are other oxidants than HO· produced at the anomalous water-splitting site, which may trigger lipid peroxidation and generation of 1O2. It was previously shown that 1O2 was not eliminated by HO· scavengers indicating that HO· is unlikely involved in 1O2 production (25). There are highly oxidizing species (such as ferryl, perferryl, and ferrous-dioxygen-ferric complex) that might initiate lipid peroxidation and thus singlet oxygen generation (24) (Fig. 10). Another metal center that is different from the water-splitting manganese complex such as the heme iron of cytochrome b559 or non-heme iron might play a role in these processes.
The transition metal ions play a critical role in the generation of HO· from H2O2 via the Fenton reaction. It is possible that the thylakoid or the PS II membranes were contaminated by the metal ions from the stroma or cytosol during the sample preparation, which affected the results of the present study. To address this problem, we pretreated the thylakoids and the PS II membranes with Chelex to remove the metal ions from the surface of the membranes. Interestingly, moderate heat stress (40 °C for 30 min) on the Chelex-treated thylakoids showed no cleavage, but instead, aggregation of the D1 protein became prominent (Fig. 9B). These results suggest that in the Chelex-treated thylakoids the moderate heat stress indeed damaged the D1 protein and the damage appeared in the form of D1 aggregation. Here we suggest that when a protease, most probably the metalloprotease FtsH, is inactivated by the removal of zinc, cleavage of the damaged D1 protein is prevented. Participation of FtsH in the heat-induced cleavage of the D1 protein was suggested previously (14).
The removal of metal ions by Chelex treatment of the thylakoids and PS II membranes had no effect on the heat-induced lipid peroxidation (Fig. 9C). Neither the addition of Fe2+ nor H2O2 affected the lipid peroxidation. These results indicate that lipid peroxidation and a possible formation of 1O2 are the events occurring inside the membrane. These results also support the view that the heat damage to the D1 protein partly takes place at the acceptor side of PS II through the action of 1O2. Sodium ascorbate, which is a membrane-impermeable reducing reagent and may eliminate 1O2, reacted at the stroma-exposed surface of the thylakoids and prevented the damage to the D1 protein. In fact, no cleavage and aggregation of the D1 protein were detected by the moderate heat stress in the presence of sodium ascorbate. At the donor side of PS II, by contrast, the HO· formed from H2O2 generated in the reaction in the heat-damaged four manganese-calcium cluster may be responsible for the damage to the D1 protein. It should be noted that removal of metal ions by Chelex treatment had apparently no significant effect on the release of the PsbO protein, indicating that the damage caused by HO· to the D1 protein is responsible for the release of the PsbO protein. Previously, we have shown with photoinhibited PS II membranes that photodamage to the D1 protein induced the release of the PsbO protein from PS II, and the released PsbO protein was subsequently damaged by the HO· produced by the Fenton reaction, which was possibly mediated by transition metals bound to the PsbO protein (39).
It is known that when light stress is superimposed on heat stress, dramatic stimulation of the damage to PS II takes place (42). However, heat is more effective than light in initiating the structural changes of PS II. It was previously shown that even a very strong illumination such as 5,000 μEm-2 s-1 induced only a partial release of extrinsic PsbO, -P, and -Q proteins from spinach PS II, in parallel with degradation of the D1 protein, although PS II activity was significantly impaired (39). By contrast, heat itself has significant effects on PS II, as even when the temperature is 40 °C the extrinsic proteins and manganese are released, followed by inhibition of the PS II activity (Figs. 6 and 7). It should be emphasized that when the heat-damaged PS II is illuminated with light, even a weak light may amplify the damage because the donor-side photoinhibition becomes prominent under these conditions. Previously, we have shown with Tris-washed spinach PS II membranes that light induced strong donor-side photoinhibition (7, 43). Under natural conditions, typically in the summer time, heat and light stresses on plants are superimposed. Therefore, the effects of combined heat and light stresses on PS II must be examined to understand the molecular responses of PS II under natural environmental conditions.
Besides cleavage of the D1 protein, the aggregation of the D1 protein with the nearby polypeptides, namely the D2 protein and CP43, is also an important event caused by the moderate heat stress on spinach thylakoid membranes (Fig. 1). Interestingly, the D1 aggregation was most prominently observed in the grana fraction where the PS II complexes are enriched (13). A similar aggregation of the D1 protein was observed by excessive illumination of spinach thylakoid and the PS II membranes and was caused by either the acceptor-side or donor-side photoinhibition (7, 44). The two cases were differentiated based on the requirement of molecular oxygen for the individual processes. The D1 aggregation induced by the acceptor-side photoinhibition of PS II was triggered by the ROS, probably 1O2, and is therefore dependent on the presence of molecular oxygen. On the other hand, that induced by the donor-side photoinhibition was triggered by cationic radicals such as P680+ and was independent of oxygen. The heat-induced aggregation of the D1 protein shares some similarities with that of the acceptor-side photoinhibition process. Presumably, disorganization of the QB-binding site is caused by moderate heat stress, which may be directly linked to aggregation, as well as cleavage, of the D1 protein. At the donor side of PS II, moderate heat stress induced release of PsbO, -P, and -Q from PS II (Fig. 7) (also see Refs. 13, 15–17) and stepwise disorganization of the manganese cluster as well (45). When the PS II membranes lacking the extrinsic proteins were treated by heat in the darkness, aggregation of the D1 protein was stimulated (13). These results suggested that the lack of the extrinsic proteins promoted the damage to the D1 protein upon heat stress, leading to its aggregation. This is similar to the D1 aggregation caused by the donor-side photoinhibition.
In summary, we demonstrated that moderate heat stress induces 1O2 and HO· in PS II, which probably damages the D1 protein, and thereby induces release of the extrinsic proteins PsbO, -P, and -Q, as well as manganese, and inhibits the PS II activity of spinach thylakoids. Interestingly, heat inhibition of PS II shares similarities with the photoinhibition of PS II in several crucial aspects.
This work was supported by Grants-in-aid for Scientific Research 18570042 and 20570039 from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Y. Y.), Ministry of Education, Youth and Sports of the Czech Republic Grant MSM 6198959215, and Grant Agency of the Czech Republic Grant 522/06/0979 (to P. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PS II, photosystem II; P680, primary electron
donor of PS II; D1, reaction center-binding protein of PS II; QA,
primary quinone electron acceptor of PS II; 1O2, singlet
oxygen;
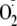 ,
superoxide anion radical; PS I, photosystem I; PsbO, -P and Q, extrinsic
proteins of PS II having apparent relative molecular masses of 33,000, 24,000,
and 18,000, respectively; ROS, reactive oxygen species; HO·, hydroxyl
radical; MES, 4-morpholineethanesulfonic acid; PVDF, poly(vinylidene
difluoride); ECL, enhanced chemiluminescence; TBA, thiobarbituric acid; MDA,
malondialdehyde; EMPO, 5-(ethoxycarbonyl)-5-methyl-1-pyrroline
N-oxide; TEMP, 2,2,6,6-tetramethylpiperidine; D2, reaction
center-binding protein of PS II forming a heterodimer with the D1 protein;
CP43 and CP47, antenna chlorophyll-binding proteins of PS II having apparent
relative molecular masses of 43,000 and 47,000, respectively; QB,
secondary quinone electron acceptor of PS II.
,
superoxide anion radical; PS I, photosystem I; PsbO, -P and Q, extrinsic
proteins of PS II having apparent relative molecular masses of 33,000, 24,000,
and 18,000, respectively; ROS, reactive oxygen species; HO·, hydroxyl
radical; MES, 4-morpholineethanesulfonic acid; PVDF, poly(vinylidene
difluoride); ECL, enhanced chemiluminescence; TBA, thiobarbituric acid; MDA,
malondialdehyde; EMPO, 5-(ethoxycarbonyl)-5-methyl-1-pyrroline
N-oxide; TEMP, 2,2,6,6-tetramethylpiperidine; D2, reaction
center-binding protein of PS II forming a heterodimer with the D1 protein;
CP43 and CP47, antenna chlorophyll-binding proteins of PS II having apparent
relative molecular masses of 43,000 and 47,000, respectively; QB,
secondary quinone electron acceptor of PS II.
References
- 1.Kyle, D. J., Ohad, I., and Arntzen, C. J. (1984) Proc. Natl. Acad. Sci. U. S. A. 81 4070-4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohad, I., Kyle, D. J., and Arntzen, C. J. (1984) J. Cell Biol. 270 14919-14927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barber, J., and Andersson, B. (1992) Trend Biochem. Sci. 17 61-66 [DOI] [PubMed] [Google Scholar]
- 4.Aro, E.-M., Virgin, I., and Andersson, B. (1993) Biochim. Biophys. Acta 1143 113-134 [DOI] [PubMed] [Google Scholar]
- 5.Andersson, B., and Aro, E.-M. (1997) Physiol. Plant. 100 780-793 [Google Scholar]
- 6.Baena-González, E., and Aro, E.-M. (2002) Philos. Trans. R. Soc. Lond. B Biol. Sci. 367 1451-1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto, Y. (2001) Plant Cell Physiol. 42 121-128 [DOI] [PubMed] [Google Scholar]
- 8.Pospíšil, P., Arató, A., Krieger-Liszkay, A., and Rutherford, A. W. (2004) Biochemistry 43 6783-6792 [DOI] [PubMed] [Google Scholar]
- 9.Asada, K. (1999) Annu. Rev. Plant Mol. Biol. 50 601-639 [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto, Y., Doi, M., Tamura, N., and Nishimura, M. (1981) FEBS Lett. 133 265-268 [Google Scholar]
- 11.Yamamoto, Y., and Akasaka, T. (1995) Biochemistry 34 9038-9045 [DOI] [PubMed] [Google Scholar]
- 12.Rokka, A., Aro, E.-M., Herrmann, R. G., Andersson, B., and Vener, A. V. (2000) Plant Physiol. 123 1525-1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komayama, K., Khatoon, M., Takenaka, D., Horie, J., Yamashita, A., Yoshioka, M., Nakayama, Y., Yoshida, M., Ohira, S., Morita, N., Velitchkova, M., Enami, I., and Yamamoto, Y. (2007) Biochim. Biophys. Acta 1767 838-846 [DOI] [PubMed] [Google Scholar]
- 14.Yoshioka, M., Uchida, S., Mori, H., Komayama, K., Ohira, S., Morita, N., Nakanishi, T., and Yamamoto, Y. (2006) J. Biol. Chem. 281 21660-21669 [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto, Y., and Nishimura, M. (1983) in The Oxygen Evolving System of Photosynthesis (Inoue, Y., Crofts, A. R., Govindjee, Murata, N., Renger, G., and Satoh, K., eds) pp. 229-238, Academic Press, Tokyo
- 16.Nash, D., Miyao, M., and Murata, N. (1985) Biochim. Biophys. Acta 807 127-133 [Google Scholar]
- 17.Enami, I., Kitamura, M., Tomo, T., Isokawa, Y., Ohta, H., and Katoh, S. (1994) Biochim. Biophys. Acta 1186 52-58 [Google Scholar]
- 18.Yamamoto, Y., Nishi, Y., Yamasaki, H., Uchida, S., and Ohira, S. (2004) Methods Mol. Biol. 274 217-227 [DOI] [PubMed] [Google Scholar]
- 19.Berthold, D. A., Babcock, G. T., and Yocum, C. F. (1981) FEBS Lett. 134 231-234 [Google Scholar]
- 20.Ford, R. C., and Evans, M. C. W. (1983) FEBS Lett. 160 159-164 [Google Scholar]
- 21.Patzlaff, J. S., and Barry, B. A. (1996) Biochemistry 35 7802-7811 [DOI] [PubMed] [Google Scholar]
- 22.Gutteridge, J. M. C., and Quinlan, G. J. (1983) J. Appl. Biochem. 5 293-299 [PubMed] [Google Scholar]
- 23.Mishra, R. K., and Singhal, G. S. (1992) Plant Physiol. 99 1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halliwell, B., and Gutteridge, J. M. (1999) Free Radicals in Biology and Medicine, 3rd Ed., Oxford University Press, New York
- 25.Pospíšil, P., Šnyrychová, I., and Nauš, J. (2007) Biochim. Biophys. Acta 1767 854-859 [DOI] [PubMed] [Google Scholar]
- 26.Sundby, C., Melis, A., Mäenpää, P., and Andersson, B. (1989) Biochim. Biophys. Acta 851 475-483 [Google Scholar]
- 27.Pospíšil, P., and Tyystäervi, E. (1999) Photosynth. Res. 62 55-66 [Google Scholar]
- 28.Cao, J., and Govindjee (1990) Biochim. Biophys. Acta 1015 180-188 [DOI] [PubMed] [Google Scholar]
- 29.Yamane, Y., Kashino, Y., Koike, H., and Satoh, K. (1998) Photosynth. Res. 57 51-59 [DOI] [PubMed] [Google Scholar]
- 30.Macpherson, A. N., Telfer, A., Barber, J., and Truscott, T. G. (1993) Biochim. Biophys. Acta 1143 301-309 [Google Scholar]
- 31.Mishra, N. P., Francke, C., van Gorkom, H. J., and Ghanotakis, D. F. (1994) Biochim. Biophys. Acta 1186 81-90 [Google Scholar]
- 32.Barber, J. (1998) Biochim. Biophys. Acta 1365 269-277 [DOI] [PubMed] [Google Scholar]
- 33.Miyao, M. (1994) Biochemistry 33 9722-9730 [DOI] [PubMed] [Google Scholar]
- 34.Miyao, M., Ikeuchi, M., Yamamoto, N., and Ono, T.-A. (1995) Biochemistry 34 10019-10026 [DOI] [PubMed] [Google Scholar]
- 35.Hideg, É., and Vass, I. (1993) Photochem. Photobiol. 58 280-283 [Google Scholar]
- 36.Buchanan, B. B., Gruissem, W., and Jones, R. L. (2000) Biochemistry & Molecular Biology of Plants, American Society of Plant Physiologists, Rockville, MD
- 37.Loll, B., Kern, J., Saenger, W., Zouni, A., and Biesiadka, J. (2005) Nature 438 1040-1044 [DOI] [PubMed] [Google Scholar]
- 38.Loll, B., Kern, J., Saenger, W., Zouni, A., and Biesiadka, J. (2007) Biochim. Biophys. Acta 1767 509-519 [DOI] [PubMed] [Google Scholar]
- 39.Henmi, T., Miyoa, M., and Yamamoto, Y. (2004) Plant Cell Physiol. 45 243-250 [DOI] [PubMed] [Google Scholar]
- 40.Wydrzynski, T., Hiller, W., and Messinger, J. (1996) Physiol. Plant. 96 342-350 [Google Scholar]
- 41.Thompson, L. K., Blaylock, R. J., Sturtevant, M., and Brudvig, G. W. (1989) Biochemistry 28 6686-6695 [DOI] [PubMed] [Google Scholar]
- 42.Ohira, S., Morita, N., Suh, H.-J., Jung, J., and Yamamoto, Y. (2005) Photosynth. Res. 84 29-33 [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto, Y., Ishikawa, Y., Nakatani, E., Yamada, M., Zhang, H., and Wydrzynski, T. (1998) Biochemistry 37 1565-1574 [DOI] [PubMed] [Google Scholar]
- 44.Ishikawa, Y., Nakatani, E., Henmi, T., Ferjani, A., Harada, Y., Tamura, N., and Yamamoto, Y. (1999) Biochim. Biophys. Acta 1413 147-158 [DOI] [PubMed] [Google Scholar]
- 45.Pospíšil, P., Haumann, M., Dittmer, J., Sole, V. A., and Dau, H. (2003) Biophys. J. 84 1370-1386 [DOI] [PMC free article] [PubMed] [Google Scholar]