

Abstract
Fibroblast growth factors (FGFs) effect cellular responses by binding to FGF receptors (FGFRs). FGF bound to extracellular domains on the FGFR in the presence of heparin activates the cytoplasmic receptor tyrosine kinase through autophosphorylation. We have crystallized a complex between human FGF1 and a two-domain extracellular fragment of human FGFR2. The crystal structure, determined by multiwavelength anomalous diffraction analysis of the selenomethionyl protein, is a dimeric assemblage of 1:1 ligand:receptor complexes. FGF is bound at the junction between the two domains of one FGFR, and two such units are associated through receptor:receptor and secondary ligand:receptor interfaces. Sulfate ion positions appear to mark the course of heparin binding between FGF molecules through a basic region on receptor D2 domains. This dimeric assemblage provides a structural mechanism for FGF signal transduction.
Fibroblast growth factors (FGFs) stimulate a variety of cellular functions by binding to cell surface FGF receptors (FGFRs) in the presence of heparin proteoglycans. FGFRs are single-chain receptor tyrosine kinases that become activated through autophosphorylation that is thought to be induced through a mechanism of ligand-mediated receptor oligomerization (1). Receptor activation gives rise to a signal transduction cascade that leads to gene activation and diverse biological responses (2, 3). Both FGFs and FGFRs are expressed in defined spatial and temporal patterns, and they are involved in differentiation of both epithelial and neuronal cells. FGFs are potent mitogens for many cell types. Aberrant signaling through FGFR can lead to tumorigenesis and skeletal disorders (4).
The FGF system has appreciable diversity in both ligands and receptors. The FGF family contains at least 15 distinct factors, highly conserved across mammalian species but divergent (30–70% sequence identity) among paralogs (2). The FGFR family includes four identified genes and numerous subtypes of alternatively spliced isoforms, particularly within the well characterized FGFR1 and FGFR2 types. Differential responses follow from this diversity.
FGFRs have an extracellular portion imbued with the ligand-binding potential, a transmembrane segment, and a tyrosine kinase domain in the cytoplasm (2). The extracellular portion comprises three Ig-like domains, D1, D2, and D3, with an acidic stretch of approximately 30 residues, the acid box, between D1 and D2. Isoforms generated by alternate splicing events include receptors that lack Ig-like domain D1 or both D1 and the acid box, as well as variants having two alternative sequences for the C-terminal half of the third Ig-like domain. The FGF binding site has been mapped to domains D2, D3, and the interdomain linker.
FGFs are secreted factors originally identified based on their mitogenicity toward fibroblasts. They are small proteins for which several FGF crystal structures have been determined; all have 12 β strands in a β-trefoil fold (2). Mutational analyses have mapped the sites of interaction with FGFRs (5). Sites of interaction between FGF and heparin have been identified in structures of complexes between heparin fragments (6–8).
We have produced a complex between human FGF1 (acidic FGF) and a fragment of human FGFR2 that comprises Ig-like domains D2 and D3. Both components were expressed as recombinant selenomethionyl proteins in Escherichia coli, and the crystal structure of the complex was determined at 2.4 Å resolution by the method of multiwavelength anomalous diffraction (MAD). The ligand-binding domains of the receptor are disposed in a unique manner with FGF bound between D2 and D3. Although the ligand and receptor are combined as a 1:1 complex in solution, two such complexes are associated as a symmetric dimer in the crystal lattice. Features of this crystallographic dimer appear to be relevant to signaling.
Materials and Methods
Expression and Purification of the FGFR2 Construct.
We cloned the D2 and D3 domains of human FGFR2 from a cDNA corresponding to a natural splice variant (9) into the pET22b secretion vector (Novagen) using PCR primers that contain the NcoI and BamHI restriction sites, respectively (5′-GGCAGAGCCCATGGATCGGCCCTCCTTCAGTTTA-3′) and (5′-GCATAGGGGGATCCTTACTCCAGGTAGTCTGGG-3′). This vector (FGFR2-D2D3) adds two N-terminal residues as a cloning artifact after signal cleavage and includes a short piece that precedes domain D1 joined directly to Ig-like domains D2 and D3 (IIIc variant), terminating at the transmembrane boundary. In sum, this gives Met-Asp followed by FGFR2 residues 22–36, a R152G replacement at the splice junction and residues 153–377. The vector was expressed in E. coli BL21 (DE3) under control of the T7 promoter. N-terminal sequencing showed that signal processing had occurred; insoluble protein accumulated, nevertheless, presumably in the periplasm. Protein was solubilized in 6 M guanidinium HCl; diluted into 0.5 M arginine, 250 mM NaCl, 0.1 mM glutathione, 0.01 mM oxidized glutathione, 1 mM EDTA, 50 mM Tris (pH 8.0), and 0.05% PEG4K at room temperature; and further diluted and/or dialyzed to reduce guanidinium and arginine to negligible concentrations. Selenomethionyl protein (93%) was produced by a nonauxotrophic protocol (10).
Expression and Purification of FGF1 and the Complex.
We cloned human FGF1 by means of PCR amplification into the pET3a vector using primers containing the NdeI and BglII restriction sites, respectively (5′-GGCCATATGAATTACAAGAAGCCCAAACTCCTCTAC-3′) and (5′-GCGAGATCTCTAATCAGAAGAGACTGGCAGGGGGAG-3′). This vector was expressed in E. coli JM109 (DE3) under control of the T7 promoter, both for natural and selenomethionyl proteins (10). Soluble, full-length (1–140) FGF1 was clarified by ultracentrifugation after cell lysis and was loaded onto a Hi-Trap heparin column (Amersham Pharmacia). After washing with buffer, this FGF1-heparin column was loaded with refolded FGFR2-D2D3. Incorrectly folded FGFR and inclusion body contaminants were washed away, and the complex was eluted at 1.5 M NaCl. We exchanged the buffer for 20 mM Tris (pH 8.0), 150 mM NaCl, and 1 mM EDTA and separated aggregated FGFR and monomeric FGF from the complex by Superdex 75 gel filtration. The complex eluted at a volume corresponding to 50 kDa.
Crystallography.
Crystals were grown by hanging-drop vapor diffusion against 1.6 M (NH4)2SO4 in 10 mM Tris (pH 7.5), starting from 1 μl of 10 mg/ml complex and 1 μl of 2:1 diluted reservoir buffer. Microseeding and macroseeding were used to achieve diffraction quality. Typical crystals had dimensions 0.35 × 0.35 × 0.15 mm. They are in space group I422 with a = b = 129.97 Å and c = 129.06 Å and contain one 1:1 complex per asymmetric unit with 61% solvent content. Crystals were frozen in a cryoprotectant composed by adding 9% sucrose (wt/vol), 2% glucose (wt/vol), 8% glycerol (vol/vol), and 8% ethylene glycol (vol/vol) to the reservoir buffer. MAD data were measured at four wavelengths near the Se K edge by using a Q4R charge-coupled device detector system at beamline X4A of the National Synchrotron Light Source. hkl (11) was used for data processing, madsys (12) was used for phase evaluation from five Se sites, and dm (13) was used for density modification. The model was built in o (14) and refined with refmac (13) against all data from spacings between 6 and 2.4 Å.
Structure Comparisons.
Receptor domains were compared against existing structural data with dali (15). Structure-based alignments were refined in toss (16) by using a superposition criterion to include segments having at least three contiguous Cα positions within 2.5 Å. For analysis of tandemly repeated Ig-like domains, each domain was transformed by superposition of a core set of Cα atoms in β strands B, C, E, F, and G onto the equivalents in telokin (17) placed in a canonical reference frame. This frame is defined by Cα positions relative to the disulfide bridge between residue n on βB and m on βF such that the origin is at the midpoint of n − 3 and m − 3, z is directed through the midpoint of n + 2 and m + 2, y = z × x′, where x′ is from m(F) to n(B), and x = y × z.
Results
Structure Determination.
Although our D2D3 fragment of FGFR2 was ill-behaved in isolation, it formed a stable complex when captured by heparin-associated FGF1. This is a 1:1 complex at ≈20 μM as judged by gel-filtration chromatography. Analysis of MAD data from a crystal of the selenomethionyl proteins (Table 1) gave a readily interpreted map. There is one FGF1:FGFR2 complex in the asymmetric unit. The atomic model refined at 2.4 Å resolution (Table 1) includes residues 5–140 from FGF1, residues 147(32)–296 and 307–362 from FGFR2, 319 water molecules, and 9 sulfate ions. The intimate association in this FGF1:FGFR2 pair makes identification with the complex in solution plausible; however, a diad axis of the crystal relates two such complexes through both receptor:receptor contacts and second-site receptor:ligand contacts. This crystallographic dimer appears to be relevant to the signaling dimer that naturally requires heparin as well as FGF for activation.
Table 1.
Crystallographic analysis
| Diffraction data statistics, 20 < d < 2.4 Å | |||||
|---|---|---|---|---|---|
| Wavelength | Reflections
|
Completeness, % | <I/σ(I)> | Rsym, % | |
| Total | Unique | ||||
| λ1 (0.9919 Å) | 180,297 | 41,549 | 99.8 | 11.8 | 6.4 |
| λ2 (0.9793 Å) | 175,453 | 41,568 | 99.8 | 11.4 | 6.7 |
| λ3 (0.9791 Å) | 174,872 | 41,632 | 99.8 | 11.5 | 6.7 |
| λ4 (0.9641 Å) | 183,114 | 41,582 | 100.0 | 11.0 | 7.0 |
| MAD diffraction difference ratios | |||||
| 20<d<3.4 Å | 3.4<d<2.4 Å | |||||||
|---|---|---|---|---|---|---|---|---|
| λ1 | λ2 | λ3 | λ4 | λ1 | λ2 | λ3 | λ4 | |
| λ1 | 0.039 (0.038) | 0.044 | 0.038 | 0.031 | 0.101 (0.092) | 0.098 | 0.096 | 0.095 |
| λ2 | 0.054 (0.039) | 0.033 | 0.048 | 0.107 (0.098) | 0.091 | 0.100 | ||
| λ3 | 0.064 (0.040) | 0.041 | 0.113 (0.092) | 0.098 | ||||
| λ4 | 0.054 (0.040) | 0.110 (0.088) | ||||||
| MADSYS phasing statistics, 20<d<2.4 Å | ||||||||
| R(|°FT|)=0.049 | <Δ(Δφ)>=50.94° | |||||||
| R(|°FA|)=0.475 | <σ(Δφ)>=18.03° | |||||||
| Refinement statistics, 6<d<2.4 Å | ||||||||
| Atoms | 3,081 | |||||||
| R/Rfree (all), % | 22.1/31.5 | |||||||
| Reflections (all) | 19,354/1,040 | |||||||
| R/Rfree(|F|>2σ(|F|)), % | 19.9/26.8 | |||||||
| Reflections (|F|>2σ(|F|)) | 18,908/1,010 | |||||||
| rms bond ideality, Å | 0.009 | |||||||
| rms B, bonds/angles, Å2 | 1.32/2.26 | |||||||
Receptor Structure in the Complex.
The extracellular portion of the FGFR in our complex has an extended disposition of its Ig-like domains (Fig. 1). Domains D2 (residues 152–249) and D3 (residues 254–360) are separated by a four-residue linker peptide extended such that there are no contacts between the domains. The overall structure appears to be dictated by contacts of the two domains with the FGF ligand, in absence of which a flexible linkage would be expected. Only the first two residues are ordered in the 17-residue C-terminal extension from D3 to the transmembrane segment of FGFR2. This suggests a flexible tethering to the membrane, which can be viewed as at the bottom of Fig. 1. Most of the 17-residue N-terminal extension before D2 in the construct is also disordered. Five ordered residues extend away from D2D3 in a conformation that may arise from crystal lattice contacts.
Figure 1.
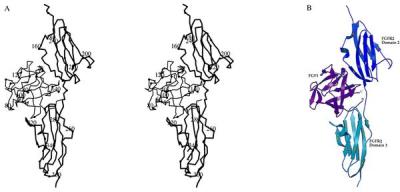
Structure of the 1:1 complex between FGF1 and FGFR2. The orientation has D3 in our canonical reference frame with z vertical and x rotated 20° from directly out of the page. FGFR2 is bold. (A) Stereodiagram of Cα backbones (molscript) (35). (B) Ribbon drawing (raster3d) (36).
Domains D2 and D3 are folded as shown in Fig. 1B. Precise boundaries of the elements of secondary structure are given in Fig. 2. Both domains adopt Ig-like folds as expected (18), despite little sequence similarity beyond the conventional disulfide bond; however, aspects of the topology are not as predicted from telokin (19). In particular, D2 has a helical turn between βA and βA′ and D3 has βC′ in place of strand D and a short helix (αE) before βE. This produces a cleft between the C′E and BC loops, where many interactions with the FGF ligand occur. The CC′ loop of D3 also flares out away from the body of the domain and is apparently flexible as residues 297–306 are disordered. The pattern of fractional surface accessibility in FGFR2 is shown in Fig. 2.
Figure 2.

Structure-based sequence alignment of FGFR2 with FGFR1, IL-1 receptor, and VCAM (human sequences, GenBank). Secondary structure assignments for FGFR2 are above the sequences, and fractional solvent accessibilities for isolated FGFR2 are shown below (○, f > 0.4; ●, f < 0.1, ◒, 0.1 < f < 0.4). FGFR2 residues are marked for contacts with the other receptor (r), FGF1 in the primary (p) and secondary (s) interfaces, and specified sulfate ions (1, 2, 3).
Systematic database searches find structural similarities to numerous Ig-like domains, and the best dali scores for both D2 and D3 are with telokin. After refined alignments, telokin superimposes remarkably well with an rms deviation of 1.01 Å for 89 equivalent Cα atoms in D2 and an rms deviation of 1.06Å for 78 matches onto D3. Vascular cell adhesion molecule (VCAM) (20) scores the highest among structures with tandemly repeated Ig-like domains, and the IL-1 receptor (21) scores best among growth factor receptors. Refined structure-based alignments of the two FGFR2 domains with VCAM and the IL-1 receptor are shown in Fig. 2. D2 is more similar to D1 of VCAM (69 matches, 1.24 Å rms deviation) than it is to D2 of the IL-1 receptor (59 matches, 1.32 Å rms deviation), whereas D3 is more similar to D3 of the IL-1 receptor (67 matches, 1.26 Å rms deviation) than to D2 of VCAM (56 matches, 1.43 Å rms deviation). This is somewhat surprising because similarities in topology with D3 are the opposite.
Ligand Structure in the Complex.
The structure of FGF1 in the complex has the expected β-trefoil fold (22), and in detail the cores from β1 to β12 (residues 11–137) of free FGF1 (22), the heparin complex (8), and the receptor complex are indistinguishable except at loops β4/β5 and β8/β9, where there are receptor contacts, and at loop β11/β12 because of heparin contacts. Otherwise, deviations (0.41 Å rms) are within variations seen before (8). The ends extending from the core do differ, however, both in the extent of ordering and in conformation. The N terminus is ordered starting at residue 5, and the C terminus is fully ordered. Both of these extensions make contacts with the receptor.
Dimeric Assemblage.
The receptor and ligand are associated in the crystal as a symmetric 2:2 complex, as shown in Fig. 3A, although the complex has a 1:1 composition in solution. The diad axis of symmetry is roughly parallel with the long axis of the receptor, bringing the receptors into apposition at one face of domain D2. There are no ligand:ligand contacts, but each receptor molecule makes contacts with both ligand molecules. Thus, a dissection of this crystallographic dimer into the constituent 1:1 complexes is not formally possible. The dimeric assemblage does have two distinct sides, however, and it seems likely that the complex in solution corresponds to the receptor:ligand pair seen in Fig. 1 and at each side in Fig. 3A. We describe this as the ipsilateral protomer. Contacts of the receptor with the ipsilateral ligand are both much more extensive and better disposed for stable association than those with the contralateral ligand.
Figure 3.
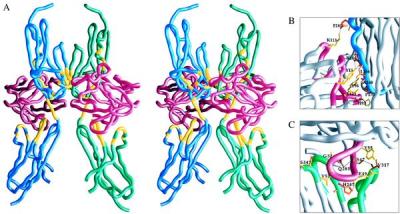
Dimeric assemblage. (A) Stereoview of a worm diagram oriented with the diad axis vertical and viewed with the two FGF1:FGFR2 complexes at either side. FGF1 chains are in red, the ipsilateral receptor chain (as rotated ≈−40° from Fig. 1) is in blue, and the contralateral chain is in green. Receptor segments in ligand contacts are in yellow. (B) Close-up of a primary D2 contact viewed at 70° from A. (C) Close-up of a D3 contact viewed at 40° from A. Drawings were made by grasp (37).
A total of 2,388 Å2 in solvent-accessible surface area is buried into the primary, ipsilateral interface as compared with a total of only 565 Å2 in the secondary, contralateral receptor:ligand interface. Moreover, the ipsilateral protomer is compact with extensive contacts from both D2 and D3 to the ligand, whereas the contralateral protomer has only D2 in contact with the ligand, making it tenuously extended as an isolated complex. It is plausible to view dimerization as a rigid-body docking of two preformed ipsilateral complexes. Receptor molecules of the dimer are in contact at the AB loops and between βB and βE of D2. This receptor:receptor interface buries 378 Å2 in surface area. The entire buried area in the interface between two ipsilateral protomers, including the two secondary ligand contacts, is 1,508 Å2. Receptor residues in intercomponent contacts in the dimer are marked on Fig. 2.
Ligand:Receptor Interactions.
The primary ligand:receptor interaction, at the ipsilateral interface, has FGF1 embraced by the receptor at the juncture of domains D2 and D3, which is as expected from mutational analyses. Contacts with D3 predominate, but there are substantial interactions with D2 and the interdomain linker segment as well. The receptor surface buried into this interface is apportioned into 350 Å2 from D2 (13 residues), 164 Å2 from the linker (residues 250–253), and 659 Å2 from D3 (23 residues). A substantial fraction of the ligand surface is involved in receptor binding, with portions of 39 of the 136 FGF1 residues buried into the primary interface and another 9 into the secondary site at the contralateral interface. These residues are in the loops connecting or extending from all 12 strands of the β-trefoil or in the strands themselves. The heparin binding site on FGF1 (8) remains free however.
The primary FGF binding site on D2 mainly involves the A′ strand and the C-terminal portion of the G strand. This is at a side on the body of D2, in contrast to the binding site on D3, where interactions are at loops. A portion of the interaction is shown in Fig. 3B. Much of it has a hydrophobic character, but there are several hydrogen bonds as well. FGF1 residues at this interface are from β1, the β1/β2 turn, the β3/β4 loop, β9, the β10/β11 loop, β12, and the C terminus. The linker segment of the receptor interacts with FGF1 residues at the N terminus, β8, β9, and β12. Ligand residues that interact with D2 and the linker are conserved in character among most members of the FGF family, suggesting that little discrimination in FGF signaling comes from this interface.
Interactions of FGF1 with D3 are at the N-terminal end of the domain. The β4/β5 hairpin (residues 46–57) and N-terminal extension of FGF1 (residues 5–9) are insinuated into the large cleft that is created between the BC and C′E loops as helix αE flares away from the domain body (Fig. 3C). Loop FG on the receptor and ligand residues from β6, the β7/β8 loop, and β8 of FGF1 are also involved. The binding cleft includes many water molecules, several direct hydrogen bonds, and a few hydrophobic interactions. Conserved ligand residues are engaged here as with D2; notably, Glu87, which was sensitive to mutation in FGF2 (Glu96) (23), makes a central hydrogen bond to the BC backbone. Many other interactions involve nonconserved residues, making it likely that this interface contributes to binding specificity. For example, the respective changes of Asn7 and Glu49 to glycine and valine in FGF7 would be expected to disrupt binding, as is observed. How it happens that the IIIb splicing alternative confers affinity for FGF7 while retaining FGF1 binding is unclear. Because the IIIc to IIIb change deletes two residues at the αE/βE junction, the cleft structure in the variant could be quite different.
The secondary ligand:receptor interaction mainly involves contacts of receptor residues from the C′D and EF loops of D2 with ligand residues from the β8/β9 and β11/β12 loops of FGF1. This interface is near other contacts in the dimeric assemblage. This site on FGF1 for the contralateral receptor is contiguous with its β9 bound to the ipsilateral receptor, and this site on FGFR2 is juxtaposed to a receptor:receptor contact at Ser220, where, moreover, a sulfate ion is coordinated to both receptor and ligand.
Heparin Binding Site.
We know the binding site for heparin on FGF1 from the complex with a heparin decasaccharide (8). A basic-rich segment from loop β10/β11 through loop β11/β12 (residues 112–128) encompasses most of the site. FGF1 molecules in the dimeric assemblage have these heparin binding sites exposed on the upper surface of the ligand as viewed in Figs. 1 and 3A. They are orientated such that the two associated heparin axes of elongation are within 10° of being co-linear and normal to the molecular diad. The FGF1 heparin sites are separated by the intervening pair of D2 domains, between which there is a channel lined with the basic side chains of Lys176, Arg178, Lys208, Arg210, and His213 of each receptor chain. The electrostatic potential along this surface is markedly positive (Fig. 4A).
Figure 4.
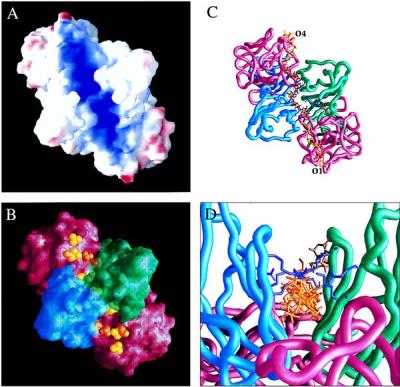
Heparin binding site. A–C have the dimeric assemblage oriented as viewed from above Fig. 3A; B–D use the same color codes as in Fig. 3. (A) Electrostatic potential surface. Potential is graded from red (−) to deep blue (>+16 kT). (B) Sulfate sites superimposed on the molecular surface. Sulfate ions in the surface channel of the complex are shown in yellow and sulfonate ions transformed from the FGF1:heparin structure (8) are in orange. (C) Heparin model (yellow) superimposed onto worm diagram. Six hexoses at each end are transformed directly from Ref. 8, and four in the middle are model built. Side chains of basic residues 176, 178, 208, and 210 are in purple. (D) End view of heparin superimposed onto worm. The view is rotated by −20° from Fig. 3A. Drawings were made by grasp (37).
Our crystals grown in 1.6 M ammonium sulfate incorporate 17 ordered sulfate ions per dimer. Nine of these are along the path of positive potential (Fig. 4B), where there are also six sulfonate groups from the decasaccharide complex. Two sulfate ions are bound to each FGF1 in the dimer, one coincides with sulfonate site 3 from the heparin complex (8) and the other is farther out from the diad axis. Three unique sulfate ions are bound to FGFR2. One (site 2) is on the diad axis coordinated symmetrically by the receptor, a second (site 1) is coordinated asymmetrically in two copies between D2 domains, and the third (site 3) is at each contralateral ligand:receptor interface. Site 3 is at level approximately coplanar with the FGF1 surface ions, but sites 1 and 2 are at a higher level out along the diad axis.
We have produced a rough model of the ternary ligand:heparin:receptor complex by combining results from the structures of the heparin and receptor complexes with FGF1. The crystallized heparin-linked dimer of FGF1 has a single six-sugar heparin fragment sandwiched between two quasisymmetric FGF molecules that bind heparin in opposite directions. Here, we have superimposed one FGF1 from a ligand:heparin complex onto an FGF1 molecule in the ligand:receptor complex and the other onto its symmetry mate in the dimeric assemblage, in each case carrying along the same heparin fragment. The two transformed heparin fragments are consequently oriented in the same polarity, which necessarily breaks the symmetry of our dimeric assemblage. Next, we aligned an NMR model heparin (24) onto the experimentally defined six-residue pieces and found that a four-sugar fragment could be oriented and fused in place by appropriate glycosidic linkages with little adjustment. The modeled segment continues the pattern of alternating l-iduronic and d-glucosamino sugars and has sulfonate groups positioned near the crystallographic sulfate sites. The completed model (Fig. 4C) has the 16-sugar heparin chain threaded through the binding channel beneath a canopy of lysine and arginine side chains (Fig. 4D), which coordinate sulfate ions in sites 1 and 2.
Discussion
Comparison with Other Ig-Like Receptors.
Structures are now known for several growth factors or hormones in complexes with ligand-binding portions of their receptors. As here, others also bind the ligand at a junction between two Ig-like domains. Structures of Ig-domain receptor:ligand complexes include representatives from the helical-cytokine, cystine-knot, and β-trefoil ligand families. They reveal various modes of binding. The cytokine receptors for growth hormone (25), erythropoietin (26), and granulocyte colony-stimulating factor (27) all bind their ligands at the outside of a sharply bent elbow between domains, the former two in 2:1 complexes and the latter in a distinctive 2:2 complex. Receptors for the cystine-knot proteins vascular epithelial growth factor (28) and nerve growth factor (29) each form complexes with just one of two implicated domains, binding distinctively. The receptor for IL-1, another β-trefoil protein, has been studied in complexes with both IL-1 (21) and the IL-1 receptor antagonist (30).
We have compared the mode of ligand binding by FGFR2 with that in other receptor:ligand complexes and also the relative disposition of the two Ig-like domains of FGFR2 with that in those receptors and in other tandem Ig-domain structures. The results are summarized in Fig. 5. The two domains of FGFR are disposed unlike others except N-cadherin (Fig. 5 A and B); and while FGF1 relative to D3 (Fig. 5C) is at the junctional loops as in several other complexes, it is uniquely placed at the A′G sandwich edge relative to D2 (Fig. 5D). As suggested (31), the FGF:receptor complex is most like the IL-1 receptor complexes. This relationship is largely superficial, however, because all interactions are arranged differently.
Figure 5.

Disposition of ligands and receptor domains in a canonical frame. Comparisons are with Protein Data Bank datasets from complexes with GH (1hwg, ref. 25), EPO (1cn4, ref. 26), G-CSF (27), IL-1 (1itb, ref. 21), IL-1RA (1ira, ref. 30), VEGF (1flt, ref. 28), and NGF (1www, ref. 29) and from repeated domains in VCAM (1vca, ref. 20), CD2 (1hng), CD4 (1cdj), FN (1fnf), and NCAD (1ncj). Each Ig domain is represented by the z-axial vector in its canonical frame with an origin dot. Each ligand is represented by the centroid of Cα positions. (A) N-terminal domains with their C-terminal domains superimposed onto canonical telokin (x, y). (B) Tandem domains as in A, including ligands (z, r). Radial positions R are +/− as x is +/−. (C) Ligands with C-terminal domains on canonical telokin (x, y). (D) Ligands with N-terminal domains on canonical telokin (x, y). Ligand z coordinates are in parentheses in C and D. Core β-strands are shown as circles at the level of the disulfide bridge + 2 in C and −3 in D.
Signaling Implications.
The FGF1:FGFR2 complex offers an elegant structural solution to the problem of transmembrane signal transduction, but it also presents some puzzles. The dimeric assemblage seen here is a plausible model for the signaling complex when heparin is bound as suggested by the course of sulfate ion sites through the electropositive channel. The C termini of FGFR2 D3 are separated by 48 Å, which is similar to other presumed signaling complexes such as growth hormone (29 Å) or CD4 dimers (28 Å). Thereby, the FGFR tyrosine kinase domains across the membrane will be brought into juxtaposition for autophosphorylation in trans. We suggest that FGF first binds to cell-surface receptors in 1:1 complexes, which then associate into signaling dimers in the presence of heparin proteoglycans.
The 1:1 FGF1:FGFR2 complex that we find in dilute solution (≈20 μM) is compatible with the Kd of 41 nM observed for the analogous FGF2:FGFR1 complex (32), to which a second FGFR1 molecule could be recruited (Kd = 2 μM) in the presence of heparin. We presume that dimeric 2:2 assemblages form here without heparin because of the high concentrations in the crystal (12 mM), and perhaps even in the crystallization medium (220 μM). Conflicts of our observations with reports of 2:1 receptor:ligand stoichiometries remain puzzling (32, 33). There are puzzles in rationalizing this work with previous results on heparin binding as well. The coincident sufficiency of eight-residue heparins both for FGF1 dimerization and for cell activation through FGFR2 seemed to suggest relevance for a heparin-linked FGF1 dimer (8), but a structural conjunction of that dimer with ipsilateral 1:1 complexes provides an implausible basis for signaling. On the other hand, the extended heparin binding site seen here (up to 16 residues) would seem to suggest greater potency for longer sugars, whereas 5-kDa and 3-kDa heparins (≈10 hexoses) were found to be equally effective (32).
Aberrant signaling because of mutations in extracellular FGFR residues is associated with severe human skeletal and craniosynostosis disorders (4). Most such defects in FGFR2 appear to cause constitutive receptor activation through inappropriate disulfide bond formation (34), often through cysteine exposure from a destabilized domain D3. A few occur at the primary ligand interface, notably S252W and P253R, which uniquely cause Apert's syndrome. These may lead to pathologically tight ligand binding.
After we had completed the structure analysis of this FGF1:FGFR2 complex, the structure of an FGF2:FGFR1 complex was reported (38). We have not made a detailed comparison because the atomic coordinates are not yet available, but the two structures are clearly similar despite differences between isoforms and lattices. This strengthens the suggested structural basis for FGF signaling.
Acknowledgments
We thank Rhett Kovall, Yee Liu, and Larry Shapiro for advice and discussions; Irit Lax and Joseph Schlessinger for cDNA clones and early discussion; Yee Liu, Peter Kwong, John Williams, and Craig Ogata for help at the Synchrotron; and present and former Hendrickson Lab members for general support and advice. This work was supported in part by National Institutes of Health Grant GM34102 and by Training Grant EY07105 (to D.J.S.). Beamline X4A at the National Synchrotron Light Source, a Department of Energy facility, is supported by the Howard Hughes Medical Institute.
Abbreviations
- FGF
fibroblast growth factor
- FGFR
FGF receptor
- MAD
multiwavelength anomalous diffraction
- VCAM
vascular cell adhesion molecule
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1DJS).
References
- 1.Ullrich A, Schlessinger J. Cell. 1990;61:203–211. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 2.Szebenyi G, Fallon J F. Int Rev Cytol. 1999;185:45–106. doi: 10.1016/s0074-7696(08)60149-7. [DOI] [PubMed] [Google Scholar]
- 3.Johnson D E, Williams L T. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. [DOI] [PubMed] [Google Scholar]
- 4.Naski M C, Ornitz D M. Front Biosci. 1998;3:D781–D794. doi: 10.2741/a321. [DOI] [PubMed] [Google Scholar]
- 5.Springer B, Pantoliano M, Barbera F, Gunyuzlu P, Thompson L, Herblin W, Rosenfeld S, Book G. J Biol Chem. 1994;269:26879–26884. [PubMed] [Google Scholar]
- 6.Ornitz D, Herr A, Nilsson M, Westman J, Svahn C, Waksman G. Science. 1995;268:432–436. doi: 10.1126/science.7536345. [DOI] [PubMed] [Google Scholar]
- 7.Faham S, Hileman R, Fromm J, Linhardt R, Rees D. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- 8.DiGabriele A D, Lax I, Chen D I, Svahn C M, Jaye M, Schlessinger J, Hendrickson W A. Nature (London) 1998;393:812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- 9.Yan G, McBride G, McKeehan W L. Biochem Biophys Res Commun. 1993;194:512–518. doi: 10.1006/bbrc.1993.1849. [DOI] [PubMed] [Google Scholar]
- 10.Doublié S. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 11.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 12.Hendrickson W A. Science. 1991;254:51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 13.Collaborative Computational Project, No 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 14.Jones T A, Kjeldgaard M O. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 15.Holm L, Sander C. Science. 1996;273:595–602. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 16.Hendrickson W A. Acta Crystallogr A. 1979;35:158–163. [Google Scholar]
- 17.Holden H M, Ito M, Hartshorne D J, Rayment I. J Mol Biol. 1992;227:840–851. doi: 10.1016/0022-2836(92)90226-a. [DOI] [PubMed] [Google Scholar]
- 18.Dionne C A, Crumley G, Bellot F, Kaplow J M, Searfoss G, Ruta M, Burgess W H, Jaye M, Schlessinger J. EMBO J. 1990;9:2685–2692. doi: 10.1002/j.1460-2075.1990.tb07454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bateman A, Chothia C. Nat Struct Biol. 1995;2:1068–1074. doi: 10.1038/nsb1295-1068. [DOI] [PubMed] [Google Scholar]
- 20.Jones E Y, Harlos K, Bottomley M J, Robinson R C, Driscoll P C, Edwards R M, Clements J M, Dudgeon T J, Stuart D I. Nature (London) 1995;373:539–544. doi: 10.1038/373539a0. [DOI] [PubMed] [Google Scholar]
- 21.Vigers G P A, Anderson L J, Caffes P, Brandhuber B J. Nature (London) 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- 22.Zhu X, Komiya H, Chirino A, Faham S, Fox G M, Arakawa T, Hsu B T, Rees D C. Science. 1991;251:90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Ramnarayan K, Anchin J, Miao W, Sereno A, Millman L, Zheng J, Balaji V, Wolff M. J Biol Chem. 1995;270:21869–21874. doi: 10.1074/jbc.270.37.21869. [DOI] [PubMed] [Google Scholar]
- 24.Mulloy B, Forster M, Jones C, Davies D. Biochem J. 1993;293:849–859. doi: 10.1042/bj2930849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Vos A M, Ultsch M, Kossiakoff A A. Science. 1992;255:306–312. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 26.Syed R S, Reid S W, Li C, Cheetham J C, Aoki K H, Liu B, Zhan H, Osslund T D, Chirino A J, Zhang J, et al. Nature (London) 1998;395:511–516. doi: 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- 27.Aritomi M, Kunishima N, Okamoto T, Kuroki R, Ota U, Morikawa K. Nature (London) 1999;401:713–717. doi: 10.1038/44394. [DOI] [PubMed] [Google Scholar]
- 28.Wiesmann C, Fuh G, Christinger H W, Eigenbrot C, Wells J A, de Vos A M. Cell. 1997;91:695–704. doi: 10.1016/s0092-8674(00)80456-0. [DOI] [PubMed] [Google Scholar]
- 29.Wiesmann C, Ultsch M H, Bass S H, de Vos A M. Nature (London) 1999;401:184–188. doi: 10.1038/43705. [DOI] [PubMed] [Google Scholar]
- 30.Schreuder H, Tardif C, Trump-Kallmeyer S, Soffientini A, Sarubbi E, Akeson A, Bowlin T, Yanofsky S, Barett R W. Nature (London) 1997;386:194–199. doi: 10.1038/386194a0. [DOI] [PubMed] [Google Scholar]
- 31.Huhtaka M T, Pentikainen O T, Johnson M S. Structure (London) 1999;7:699–709. doi: 10.1016/s0969-2126(99)80091-4. [DOI] [PubMed] [Google Scholar]
- 32.Pantoliano M W, Horlick R A, Springer B A, Dyk D E V, Tobery T, Wetmore D R, Lear J D, Nahapetian A T, Bradley J D, Sisk W P. Biochemistry. 1994;33:10229–10248. doi: 10.1021/bi00200a003. [DOI] [PubMed] [Google Scholar]
- 33.Hsu Y-R, Nybo R, Sullivan J K, Costigan V, Spahr C S, Wong C, Jones M, Pentzer A G, Crouse J A, Pacifici R E, et al. Biochemistry. 1999;38:2523–2534. doi: 10.1021/bi9821317. [DOI] [PubMed] [Google Scholar]
- 34.Robertson S C, Meyer A N, Hart K C, Galvin B D, Webster M K, Donoghue D J. Proc Natl Acad Sci USA. 1998;95:4567–4572. doi: 10.1073/pnas.95.8.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 36.Meritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 37.Nicholls A, Sharp K, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 38.Plotnikov A N, Schlessinger J, Hubbard S R, Mohammadi M. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]