

Abstract
Pathogenic bacteria experience nitrosative stress from NO generated in the host and from nitrosating species such as S-nitrosoglutathione. The food-borne pathogen Campylobacter jejuni responds by activating gene expression from a small regulon under the control of the NO-sensitive regulator, NssR. Here, we describe the full extent of the S-nitrosoglutathione response using transcriptomic and proteomic analysis of batch- and chemostat-cultured C. jejuni. In addition to the NssR regulon, which includes two hemoglobins (Cgb and Ctb), we identify more than 90 other up-regulated genes, notably those encoding heat shock proteins and proteins involved in oxidative stress tolerance and iron metabolism/transport. Up-regulation of a subset of these genes, including cgb, is also elicited by NO-releasing compounds. Mutation of the iron-responsive regulator Fur results in insensitivity of growth to NO, suggesting that derepression of iron-regulated genes and augmentation of iron acquisition is a physiological response to nitrosative damage. We describe the effect of oxygen availability on nitrosative stress tolerance; cells cultured at higher rates of oxygen diffusion have elevated levels of hemoglobins, are more resistant to inhibition by NO of both growth and respiration, and consume NO more rapidly. The oxygen response is mediated by NssR. Thus, in addition to NO detoxification catalyzed by the hemoglobins Cgb and possibly Ctb, C. jejuni mounts an extensive stress response. We suggest that inhibition of respiration by NO may increase availability of oxygen for Cgb synthesis and function.
Campylobacter species are the leading cause of gastroenteritis in the developed world (1). During the course of transmission from avian to human hosts (“food-to-fork”) and in the gut, the bacterium encounters nitric oxide (NO) and other agents of nitrosative stress. NO is a powerful weapon in the armory of mammalian cells (2) to combat microbial infections. The chief sources of NO are from host NO synthases, especially inducible NO synthase, bacterial reduction of dietary nitrate to nitrite and hence to NO, and the reactions of salivary nitrite with stomach acid to generate NO (3). Acidified nitrite kills Campylobacter jejuni, and NO synthase 2 expression is increased in macrophages on exposure to the bacterium (4). Both diarrheal and systemic C. jejuni strains are efficiently killed by γ-interferon-γ-lipopolysaccharide-stimulated macrophages in an NO synthase-dependent manner (4).
NO generated in the host readily diffuses into microbial cells and reacts
with diverse targets, particularly iron-sulfur clusters
(5), hemes
(6), and thiols
(7). The mechanism of toxicity
is complicated by the ability of NO to be oxidized to the nitrosonium cation
(NO+) or to generate nitrite
(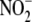 ) or peroxynitrite
(ONOO-) (8). In
nitrosation, the NO+ group is transferred from a nitrosating agent
to a nucleophilic acceptor such as an amine or thiol. Important nitrosating
agents are S-nitrosothiols, which can be generated intracellularly or
provided externally as in the case of S-nitrosoglutathione
(GSNO).3 However, NO
is not only a promiscuous toxin but also specifically targets certain cysteine
thiols, modifications of which modulate protein function.
) or peroxynitrite
(ONOO-) (8). In
nitrosation, the NO+ group is transferred from a nitrosating agent
to a nucleophilic acceptor such as an amine or thiol. Important nitrosating
agents are S-nitrosothiols, which can be generated intracellularly or
provided externally as in the case of S-nitrosoglutathione
(GSNO).3 However, NO
is not only a promiscuous toxin but also specifically targets certain cysteine
thiols, modifications of which modulate protein function.
The best-studied mechanisms for bacterial NO detoxification
(8,
9) are in enteric bacteria. The
genome-wide responses of Escherichia coli to acidified nitrite
(10), GSNO
(11) and NO
(12–14)
are dominated by enhanced transcription of hmp
(15) and norVW
(16,
17) encoding the prominent
NO-detoxifying proteins. Flavohemoglobin Hmp acts aerobically as a nitric
oxide denitrosylase (dioxygenase) converting NO to the relatively innocuous
nitrate ion ( )
(8), and its efficacy in
resisting nitrosative stress has been demonstrated by assays of viability
(18), cell respiration
(19), and macrophage killing
(20). Hmp synthesis is
regulated in response to nitrosative stresses aerobically and anaerobically by
Fnr (5), MetR
(21), NsrR
(22), and (weakly) Fur
(23). Importantly, oxygen
availability does not regulate hmp gene expression. In
Salmonella, NsrR-dependent flavohemoglobin expression
(24,
25) is a critical determinant
of NO detoxification and bacterial viability in macrophages
(26).
)
(8), and its efficacy in
resisting nitrosative stress has been demonstrated by assays of viability
(18), cell respiration
(19), and macrophage killing
(20). Hmp synthesis is
regulated in response to nitrosative stresses aerobically and anaerobically by
Fnr (5), MetR
(21), NsrR
(22), and (weakly) Fur
(23). Importantly, oxygen
availability does not regulate hmp gene expression. In
Salmonella, NsrR-dependent flavohemoglobin expression
(24,
25) is a critical determinant
of NO detoxification and bacterial viability in macrophages
(26).
Intriguingly, the genome sequence of C. jejuni contains neither flavohemoglobin nor flavorubredoxin genes (27). However, C. jejuni gene Cj1586 (cgb) encodes a single-domain hemoglobin, the mutation of which leads to hypersensitivity to GSNO and NO (28). Expression of Cgb is strongly and specifically induced after exposure to nitrosative stress. Cells expressing GSNO-induced levels of Cgb are resistant to NO (28), and Cgb is thought to catalyze a dioxygenase or denitrosylase reaction (like Hmp) that converts NO and oxygen to nitrate (29). Expression of Cgb in response to NO is mediated via NssR (Cj0466), a member of the Crp-Fnr superfamily, which acts as a positive regulator of a small regulon that includes both cgb and ctb (Cj0465c), the latter encoding the C. jejuni truncated hemoglobin (30). A ctb mutant is not compromised in its tolerance of nitrosative stress-generating agents (31), but Ctb has been implicated in oxygen delivery (31, 32) or in catalyzing a peroxidase- or P-450-type of oxygen chemistry (33). Because an nssR mutant shows increased hypersensitivity to nitrosative stress when compared with a cgb mutant, there appears to be some role for members of the regulon other than cgb in the nitrosative stress response (30).
The purpose of this work was to define the entire set of responses in C. jejuni to nitrosative stress elicited by GSNO, a nitrosating agent with wide-ranging effects on the transcriptome in enteric bacteria (11, 34). We show that, in addition to the previously recognized NssR regulon, C. jejuni responds by up-regulation of genes involved in iron metabolism, oxidative stress, and the heat-shock system. We also test the hypothesis that oxygen availability in this microaerophile compromises the efficacy of oxygen-dependent NO detoxification reactions, particularly that catalyzed by Cgb, and show that cgb expression is regulated by oxygen provision with important implications for resisting nitrosative stress in vivo.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth—All strains were derived from the sequenced C. jejuni NCTC 11168 (27). Mutants defective in the hemoglobins Cgb and Ctb were strains RKP1336 (also known as CJCGB01, cgb::Kanr (28)) and RKP1386 (ctb::Kanr (31)), respectively. The ahpC mutant (CJA01, 11168 ahpC::Cmr (28)) was kindly provided by Dr. Simon Park (University of Surrey). Mutants defective in the chu-encoded hemin uptake system (11168 chuA::Cmr and chuB::Cmr; JDR5 and JDR6, respectively) (35) and in fur (11168 fur::Kanr, AV45) (36) were kindly donated by Professor Julian Ketley (University of Leicester).
Bacteria were grown at 42 °C in Mueller-Hinton (MH) broth (Oxoid), on MH agar, or Columbia blood agar (Oxoid) supplemented with vancomycin (10 μg ml-1), kanamycin (50 μg ml-1), or chloramphenicol (20 μg ml-1) as necessary. Microaerophilic conditions (10% O2, 10% CO2, 80% N2) were generated using a MACS-VA500 microaerophilic work station (Don Whitley Scientific Ltd). Liquid cultures (50 ml) were grown in MH broth in 100-ml conical flasks in the microaerophilic work station overnight before being used as a 3% (v/v) inoculum for 150 ml of MH broth in 250-ml baffled flasks. Cultures were shaken on a Mini-Orbital Shaker SO5 (Stuart Scientific, Stone, UK) at ∼125 rpm at 42 °C, and growth was measured using A600 nm. GSNO was synthesized (37) and added to agar or liquid cultures (absorbance 0.25–0.3). The NO-releasing compounds 3-[2-hydroxy-1-(1-methylethyl)-2-nitrosohydrazino]-1-propanamine (NOC-5) and 3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-propanamine (NOC-7) have half-lives of NO release of 93 and 10 min, respectively, at pH 7.4, 22 °C (Calbiochem data). Using literature values for the rate constants of NO release at 37 °C, we also calculated half-lives at 42 °C (bacterial growth temperature) of 10.5 (NOC-5) and 3.0 (NOC-7) min. Stock solutions (0.1 m) of each were made up in 0.1 m NaOH.
Wild-type C. jejuni was grown in 50 ml of MH broth with vancomycin in a 100-ml conical flask overnight. A 10-ml sample of this culture was used to inoculate two parallel custom-built chemostats similar to those described before (38) but modified for microaerophilic growth (39). In brief, the stirred vessels have a working volume of 125 ml, with an inflow rate of 12.5 ml medium h-1; the dilution rate (which at steady state is equal to the specific growth rate) was 0.1 h-1. A gas mix of 10% O2, 10% CO2, and 80% N2 was passed into the head space of the culture vessel at a rate of 0.25 liters min-1 to obtain a microaerobic atmosphere; gas transfer was assured by the stable vortex formed on magnetic stirring (40). Silicone water jackets maintained the temperature at 42 °C. All growth experiments were repeated two or more times on separate days; typical data are presented with intra-experiment variation.
Transcriptomic Analysis of Wild-type C. jejuni and the Response to GSNO—Chemostat cultures were used to minimize perturbations in growth, particularly growth rate, that are a secondary consequence of GSNO addition (11). Cells were grown in two parallel chemostats to reach a steady state, verified by collecting >5 culture volumes, before GSNO (final concentration 0.25 mm) was added to one culture. After a further 10 min, the 125-ml volume was separated into 3 aliquots, and each was mixed immediately on ice with 3.6 ml of 100% ethanol and 185 μl of phenol to stabilize RNA. The cells were subsequently harvested by centrifugation, and total RNA was purified by using a Qiagen RNeasy mini kit. Alternatively, to delineate the time course of GSNO-elicited changes, cells were grown in batch culture to an optical density of ∼0.3. GSNO (0.25 mm) was then added, and the cultures were incubated in the microaerophilic work station for up to 45 min. Cells were subsequently harvested, and RNA was isolated as described above. Two independent RNA preparations (biological replicates) of each sample were labeled and hybridized to glass-slide microarrays, designed and fabricated at the Institute of Food Research. Equal quantities of RNA from control and GSNO-treated cultures were labeled by using nucleotide homologues of dCTP containing either Cy3 or Cy5 fluorescent dye (PerkinElmer Life Sciences). For each microarray slide, one sample was labeled with Cy3-dCTP, whereas another sample was labeled with Cy5-dCTP. Methods for reverse transcription and hybridization, scanning, and data analysis were as before (41). A control experiment (comparison of two mRNA samples from replicated independent cultures) was used to estimate the boundaries between genes that were equally and differentially expressed in the two samples. For the analyses described here, this boundary would detect changes equivalent to about 3·3-fold greater intensity in one of the fluorescence channels. Applying this boundary to the control dataset gave an error rate for misclassification of ∼0·43% of the gene features that give a fluorescence signal above background. Genes were annotated using on-line databases at the Wellcome Trust Sanger Institute and CampyDG. All microarray raw data and protocols are deposited at Gene Expression Omnibus (NCBI) (accession number GSE7048).
Real-time PCR—cDNA synthesis from RNA was carried out as before using an ABI 7700 thermocycler (38). The primers used are listed in supplemental Table S1. All assays were done on duplicate samples from triplicate cultures. Coefficient of variation (normalized S.D.) was calculated as described in the ABI User Bulletin as square root of (control gene S.D./fold mean)2 + (test gene S.D./fold mean)2.
Proteome Analysis by Two-dimensional Gel Electrophoresis—During steady state growth, GSNO (0.25 mm) was added to one chemostat vessel and to the medium reservoir to give a constant concentration of GSNO for 30 min. Cells harvested by centrifugation (3000 × g for 10 min) from both chemostats were washed with 10 mm Tris-buffered saline, pH 7·5, and lysed by four 1-min periods of beating with glass beads (≤106-μm diameter) (Sigma) in a buffer containing 50 mm Tris, pH 7.5, 0.3% SDS, 0.2 m dithiothreitol, 3.3 mm MgCl2, 16.7 μg of RNase ml-1, and 1.67 units of DNase ml-1. Extracts were kept on ice for 20 min before centrifuging at 18,500 × g for 20 min; supernatants were analyzed by two-dimensional proteomic analysis (41). Protein concentrations were determined using the 2D Quant Kit (GE Healthcare).
Determination of Respiration Rates and NO Consumption—Samples from chemostats or batch cultures (mid-exponential growth) were removed before GSNO addition (final concentration 0.25 mm), and the cultures were incubated in the microaerophilic work station with shaking (125 rpm) for a further 5, 10, 15, or 45 min or maintained in the chemostat for 10 min. Chloramphenicol (100 μm) was added to the cultures for 5 min to halt protein synthesis before centrifugation (5500 × g, 4 °C, 10 min). The pellet was resuspended in buffer (2 ml of 50 mm Tris, pH 7). Respiration rates were measured in a digital Clark-type O2 electrode system (Rank Brothers, Bottisham, UK) with a working volume of 1.5 ml. The electrode was calibrated with air-saturated 50 mm Tris buffer (assuming an oxygen concentration at air saturation of 200 μm O2), and anoxia was achieved with the addition of a few grains of sodium dithionite. Sodium formate (5 mm) was added as a reductant, and the oxygen concentration was allowed to decrease by ∼50%. An anoxic NO-saturated solution, prepared as in (15), was then added using a Hamilton microsyringe to give a final concentration of 10 μm. The initial respiratory rate, the inhibited rate, and the rate when oxygen uptake was spontaneously reinitiated were calculated. The period of inhibition of respiration was calculated as the period between the addition of the NO solution (when respiration becomes inhibited) and the point where oxygen uptake was reinitiated. All respiratory data are the means of 5 measurements of cells from a single culture. Similar data were obtained in duplicate cultures.
For respiratory analyses of cells grown under different aeration conditions, overnight cultures grown in MH broth (50 ml in 100 ml flasks) were used as a 2% (v/v) inoculum for either 100-ml or 200-ml volumes in 250-ml baffled flasks, thus altering the oxygen diffusion rate, K, from gas to liquid (31, 40). The cultures were shaken (125 rpm) in the microaerophilic work station at 42 °C for 9 h. Samples were removed either immediately or after a further 20-min incubation with 0.25 mm GSNO, then treated with 100 μm chloramphenicol and adjusted to equal cell densities. NO was measured using an NO electrode (World Precision Instruments) inserted through the lid of the chamber (42) and calibrated as described by the manufacturer.
Detection of C. jejuni Globins by Immunoblotting—Cultures were exposed to 0.25 mm GSNO for either 10 min or 1 h or left untreated, then harvested by centrifugation. Cells were resuspended in 0.4 ml of 50 mm Tris buffer, pH 7.5, then disrupted by ultrasonication using an MSE/Sanyo Soniprep 150 sonicator delivering 4 × 20 s bursts at an amplitude of 10 μm. Suspensions were clarified by centrifugation at 8000 × g for 30 min before freezing at -70 °C. For Western blotting, crude extracts were resolved by SDS-PAGE using Novex® 10–20% Tricine Ready Gels (Invitrogen). The Bio-Rad protein assay was used with bovine serum albumin standards to ensure that 20 μg of protein was loaded per lane. Proteins were transferred onto a nitrocellulose membrane in a mini-transblot apparatus (Bio-Rad) at 11 mA overnight. Immobilized proteins were probed with primary anti-Ctb (31) or anti-Cgb (28) antibody followed by a secondary antibody (horseradish peroxidase-conjugated anti-rabbit IgG, Sigma) and developed using ECL detection reagents (Amersham Biosciences). Blots were quantified using Imagemaster Pro 6.0 software (Invitrogen).
RESULTS
Pretreatment with GSNO Protects Respiration against Inhibition by NO—C. jejuni exhibits a response to nitrosative stress that is inducible by GSNO and protects growth and respiration from inhibition (28). To determine the speed of this response and identify time points for transcriptomic analyses in both batch and continuous culture, wild-type cells were exposed to 0.25 mm GSNO for 5, 10, 15, and 45 min; chloramphenicol was then added to prevent further protein synthesis, and samples were harvested for assays of respiration. This concentration is lower than the cytostatic concentration (0.5 mm) in Salmonella typhimurium (43) where even 4 mm GSNO is not cytocidal in vitro. The level of GSNO used here will also produce only submicromolar concentrations of NO (34). We then tested the consequences of adaptation by measuring inhibition of respiration by NO. For the wild-type strain grown in continuous culture (dilution rate, D = 0.1 h-1), NO instantly inhibited respiration, which did not recover before depletion of oxygen (Fig. 1A). In contrast, cells treated with 0.25 mm GSNO for 10 min recovered the initial respiratory rate after a transient period of inhibition (Fig. 1B). The period during which respiration was inhibited decreased as the length of GSNO pre-exposure increased (data not shown). For batch-grown wild-type cells, the inhibited period (mean of 10 determinations with S.D. in parentheses) was 4.2 (0.41) min after 5 min of pretreatment with GSNO, decreasing to 4.1 (0.19), 3.7 (0.25), and 2.5 (0.29) min when pretreated with GSNO for 10, 15, and 45 min, respectively. Fig. 1C shows that the cgb mutant grown in continuous culture failed to recover from the inhibition exerted by NO, even after 10 min of pre-exposure to 0.25 mm GSNO (Fig. 1D). We conclude that C. jejuni cells in both batch and continuous cultures elicit an adaptive response to GSNO that is evident after 5 min of exposure but increases up to 45 min.
FIGURE 1.

GSNO pre-exposure elicits adaptive protection of respiration in chemostat-cultured C. jejuni. Respiration was measured in a Clark-type O2 electrode; 10 μm NO was added as indicated by the vertical arrows. Values on the traces represent mean oxygen consumption rates (nmol of O2 min-1 mg of protein-1) in three separate experiments (five replicates in each). A and B, wild-type cells before (A) and after (B) exposure to 0.25 mm GSNO for 10 min. C and D, cgb mutant cells before (C) and after (D) exposure to 0.25 mm GSNO for 10 min.
Dynamics of Transcriptome Changes on Exposure to GSNO—Although Cgb is pivotal to the C. jejuni response to nitrosative stress (28, 30), the global cellular responses are unknown. We established the dynamics of stress response by extracting total RNA from batch-grown wild-type cultures after exposure to 0.25 mm GSNO for 5, 15, and 45 min, consistent with the respiratory assays in Fig. 1. Fluorescently labeled cDNAs were used to probe microarrays. A rapid response was observed with a large number of genes up-regulated within 5 min of GSNO treatment. The hemoglobin gene cgb and Cj0761 (encoding an unknown gene product) both belong to the NssR regulon (30) and increased in transcription, being virtually undetectable in the control samples and highly up-regulated within 5 min of GSNO addition; transcription was elevated for at least 45 min after GSNO treatment (not shown). Fig. 2 shows a selection of other genes that demonstrated different patterns over time: trxA (Cj0147c), ahpC (Cj0334), Cj0414 and Cj0415 (encoding a putative oxidoreductase), and p19 (Cj1659) each showed a coordinate increase in transcription with increasing exposure time. However, trxB (Cj0146c), Cj0176c (encoding a putative lipoprotein), ctb (Cj0465c), and chuA (Cj1614) either retained approximately the same transcriptional increase between 5 and 45 min exposure or decreased transcription with increasing time. The transcription of Cj0437 (sdhA) decreased in the presence of GSNO.
FIGURE 2.

Time-dependent transcriptional changes on exposure to GSNO. Raw microarray data were background-corrected and normalized for pin-specific effects. Data are plotted as Cy3/Cy5 ratios against GSNO exposure time for each of 10 genes. Four stable genes identified from previous microarray studies (data not shown) were used to assess the array quality. Normalized Cy3/Cy5 ratio for these control genes across all arrays used were: Cj1027c (gyrA), 1.073, S.D. 0.246 (n = 32); Cj1290c (aac), 1.095, S.D. 0.222 (n = 24); Cj1437c, 1.129, S.D. 0.180 (n = 24); Cj1494c (carA), 1.033, S.D. 0.277 (n = 24).
The Transcriptome of Continuously Cultured C. jejuni after Exposure to GSNO Reveals NssR-regulated Genes—For detailed analysis of the transcriptome of cells grown for 10 min with 0.25 mm GSNO, we exploited continuous culture (11) to eliminate differences between the cultures that are only indirect effects of GSNO treatment. Of the 1632 genes arrayed, 97 displayed statistically significant ≥2-fold increases in mRNA levels (p < 0.05). Table 1 lists those genes that were up-regulated >2.5-fold when exposed to GSNO, with the functions of their products and levels of regulation. A full data set (>2.0-fold) is presented in supplemental Table S2. Only 7 genes (see supplemental Table S3) were down-regulated, all <2.6-fold.
TABLE 1.
Genes with increased transcript levels (≥2.5-fold) in wild-type C. jejuni NCTC 11168 after steady state growth and exposure to 0.25 mm GSNO for 10 min.
| Gene number | Alternative gene name, protein, and function | p value (<0.05) | -Fold increase |
|---|---|---|---|
| 1. Gene implicated in nitrosative stress tolerance | |||
| Cj1586a | cgb, bacterial hemoglobin, detoxification of NO, and reactive nitrogen species | 9.16 × 10–6 | 320.0 |
| 2. Stress responses other than reactive nitrogen species | |||
| Cj0758 | grpE, heat shock protein, acts with DnaJ to stimulate ATPase activity of DnaK | 8.27 × 10–9 | 10.30 |
| Cj0757 | hrcA, putative heat shock regulator, negative regulator of class I heat shock genes (grpE-dnaK-dnaJ and groELS operons) | 6.40 × 10–6 | 8.38 |
| Cj0759 | dnaK, heat shock protein, chaperone of the HSP70 family | 1.56 × 10–7 | 6.12 |
| Cj0146c | trxB, thioredoxin reductase, role in oxidative stress tolerance with thioredoxin and possibly AhpC | 1.42 × 10–7 | 4.15 |
| Cj0779 | Probable thiol peroxidase, has antioxidant activity due to its ability to remove peroxides or H2O2 | 1.27 × 10–3 | 3.90 |
| Cj0334 | ahpC, alkyl hydroperoxide reductase subunit C involved in oxidative stress | 3.33 × 10–7 | 3.80 |
| Cj0311 | Ctc protein homologue, involved in general stress tolerance | 3.35 × 10–5 | 2.90 |
| Cj0147c | trxA, thioredoxin, involved in peroxide stress tolerance with trxB | 9.36 × 10–6 | 2.46 |
| 3. Binding and transport, particularly of iron | |||
| Cj1659 | p19, probably involved in iron uptake | 2.62 × 10–3 | 7.94 |
| Cj0753c | tonB, required for hemin uptake, utilization of hemin, ferrichrome, and enterochelin as iron sources | 4.93 × 10–5 | 5.82 |
| Cj1614 | chuA, hemin uptake system outer membrane receptor | 4.12 × 10–2 | 3.79 |
| Cj0755 | cfrA, putative iron uptake protein (ferric receptor) | 9.08 × 10–3 | 3.70 |
| Cj0175c | Putative iron uptake ABC-type transport periplasmic iron-binding protein | 9.61 × 10–3 | 3.41 |
| Cj1352 | ceuB, enterochelin uptake permease | 2.86 × 10–2 | 3.06 |
| Cj1658 | Putative integral membrane protein | 5.68 × 10–3 | 2.86 |
| Cj0174c | Putative iron uptake ABC-type transport permease, part of binding protein-dependent transport system | 1.43 × 10–4 | 2.85 |
| Cj1528 | Probable transmembrane transport protein pseudogene | 1.03 × 10–3 | 2.72 |
| Cj1353 | ceuC, enterochelin uptake permease | 5.83 × 10–3 | 2.57 |
| 4. Respiration, metabolism, and biosynthesis | |||
| Cj0465ca | ctb, truncated globin, possible role in facilitated diffusion of oxygen to terminal oxidase(s) | 8.97 × 10–7 | 63.80 |
| Cj0414 | Putative oxidoreductase subunit, similarity to Erwinia cypripedii gluconate dehydrogenase subunit III precursor, function unknown | 1.14 × 10–7 | 6.26 |
| Cj0415 | Putative oxidoreductase subunit, similarity to Erwinia cypripedii gluconate dehydrogenase subunit III precursor, function unknown | 1.04 × 10–4 | 5.70 |
| Cj0240c | Putative aminotransferase (NifS protein homolog), possible role in biotin synthesis | 5.65 × 10–8 | 4.48 |
| Cj0239c | nifU, NifU protein homolog, possible role in biotin synthesis | 8.09 × 10–7 | 3.81 |
| Cj0470 | tuf, probable elongation factor | 8.74 × 10–4 | 3.53 |
| Cj1382c | fldA, flavodoxin, electron acceptor of the pyruvate-oxidoreductase complex that catalyzes pyruvate oxidative decarboxylation | 6.01 × 10–4 | 3.43 |
| Cj0915 | Putative hydrolase | 1.75 × 10–3 | 2.99 |
| Cj0701 | Putative protease | 1.53 × 10–2 | 2.98 |
| Cj0835c | acnB, aconitate hydratase | 2.69 × 10–3 | 2.69 |
| Cj1534c | pgi, probable glucose-6-phosphate isomerase involved in glycolysis and gluconeogenesis | 6.73 × 10–3 | 2.62 |
| Cj0473 | nusG, transcription anti-termination protein, acts as component of transcription complex, and interacts with termination factor rho and RNA polymerase | 1.04 × 10–4 | 2.59 |
| 5. Membrane and periplasmic components | |||
| Cj0830a | Putative integral membrane protein | 5.40 × 10–8 | 12.30 |
| Cj1660 | Putative integral membrane protein | 9.40 × 10–5 | 11.60 |
| Cj0772c | Putative periplasmic protein | 3.39 × 10–7 | 4.61 |
| Cj0176c | Putative lipoprotein | 9.04 × 10–4 | 4.40 |
| Cj0770c | Putative periplasmic protein | 3.59 × 10–6 | 3.84 |
| Cj1159c | Small hydrophobic protein | 4.61 × 10–3 | 3.57 |
| Cj0771c | Putative periplasmic protein | 3.06 × 10–6 | 3.05 |
| Cj1513c | Possible periplasmic protein | 5.93 × 10–4 | 2.82 |
| 6. Motility, chemotaxis, and signaling | |||
| Cj0262c | Putative methyl-accepting chemotaxis signal transduction protein | 1.67 × 10–6 | 2.62 |
| Cj1179c | fliR, flagellar biosynthesis protein | 5.05 × 10–4 | 2.55 |
| 7. Protein synthesis | |||
| Cj0094 | rplU, 50 S ribosomal protein L21, binds to 23 S ribosomal RNA in the presence of protein L20 | 5.93 × 10–4 | 3.48 |
| Cj0477 | rplL, 50S ribosomal protein L7/L12; possible binding site for several factors involved in protein synthesis and appears to be essential for accurate translation | 7.50 × 10–3 | 2.89 |
| Cj0475 | rplA, 50 S ribosomal protein L1, binds directly to 23 S rRNA | 6.13 × 10–4 | 2.70 |
| Cj1707c | rplC, 50 S ribosomal protein L3, a primary rRNA-binding protein that nucleates assembly of the 50 S subunit | 2.67 × 10–3 | 2.66 |
| Cj0474 | rplK, 50 S ribosomal protein L11, binds directly to 23 S ribosomal RNA | 1.87 × 10–5 | 2.57 |
| Cj0476 | rplJ, 50S ribosomal protein L10 | 3.31 × 10–3 | 2.56 |
| Cj1706c | rplD, 50 S ribosomal protein L4, a primary rRNA binding protein, important during early stages of 50 S assembly | 2.00 × 10–3 | 2.56 |
| Cj1702c | rplV, 50 S ribosomal protein, binds specifically to 23 S rRNA on stimulation by other ribosomal proteins | 1.77 × 10–3 | 2.55 |
| 8. Hypothetical gene products | |||
| Cj0761 | Function unknown | 1.72 × 10–8 | 49.70 |
| Cj1384c | Hypothetical | 5.43 × 10–3 | 4.05 |
| Cj1383c | Function unknown | 1.73 × 10–3 | 3.81 |
| Cj0849c | Hypothetical | 3.94 × 10–8 | 3.53 |
| Cj0724 | Hypothetical | 9.49 × 10–4 | 4.18 |
| Cj1656c | Hypothetical | 8.82 × 10–4 | 3.48 |
| Cj1514c | Hypothetical | 4.27 × 10–6 | 2.99 |
| Cj1172c | Hypothetical | 2.34 × 10–4 | 2.80 |
| Cj0794 | Hypothetical | 4.42 × 10–2 | 2.68 |
Signifies regulation by NssR
The most highly up-regulated gene (320-fold) was cgb. Other genes under the regulation of the NO-responsive regulator NssR (30) and observed to be up-regulated were ctb (63.8-fold) encoding the truncated globin (31), Cj0830 encoding a membrane protein (12.3-fold), and Cj0761 encoding a protein of unknown function (49.7-fold). Interestingly, Cj0466 encoding the NssR regulator itself was also up-regulated (2.2-fold, supplemental Table S2).
We considered the possibility that the addition to cultures of GSNO elicits responses to GSNO-derived species rather than to this nitrosating agent per se. In redox homeostasis, the product of GSH oxidation is the disulfide GSSG, which can also arise from the reaction of HNO (nitroxyl) with GSH (44). However, in bacteria, extracellular GSNO is thought to exert intracellular nitrosation effects by import of S-nitrosocysteinylglycine, not GSNO (43). Periplasmic γ-glutamyltranspeptidase generates the S-nitrosodipeptide, which is inwardly transported by a dipeptide permease (34, 43). Thus, GSSG is not a product of GSNO-mediated nitrosation in these experiments, and experiments with the disulfide were not performed.
Transcriptome Analysis Suggests Roles for Diverse Stress Response Genes—Bacterial exposure to stress may lead to induction of heat-shock proteins that are either chaperones or ATP-dependent proteases (45), but such changes are not observed in E. coli chemostat cultures treated with either GSNO (11) or NO (13). Nevertheless, in the present study the three most highly induced stress-related genes, apart from members of the NssR operon, were Cj0757, Cj0758, and Cj0759, which constitute an operon encoding homologues of HrcA, GrpE, and DnaK, respectively (Table 1), all implicated in the heat-shock response (27). A number of other stress-related genes were identified including Cj0311 that encodes a Ctc protein homologue involved in general stress tolerance (46, 47).
Further stress-related genes included several involved in oxidative stress tolerance. Cj0147c encodes a thioredoxin (TrxA), which, with its cognate reductase TrxB (encoded by Cj0146c, also up-regulated) is involved in antioxidant defense. Also up-regulated was Cj0334, encoding alkylhydroperoxide reductase subunit C, a member of the peroxiredoxin family. Genes with diverse roles in metabolism, biosynthesis, motility, and protein synthesis, but whose roles in resisting nitrosative stress are not evident, were also up-regulated. These include genes (Cj0414, Cj0415) thought to encode subunits of gluconate dehydrogenase. Nine genes encoding membrane proteins and six encoding periplasmic proteins were also identified, including Cj0830 (12.3-fold up-regulated), which is under the control of the NO responsive regulator NssR (30) and predicted to encode a membrane protein.
Nitrosative Stress Elicits Changes in Iron-responsive Genes—A prominent feature of the list of up-regulated genes (Table 1) is the abundance of genes encoding proteins involved in iron transport. Indeed, 18 up-regulated genes (9 of which are in Table 1) were previously reported (41) to undergo significant transcriptional changes under low iron conditions. Furthermore, katA, ahpC, and the genes encoding the thioredoxin and thioredoxin reductase (see below) are all iron-regulated by the two C. jejuni Fur homologues, Fur and PerR (41, 48). In C. jejuni, perR adopts the role of oxyR of E. coli, having a role in peroxide stress (48, 49). Most C. jejuni strains do not secrete siderophores but acquire iron from foreign siderophores (enterochelin, ferrichrome), hemin, and hemoglobin as well as free ferric and ferrous ions (50). The most highly up-regulated iron-related gene was Cj1659, encoding periplasmic protein p19 (51), which with Cj1658 (Table 1) might constitute an iron uptake system. The tonB gene (Cj0753c) (Table 1) encodes the periplasm-bridging protein that transduces the protonmotive force into the energy required to drive transport and is required for utilization of ferrichrome, hemin, or enterochelin as iron sources (52). The C. jejuni genome encodes two other TonB homologs, but these were not observed in this study. Divergently transcribed from Cj0753c is the cfrA gene, encoding an outer membrane TonB-dependent siderophore receptor. Also up-regulated (Table 1) were chuA (Cj1614), required for growth on hemin as an iron source, Cj0175c (encoding a component of a ferric ion transporter designated Cfbp (50)), Cj1352 (encoding a component of an ABC transporter probably involved in enterochelin uptake), Cj0174c (encoding a putative iron-uptake ABC transport system permease protein that is part of a binding-protein-dependent transport system), and Cj1658, probably encoding a ferric iron receptor (51). Other iron-related up-regulated genes included Cj1398 (2.3-fold; supplemental Table S2) encoding a FeoB homolog with an uncertain role in ferrous ion transport (53).
Quantitative Real-time PCR Analyses of the Response to GSNO, Its Products and NO-releasing Compounds—To substantiate results from the microarrays from GSNO-treated cultures, several up-regulated genes were examined by quantitative real-time PCR to determine mRNA levels relative to gyrA mRNA. The data for GSNO in Table 2 support the microarray analysis (Table 1), but quantitative variation is expected due to the differences between the two methodologies. The large discrepancy in the case of Cj1586 (cgb; 38-fold by PCR, 320-fold in the microarrays) is presumably a consequence of the extremely low levels of gene expression (and, hence, reliability of the data) in the absence of GSNO.
TABLE 2.
Gene up-regulation in response to GSNO, its potential products, and NO-releasing compounds, and the effects of a cgb mutation cgb transcripts were not measured in the cgb mutant, ND, not done.
|
Gene number
|
Gene name,
functiona
|
-Fold increase in transcript level
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
GSNOb
|
NOC-5 and
-7c
|
GSHb
|
GSSGb
|
||||||
| Wild type | cgb | Wild type | cgb | Wild type | Wild type | ||||
| ±normalized S.D. | ±normalized S.D. | ±normalized S.D. | ±normalized S.D. | ||||||
| Cj0146c | trxB | 3.7 (±0.04) | 2.1 (±0.18) | 4.6 (±0.08) | 2.6 (±0.37) | ND | ND | ||
| Cj0240c | NifS homolog | 4.0 (±0.69) | 2.6 (±0.34) | 2.6 (±0.53) | 2.7 (±0.15) | ND | ND | ||
| Cj0414 | Dehydrogenase subunit | 3.9 (±0.46) | 4.2 (±0.28) | 4.6 (±1.34) | 2.2 (±0.16) | ND | ND | ||
| Cj1586 | cgb | 38 (±0.70) | ND | 47 (±0.4) | ND | 8.1 (±0.36) | 1.4d | ||
| Cj1659 | p19 | 4.9 (±0.78) | 7.5 (±0.25) | 5.6 (±1.74) | 9.1 (±0.26) | 2.1 (±0.3) | 2.0d | ||
Fuller details of genes are given in Table 1
GSNO, GSH, and GSSG exposure was to 0.25 mm (final concentrations) for 10 min
NOC exposure was to 10 μm (final concentration of each of NOC-5 and NOC-7) for 10 min
Means of two determinations
GSNO, when added to cultures or in vivo, may form other products or generate NO in submicromolar concentrations (34), so the changes in gene regulation may reflect either intracellular nitrosation events (which could arise from either GSNO or NO), reaction of intracellular sensors with NO per se, or a response to the products of GSNO degradation. First, we assessed the stability of GSNO at 0.25 mm in medium over 10 min, as used in the transcriptomics. GSNO solutions have an absorbance maximum at 336 nm (ε = 0.77 mm-1 cm-1); in growth medium (diluted 1 in 2 to minimize UV interference) the absorbance (measured in a dual-wavelength spectrophotometer, reference wavelength of 400 nm) was stable over 25 min, indicating no measurable degradation to GSH or GSSG under these conditions. Second, we repeated the quantitative real-time PCR determinations with GSH and GSSG (both 0.25 mm) over 10 min. Expression levels of cgb (as an example of the NssR regulon) and of p19 (Cj1659, a representative of the iron-related genes) by both compounds are shown in Table 2. Even at 0.25 mm, GSH was a weak inducer of cgb (one-fifth of the effect of GSNO measured by PCR) and did not significantly up-regulate p19. GSSG was without effect on either gene. Because GSNO produces undetectable levels of either thiol under our conditions, it can be concluded that the transcriptomic and quantitative real-time PCR determinations report on the nitrosating agent GSNO.
Real-time PCR was also used to compare the effects of GSNO and NO. To provide cultures with a sustained input of NO over several minutes, we used in combination a fast (NOC-7, half-life 3 min) and a slower NO-releasing compound (NOC-5, half-life 10.5 min) as described under “Experimental Procedures” and previous studies (5). Table 2 shows that the five genes compared all showed similar extents of up-regulation with both GSNO and NO.
We also tested the hypothesis that in the absence of Cgb the extents of transcriptional changes may be higher as, in a cgb mutant, NO is not removed rapidly by the action of the hemoglobin. The data (Table 2) show that expression levels were not markedly different from the wild-type strain.
A Role for Derepression of Fur-regulated Genes in Nitrosative Stress Tolerance—Up-regulation of genes involved in iron acquisition in response to nitrosative stress has been observed in other bacteria (10, 23, 54, 55). It has been suggested (55) that responses of the Fur regulon to nitrosative stress in Staphylococcus aureus are a pathological consequence of, rather than an adaptive response to, the stress. Nevertheless, NO reacts with Fur-bound ferrous iron forming dinitrosyl iron complexes, thus rendering Fur inactive and derepressing Fur-repressed genes (49). To test whether derepression of iron-regulated genes via Fur is an adaptive response to nitrosative stress, a fur mutant was studied. In these experiments, NO released from NOC compounds was used in preference to GSNO as the former is anticipated to target Fur more effectively (13). A fur mutant grew more slowly than the wild-type strain (Fig. 3). Growth approximated to a linear (not an exponential) pattern, consistent with oxygen-limited growth (56) and the data are plotted in this form; before adding NOCs, the growth rates were 0.054 and 0.019 absorbance units h-1 for wild-type and fur strains, respectively. For the wild-type strain, increasing NOC concentrations significantly retarded growth, the growth rate falling to 0.009 absorbance units h-1 at 15 μm NO; in contrast, the fur mutant was relatively insensitive to NOC compounds, the rate at 15 μm NO being reduced only 20% to 0.015 absorbance units h-1. Growth data are the means of triplicate flasks for each condition in two separate experiments.
FIGURE 3.

A fur mutant is resistant to inhibition by NOC compounds. Cultures of the wild-type (A) and fur mutant (B) strains were grown to mid-exponential phase, and a mixture of NOC-5 and NOC-7 was added at the arrows to the final concentrations shown (μm) against each growth curve. Note the different absorbance and time scales in A and B. Individual growth curves are the means of 3 replicates and have been offset vertically for clarity.
We considered the possibility that mutation of fur has polar effects on neighboring genes. In the C. jejuni chromosome, the gene order is Cj0399-fur(Cj0400)-lysS; promoters have been identified upstream of Cj0398 and Cj0399 (57). Because the insertion is such that the cassette promoter maintains downstream expression, lysS and its downstream gene (glyA) are expressed in this construct. It has not been possible to construct on the chromosome an insertion that is oriented in the opposite direction (36), but the present data strongly suggest that derepressed expression of Fur-regulated genes abrogates nitrosative stress.
Mutants defective only in chuA and chuB were also tested with GSNO at a concentration that gave no apparent inhibition of growth in the wild-type strain but did affect the growth rate of the hypersensitive cgb mutant. A growth comparison of the wild-type strain and the chuA and chuB mutant strains with 0.75 mm GSNO revealed unchanged sensitivity of the mutants (not shown). This result was corroborated in disc diffusion assays in which GSNO (3 μl of a 100 mm stock solution) was applied to filter discs laid on an agar lawn of cells (not shown). We also confirmed (not shown) the earlier finding (28) that the ahpC mutant is not hypersensitive to GSNO despite its up-regulation in the transcriptomic experiments.
Proteomic Analysis—The response to GSNO was also characterized by a proteomic analysis. Fig. 4, left, shows an overlay of two-dimensional gels obtained from control and GSNO-treated, continuously cultured C. jejuni and identifies segments of the gel (A–D) shown in greater detail (right). Only a few proteins were affected by GSNO in triplicate experiments. Notably, and consistent with the transcriptomic data, the levels of the globins Cgb and Ctb (Fig. 4, panel B) showed -fold increases of 5.2 (note that the p value is not applicable as Cgb was reliably detected in only one of three control gels) and 2.5 (p = 0.04), respectively. DnaK increased 2.5-fold (p = 0.04, three gels) (Fig. 4, panel A), also consistent with the transcriptomic study. GroEL increased (2.1-fold) in duplicate GSNO-treated cells. Further proteins that increased in the presence of GSNO included those encoded by Cj0383c (6,7-dimethyl-8-ribityllumazine synthase, RibH) and Cj0509c (ATP-dependent CLP protease ATP-binding subunit) that were 2.1 (p = 0.01)- and 2.7 (p = 0.02)-fold increased, respectively. Cj0509c was significantly up-regulated in array analysis of batch cultures (not shown), but RibH was not seen in the array data.
FIGURE 4.

Proteomic analysis of changes elicited by GSNO. Left, overlaid two-dimensional gels of protein extracted from control, untreated cultures (orange spots) and cultures exposed to 0.25 mm GSNO for 30 min (blue spots). Right, selected areas (A–D) are enlarged and resolved into gel images of the control cells (orange background) and GSNO-treated cells (blue background).
Proteins that decreased in abundance were Cj1626c (putative periplasmic protein), Cj1478c (CadF, outer membrane fibronectin-binding protein), Cj0449c (hypothetical), Cj0437 (succinate dehydrogenase flavoprotein subunit, SdhA), and Cj0438 (succinate dehydrogenase iron-sulfur protein, SdhB). These proteins were 3.1 (p = 0.014)-, 2.5 (p = 0.006)-, 2.1 (p = 0.004)-, 4.8 (p = 0.075)-, and 4.8 (p = 0.084)-fold higher in the control samples that were not exposed to GSNO. In duplicate gels, SdhA and SdhB levels were significantly higher in the control samples than in GSNO-treated cells (Fig. 4, panels C and D).
Oxygen Tension Influences Respiratory Inhibition by NO and Cgb and Ctb Expression—C. jejuni is a microaerophile (58), requiring optimally 5–7% oxygen and growing in the laboratory at 3–15% oxygen (about 40–200 μm (31)). Cgb probably catalyzes an O2-dependent dioxygenase or denitrosylase reaction (29), suggesting that O2 provision may be critical in nitrosative stress and NO detoxification. Conversely, inhibition of respiration by NO (Fig. 1) is expected to increase O2 concentrations under conditions of constant O2 diffusion (see Fig. 8 and “Discussion”). We, therefore, investigated the dependence of NO detoxification on oxygen availability. Inhibition of respiration by NO was dependent upon oxygen availability within the range of oxygen concentrations relevant to growth of this microaerophile (supplemental Fig. S1). When NO was added to respiring suspensions of cells from microaerobic conditions, respiration was inhibited at 20–60 μm oxygen, but the extent of inhibition diminished as NO was added at progressively higher oxygen concentrations. For example, the degrees of inhibition of initial respiration by NO added at 20, 40, 60, and 80 μm O2 were (%, mean values of 5 assays with S.D. in parentheses): 95 (±0.08), 88 (±0.39), 80 (±0.71), and 74 (±0.99), respectively. Only at the upper limits of relevant oxygen concentrations (160 μm) was inhibition transient, with recovery of cell respiration to near-control values. Similar observations have been made for E. coli (19, 59) and are attributable to competition for the oxidase hemes by NO and O2 (60), the oxygen dependence of the detoxification reaction (42), and/or the propensity for NO to persist in solutions at low oxygen concentrations.
FIGURE 8.

Model of NO sensing and the effects of iron and oxygen. NO is shown as the major intracellular stress, but GSNO may influence the same processes either by prior NO release or nitrosation reactions. Proteins are indicated by circles/ovals, and genes are indicated by broad open arrows preceded by their promoters (thick lines). Inhibitory effects of NO on respiration and metalloproteins are shown by short dashed lines, and the proposed interactions of NO and O2 are shown with transcription factors NssR and Fur by long dashed lines. NO or GSNO are sensed by NssR, which positively regulates transcription of cgb and other members of this regulon. The cgb gene is also positively regulated by O2, in part due to NssR. O2 levels rise on inhibition of respiration by NO (metabolic hypoxia). Cgb protein catalyzes an NO detoxification reaction, thought to result in nitrate formation. NO reacts with iron centers in key proteins but also with Fe(II) in Fur, inactivating it and causing derepression of iron-repressed genes (only tonB is shown as an example). Enhanced iron acquisition may be an adaptive response to NO toxicity.
To test whether the nitrosative stress response is subject to oxygen availability, we grew cells in baffled flasks (250 ml) containing 75, 100, 150, or 200 ml of MH broth to vary the first-order constant, K, for oxygen diffusion from gas phase to liquid (40). At culture volumes of 100, 150, and 200 ml under our conditions, the K values are 0.43, 0.16, and 0.06 min-1, respectively (31). After 9 h of growth, cultures were either harvested or supplemented with 0.25 mm GSNO for 10 or 60 min before harvest. Western blot analysis showed that, in the absence of nitrosative stress, Cgb was barely expressed at any of the K values tested (Fig. 5A), whereas Ctb was readily detected under all aeration conditions (Fig. 5B). Cgb levels were markedly and progressively increased after 10 and 60 min GSNO treatment and were clearly oxygen-dependent (Fig. 5A); at a K value of 0.43 min-1, Cgb levels at 10 min were ∼4.8-fold higher than at K = 0.06 min-1. At 60 min after GSNO treatment, the value was 5.5-fold. Similar results were obtained in two experiments. After treatment with GSNO for 10 min, Ctb levels were raised independently of oxygen availability (Fig. 5B), but after 60 min of exposure, significantly more Ctb was expressed at a K value of 0.43 min-1. These results reveal an unexpected oxygen dependence of Cgb and Ctb expression and provide clues to the mechanism of Cgb-mediated NO detoxification.
FIGURE 5.

Oxygen regulation of the hemoglobins Cgb and Ctb. Immunoblotting was used to quantify Cgb (A) and Ctb (B and C) at the aeration rates indicated by the first-order rate constants (K) for oxygen diffusion to the cultures. A and B show blots from the wild-type strain; C shows blots from an nssR mutant. Extracts were prepared from cells grown in the absence of GSNO (top row) or from cells pretreated with 0.25 mm GSNO for 10 or 60 min (below). In the control and 10-min samples, 20 μg of protein was loaded, and in the 60 min samples 5 μg of protein was loaded; the loading was 20 μg of protein for all nssR samples.
Oxygen dependence of globin expression was verified by growing cultures at various K values and removing samples before and for 1 h after GSNO addition. Cultures grown at K values of 0.16 min-1 and higher showed no growth inhibition on adding GSNO. However, the culture grown at a K value of 0.06 min-1 exhibited marked growth arrest (Fig. 6A); in the absence of GSNO, growth at K = 0.06 proceeded at the rate shown in Fig. 6A before GSNO addition (not shown). These results suggest that the decreased levels of Cgb and Ctb are detrimental to GSNO tolerance. Fig. 6B shows that, when cultures grown without GSNO at K values of 0.43 or 0.06 min-1 were challenged with NO, respiration was inhibited and did not recover during the time course of the experiment (traces 1 and 2). NO inhibited respiration more extensively (to 19% ± 0.07 of the initial rate; mean ± S.D.) in the poorly aerated culture than in the highly aerated culture (33% ± 0.18 of the initial rate). In contrast, when the cultures were pre-exposed to GSNO for 20 min (Fig. 6B, traces 3 and 4) the respiration of the highly aerated culture was inhibited to only 51% ± 0.91 that of the control value, and respiration recovered to the initial rate with an inhibitory period of less than 2 min (trace 3). In contrast, the poorly aerated culture was inhibited to 17% that of the control value, and respiration rate did not recover to pre-NO levels (trace 4). Thus, both oxygen provision and pretreatment of cells with GSNO confer resistance to NO.
FIGURE 6.

Oxygen availability during growth determines GSNO tolerance. A, growth at K values (first-order rate constants for oxygen diffusion to the cultures) of >0.43 (top) and 0.43, 0.16, and 0.06 (bottom) min-1 with GSNO (0.25 mm) added at the arrows. The values are means of three experiments with S.D. In the absence of GSNO, growth at K = 0.06 proceeded at the rate shown before GSNO addition. B, O2 uptake for cultures without (traces 1 and 2) or with GSNO (0.25 mm) for 20 min (traces 3 and 4) at the oxygen diffusion rates (K, min-1) shown. NO (10 μm) was added to each trace as shown by the arrows. Values on the traces represent mean oxygen consumption rates (nmol of O2 min-1 mg of protein-1) calculated from five replicate traces in a single experiment, which was repeated three times with similar results.
Cells cultured with elevated oxygen provision also consumed NO more effectively. Fig. 7A shows that cultures grown with higher oxygen availability (K = 0.43 min-1; traces 3 and 4) removed added NO faster than cultures grown at low oxygen availability (K = 0.06 min-1, traces 1 and 2) consistent with the globin quantification (Fig. 5). Furthermore, the highly aerated cultures showed a further significant increase in the rate of NO removal when pre-exposed to nitrosative stress (trace 4). In contrast, cultures exposed to GSNO when grown at low aeration exhibited a decreased ability to remove NO (trace 2).
FIGURE 7.

Oxygen availability influences the efficiency of NO consumption in an NssR-dependent manner. A, wild-type; B, nssR mutant. Traces 1 and 2 were obtained with cells grown with an aeration rate of K = 0.06 min-1, and traces 3 and 4 were obtained with cells grown with an aeration rate of K = 0.43 min-1. Traces 2 and 4 were from cells pre-grown with GSNO for 20 min. Each trace was repeated four times in duplicate experiments; typical traces are shown.
NssR Is Required for O2-mediated Gene Regulation and Globin Expression—Because immunoblotting (Fig. 5) showed that the globins are regulated by oxygen availability and transcriptomic analyses (Table 1 (30)) showed that the globin genes are part of the NssR regulon, we investigated whether the oxygen- and GSNO-dependent globin expression was mediated by NssR. Fig. 5C shows immunoblots of Ctb levels in nssR mutant cells cultivated with different aeration rates and after GSNO exposure. The marked aeration-dependent increase in Ctb in cells exposed to GSNO for 60 min is clearly missing. Cgb was not detectable in nssR mutant cells irrespective of the presence of GSNO or oxygen transfer rate (blots not shown). Furthermore, assays of NO consumption rates in the nssR mutant (Fig. 7B) showed that the rapid NO disappearance that is characteristic of cells grown with GSNO and at high aeration (Fig. 7A, trace 4) is missing. Thus, both oxygen provision during growth and pre-exposure to nitrosative stress are required for full NssR-mediated tolerance of NO.
To determine whether aeration rates influenced NssR-mediated regulation of other genes in the regulon, expression of five genes was tested by RT-PCR in cultures grown at K values of 0.43 and 0.06 min-1 in the presence or absence of GSNO. In cultures grown at the higher aeration, the addition of GSNO led to an increase in transcription of all genes tested except NssR itself, and the increases in expression (mean of duplicate determinations in three experiments ± coefficient of variation) were as follows: Cj0830, 2.4 (±0.08); Cj0761, 4.1 (±0.20); ctb, 3.2 (±0.09); cgb, 12.0 (±0.6); nssR, 1.2 (±0.40). In contrast, the -fold changes in the cultures grown with poor aeration were Cj0830, 1.2 (±0.25); Cj0761, 1.2 (±0.26); ctb, 2.6 (±0.19); cgb, 4.2 (±0.18); nssR, 0.3 (±0.98). Thus, the presence of oxygen favors the transcription of genes required for tolerance of nitrosative stress.
DISCUSSION
In pathogenic bacteria nitrosative stress elicits adaptive responses at the transcriptional level, resulting in enhanced synthesis of the NO-detoxifying hemoglobin Hmp, sometimes augmented by flavorubredoxin. Significantly, transcription of hmp and other genes implicated in NO tolerance is evident in E. coli from nitrate-rich urine in patients with urinary tract infections (61) and in Yersinia pestis isolated from the rat bubo (62). The human pathogen C. jejuni lacks both these NO-detoxifying proteins, and the full extent of the responses mounted by C. jejuni to nitrosative stresses has not been previously described. Here we show that both Cgb and Ctb are regulated not only by nitrosative stress but also by oxygen availability, perhaps indicating (i) its requirement as a co-substrate in detoxifying NO and/or (ii) a role for oxygen in NssR activity. Combining transcriptomic and proteomic approaches has provided a fuller picture of this response. The relative paucity of proteins shown to respond to GSNO may reflect a bias by the proteomic methods against membrane proteins. Previous comparisons of microarray and proteomic data in Campylobacter (41, 45) also report numerous transcriptome changes relative to the proteome, perhaps suggesting unstable transcripts. However, some of the differences observed between the two-dimensional gel and the microarray results are not explained by membrane protein issues. P19 (Cj1959) is readily visible on a protein gel and changes in this and other iron associated proteins occur under iron-limiting conditions (41). The changes in transcript for P19 were much lower (7.94-fold) in this study with no observed difference in protein compared with the iron depletion study (110-fold) where an 11-fold increase in protein was also found.
In previous studies of the effects of NO and related reactive nitrogen species on gene expression, a wide range of reagents have been used, including NO gas, NO generated from NOCs and related donors, nitrosating agents such as GSNO, and poorly defined mimics of the macrophage environment, such as acidified nitrite. It has been reported before that acidified nitrite has effects on C. jejuni that resemble those of macrophage-generated NO (4) and that the effects of nitrite are attributable to NO in solution. Given the uncertainty of the composition of solutions of acidified nitrite and the difficulties of interpreting earlier transcriptomic studies with this reagent (10), we have focused instead on a well characterized nitrosating agent (GSNO) and extensively characterized NOCs with defined half-lives. These reagents have been well characterized, and studies have confirmed the distinct effects that these agents have on gene expression in E. coli (11, 13, 14, 34). In this study a subset of the genes up-regulated by GSNO and revealed in the microarrays was also shown to be regulated by NO released from NOCs (Table 2). Deletion of Cgb had little effect on these transcriptional patterns (Table 2), suggesting that the effects of GSNO are not wholly explained by prior conversion of GSNO to NO. The effect of GSH (albeit at 0.25 mm, vastly greater than could have arisen in the media from GSNO breakdown) on cgb expression (Table 2) may result from a redox effect on NsrR but is outside the scope of the present work.
The adaptive responses of bacteria to nitrosative stresses have previously been shown to include expression not only of the NO-detoxifying hemoglobins and flavorubredoxin (9) but also genes involved in iron metabolism and oxidative stress (10, 12, 13, 24, 55). Here, transcriptome and proteome profiling have implicated thioredoxin reductase and alkylhydroperoxide reductase as well as heat shock proteins in the response. Significantly, at least seven genes implicated in iron metabolism and transport were identified. The transcriptome data provide evidence for enhanced iron uptake after GSNO stress, presumably in response to disruption of iron protein function. Likely roles for iron proteins are in respiration, a key target of nitrosative damage (Figs. 1 and S1), and in globin-mediated NO detoxification. The necessity of maintaining or enhancing iron uptake may also be related to functions of NO-sensitive Fe-S clusters required in respiration or gene regulation (5). Decreased succinate dehydrogenase proteins (Fig. 4) reflect down-regulation of sdhA (Fig. 2) and sdhB (supplemental Table S3) and might also be caused by reaction of reactive nitrogen species with flavin (SdhA) or Fe-S clusters (SdhB). The effects on succinate dehydrogenase might reflect reduced electron flux into the respiratory chain on inhibition of respiration. Other iron targets are in transcriptional regulators and in l-serine dehydratase; catabolism of serine is important for the growth of this pathogen in vivo (63).
However, unlike earlier studies that have attributed iron-responsive changes to an adventitious or pathological consequence of the reaction of NO with Fur and other iron-responsive regulators, we show that the Fur regulon plays a role in resisting nitrosative stress. Fig. 3 illustrates the resistance of a fur mutant to growth inhibition by NOCs; although the component(s) of the regulon responsible for this protection has not been identified, we have ruled out the chuAB hemin uptake system. It appears paradoxical that the iron response is triggered in rich media, but the poor bioavailability of iron in broth media lacking chelators is well documented (64).
Transcriptome analysis also revealed roles for diverse stress response genes, particularly the heat shock response. HrcA is widely distributed in eubacteria and, with GroEL, is a negative regulator of Class I heat-shock genes including grpE and dnaK (65). The rationale for involvement of heat-shock proteins in response to GSNO is unclear, but in Helicobacter pylori, which shares the same operon structure (hrcA-grpE-dnaK) (66), GroEL and DnaK are increased after acid shock (67). However, our analysis did not reveal all temperature-responsive genes; thus, gene cluster B (68) comprises 33 highly up-regulated genes, evident 10 min after a temperature shift, and a further study (69) showed the preferential synthesis of 24 proteins on increasing temperature. We surmise that the limited expression of heat-shock genes is a response to an accumulation of unfolded or damaged proteins after application of the nitrosating agent, perhaps in turn elicited by protein S-nitrosation. The mammalian inducible heat shock protein 70 (Hsp70) has a broad role, protecting cells against diverse injuries including nitrosative stress (70).
Oxidative stress genes were also evident and may reflect the fact that the products of NO synthase 2 in vivo (4) include not only NO but also superoxide and peroxide (71). Examples of oxidative stress responses in this study were up-regulation of trxA, trxB, and ahpC. TrxA and other members of the thioredoxin superfamily are characterized by a conserved CXXC motif. The role of thioredoxin in nitrosative stress is not obvious, but the glutaredoxin-like proteins have been implicated in nitrosative stress tolerance. These proteins (NrdH-redoxins or NrdH) are sometimes found in organisms lacking glutathione, like C. jejuni. Intriguingly, NrdH has a thioredoxin-like activity and structure (72). In C. jejuni, limiting iron levels increase transcription of ahpC and expression of the 26-kDa protein (73). Insertional mutagenesis of ahpC leads to increased sensitivity to cumene hydroperoxide and atmospheric oxygen but not hydrogen peroxide (73). As in H. pylori, the C. jejuni genome lacks ahpF, encoding the large subunit of alkylhydroperoxide reductase and, as in H. pylori (74), thioredoxin and its reductase (up-regulated in C. jejuni, see above) probably constitute a reduction pathway for AhpC. In H. pylori the thioredoxin system mediates resistance to oxidative and nitrosative (NO, GSNO) stresses (75), presumably by its role in electron transfer to AhpC. In other bacteria AhpC confers resistance to nitrite, GSNO, and peroxynitrite (76). Cj0779, predicted to encode a thiolperoxidase (Tpx) or scavengase p20 and also up-regulated (Table 1), shows the highest similarity with thiolperoxidases of H. pylori, Aquifex aeolicus, and other bacteria. The C. jejuni protein has 43% sequence identity with the tpx (also called tagD) gene products of H. pylori and a similar degree of identity with Tpx of other campylobacters and p20 oxidoreductase of Wolinella succinogenes. The purified H. pylori enzyme has a thioredoxin-linked peroxidase activity (77), consistent with the concomitant up-regulation of thioredoxin genes in this study. The scavengase p20s appear important in pathogenesis (78), where nitrosative stress may be encountered. Several genes identified as up-regulated by GSNO in the present study (notably cgb and putative oxidoreductase genes Cj0414 and Cj0415) were also increased in a recent transcriptomic analysis of C. jejuni growing under mildly acidic conditions (79), but the significance of this cannot be explained at present.
These findings together with the up-regulation of the NssR regulon expression by GSNO are rationalized in Fig. 8. In this model, NO derepresses Fur-regulated genes via the mechanism previously proposed for E. coli Fur (49). These genes include tonB, p19, and others, one or more of which may contribute to tolerance to GSNO and NO. Protection may be explained by enhanced iron uptake to repair or reassemble heme proteins or Fe-S clusters damaged by NO. However, the iron may also be required for full activity of NssR, which is triggered by exposure to NO, GSNO, or related species. The nature of the sensing mechanism has not been elucidated, but in NO-sensing proteins, there are precedents for important roles for iron in Fe-S clusters (5, 80, 81), a mononuclear non-heme iron center (82), and heme (83–85). Because NssR is a positive regulator of cgb and the other NssR regulon genes, we suggest that there might exist an iron requirement either for interaction of NO with NssR or for reconstitution of NssR after interaction with NO.
In either event oxygen also plays a key role in the transcriptional regulation and probably also in the Cgb-catalyzed NO detoxification reaction. It is worth noting that the potent inhibition of oxygen consumption elicited by NO might in itself raise oxygen concentrations in vivo so as to promote transcription of protective genes. This effect is reminiscent of the “metabolic hypoxia” reported in mitochondria, in which, due to increases in NO, the available O2 cannot be used (86). Upon NO inhibition of respiration, O2 may be redistributed toward non-respiratory O2-dependent roles (87), which might include in bacteria NO detoxification.
Supplementary Material
Acknowledgments
We thank Julian Ketley (University of Leicester) and Simon Park (University of Surrey) for strains and helpful discussions. Martin Hughes generously provided GSNO, a protocol for its synthesis, and advice on the use of GSNO and NOCs. The matrix-assisted laser desorption ionization time-of-flight protein identification work was carried out by Mike Naldrett and Andrew Botrill at the Institute of Food Research-John Innes Centre proteomic facility, Norwich, UK.
This work was supported by the Biotechnology and Biological Sciences Research Council, Swindon, United Kingdom. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S3 and Fig. S1.
Footnotes
The abbreviations used are: GSNO, S-nitrosoglutathione; Cm, chloramphenicol; GSSG, glutathione disulfide; Kan, kanamycin; MH, Mueller-Hinton growth medium; NOC-5, 3-[2-hydroxy-1-(1-methylethyl)-2-nitrosohydrazino]-1-propanamine; NOC-7, 3-(2-hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1--propanamine.
References
- 1.Friedman, C. R., Neimann, J., Wegener, H. C., and Tauxe, R. V. (2000) in Campylobacter (Nachamkin, I., and Blaser, M. J., eds) 2nd Ed., pp. 121-138, American Society for Microbiology, Washington, DC
- 2.Shiloh, M. U., and Nathan, C. F. (2000) Curr. Opin. Microbiol. 3 35-42 [DOI] [PubMed] [Google Scholar]
- 3.Lundberg, J. O., Weitzberg, E., Cole, J. A., and Benjamin, N. (2004) Nat. Rev. Microbiol. 2 593-602 [DOI] [PubMed] [Google Scholar]
- 4.Iovine, N. M., Pursnani, S., Voldman, A., Wasserman, G., Blaser, M. J., and Weinrauch, Y. (2008) Infect. Immun. 76 986-993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cruz-Ramos, H., Crack, J., Wu, G., Hughes, M. N., Scott, C., Thomson, A. J., Green, J., and Poole, R. K. (2002) EMBO J. 21 3235-3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hausladen, A., Gow, A., and Stamler, J. S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10108-10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hess, D. T., Matsumoto, A., Kim, S.-O., Marshall, H. E., and Stamler, J. S. (2005) Nat. Rev. Mol. Cell Biol. 6 150-166 [DOI] [PubMed] [Google Scholar]
- 8.Poole, R. K., and Hughes, M. N. (2000) Mol. Microbiol. 36 775-783 [DOI] [PubMed] [Google Scholar]
- 9.Poole, R. K. (2005) Biochem. Soc. Trans. 33 176-180 [DOI] [PubMed] [Google Scholar]
- 10.Mukhopadhyay, P., Zheng, M., Bedzyk, L. A., LaRossa, R. A., and Storz, G. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 745-750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flatley, J., Barrett, J., Pullan, S. T., Hughes, M. N., Green, J., and Poole, R. K. (2005) J. Biol. Chem. 280 10065-10072 [DOI] [PubMed] [Google Scholar]
- 12.Justino, M. C., Vicente, J. B., Teixeira, M., and Saraiva, L. M. (2005) J. Biol. Chem. 280 2636-2643 [DOI] [PubMed] [Google Scholar]
- 13.Pullan, S. T., Gidley, M. D., Jones, R. A., Barrett, J., Stevanin, T. A., Read, R. C., Green, J., and Poole, R. K. (2007) J. Bacteriol. 189 1845-1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyduke, D. R., Jarboe, L. R., Tran, L. M., Chou, K. J. Y., and Liao, J. C. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 8484-8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poole, R. K., Anjum, M. F., Membrillo-Hernández, J., Kim, S. O., Hughes, M. N., and Stewart, V. (1996) J. Bacteriol. 178 5487-5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardner, A. M., Helmick, R. A., and Gardner, P. R. (2002) J. Biol. Chem. 277 8172-8177 [DOI] [PubMed] [Google Scholar]
- 17.Gomes, C. M., Giuffre, A., Forte, E., Vicente, J. B., Saraiva, L. M., Brunori, M., and Teixeira, M. (2002) J. Biol. Chem. 277 25273-25276 [DOI] [PubMed] [Google Scholar]
- 18.Membrillo-Hernández, J., Coopamah, M. D., Anjum, M. F., Stevanin, T. M., Kelly, A., Hughes, M. N., and Poole, R. K. (1999) J. Biol. Chem. 274 748-754 [DOI] [PubMed] [Google Scholar]
- 19.Stevanin, T. M., Ioannidis, N., Mills, C. E., Kim, S. O., Hughes, M. N., and Poole, R. K. (2000) J. Biol. Chem. 275 35868-35875 [DOI] [PubMed] [Google Scholar]
- 20.Stevanin, T. A., Read, R. C., and Poole, R. K. (2007) Gene 398 62-68 [DOI] [PubMed] [Google Scholar]
- 21.Membrillo-Hernández, J., Coopamah, M. D., Channa, A., Hughes, M. N., and Poole, R. K. (1998) Mol. Microbiol. 29 1101-1112 [DOI] [PubMed] [Google Scholar]
- 22.Bodenmiller, D. M., and Spiro, S. (2006) J. Bacteriol. 188 874-881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez-Urzua, E., Zamorano-Sanchez, D. S., Ponce-Coria, J., Morett, E., Grogan, S., Poole, R. K., and Membrillo-Hernandez, J. (2007) Arch. Microbiol. 187 67-77 [DOI] [PubMed] [Google Scholar]
- 24.Bang, I. S., Liu, L. M., Vazquez-Torres, A., Crouch, M. L., Stamler, J. S., and Fang, F. C. (2006) J. Biol. Chem. 281 28039-28047 [DOI] [PubMed] [Google Scholar]
- 25.Gilberthorpe, N. J., Lee, M. E., Stevanin, T. M., Read, R. C., and Poole, R. K. (2007) Microbiology 153 1756-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevanin, T. M., Poole, R. K., Demoncheaux, E. A. G., and Read, R. C. (2002) Infect. Immun. 70 4399-4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parkhill, J., Wren, B. W., Mungall, K., Ketley, J. M., Churcher, C., Basham, D., Chillingworth, T., Davies, R. M., Feltwell, T., Holroyd, S., Jagels, K., Karlyshev, A. V., Moule, S., Pallen, M. J., Penn, C. W., Quail, M. A., Rajandream, M. A., Rutherford, K. M., vanVliet, A. H. M., Whitehead, S., and Barrell, B. G. (2000) Nature 403 665-668 [DOI] [PubMed] [Google Scholar]
- 28.Elvers, K. T., Wu, G., Gilberthorpe, N. J., Poole, R. K., and Park, S. F. (2004) J. Bacteriol. 186 5332-5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu, C. Y., Mukai, M., Lin, Y., Wu, G. H., Poole, R. K., and Yeh, S. R. (2007) J. Biol. Chem. 282 25917-25928 [DOI] [PubMed] [Google Scholar]
- 30.Elvers, K. T., Turner, S. M., Wainwright, L. M., Marsden, G., Hinds, J., Cole, J. A., Poole, R. K., Penn, C. W., and Park, S. F. (2005) Mol. Microbiol. 57 735-750 [DOI] [PubMed] [Google Scholar]
- 31.Wainwright, L. M., Elvers, K. T., Park, S. F., and Poole, R. K. (2005) Microbiology 151 4079-4091 [DOI] [PubMed] [Google Scholar]
- 32.Wainwright, L. M., Wang, Y. H., Park, S. F., Yeh, S. R., and Poole, R. K. (2006) Biochemistry 45 6003-6011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu, C. Y., Egawa, T., Wainwright, L. M., Poole, R. K., and Yeh, S.-R. (2007) J. Biol. Chem. 282 13627-13636 [DOI] [PubMed] [Google Scholar]
- 34.Jarboe, L. R., Hyduke, D. R., Tran, L. M., Chou, K. J. Y., and Liao, J. C. (2008) J. Biol. Chem. 283 5148-5157 [DOI] [PubMed] [Google Scholar]
- 35.Ridley, K. A., Rock, J. D., Li, Y., and Ketley, J. M. (2006) J. Bacteriol. 188 7862-7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Vliet, A. H. M., Wooldridge, K. G., and Ketley, J. M. (1998) J. Bacteriol. 180 5291-5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hart, T. W. (1985) Tetrahedron Lett. 26 2013-2016 [Google Scholar]
- 38.Lee, L. J., Barrett, J. A., and Poole, R. K. (2005) J. Bacteriol. 187 1124-1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pullan, S. T., Monk, C. E., Lee, L., and Poole, R. K. (2008) in Methods Enzymol. (Poole, R. K., ed) pp. 499-519, Elsevier, Inc., San Diego, CA [DOI] [PubMed]
- 40.Pirt, S. J. (1985) Principles of Microbe and Cell Cultivation, pp. 81-93, Blackwell Scientific Publications, Oxford, UK
- 41.Holmes, K., Mulholland, F., Pearson, B. M., Pin, C., McNicholl-Kennedy, J., Ketley, J. M., and Wells, J. M. (2005) Microbiology 151 243-257 [DOI] [PubMed] [Google Scholar]
- 42.Mills, C. E., Sedelnikova, S., Sφballe, B., Hughes, M. N., and Poole, R. K. (2001) Biochem. J. 353 207-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Groote, M. A., Granger, D., Xu, Y. S., Campbell, G., Prince, R., and Fang, F. C. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 6399-6403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donzelli, S., Espey, M. G., Thomas, D. D., Mancardi, D., Tocchetti, C. G., Ridnour, L. A., Paolocci, N., King, S. B., Miranda, K. M., Lazzarino, G., Fukuto, J. M., and Wink, D. A. (2006) Free Radic. Biol. Med. 40 1056-1066 [DOI] [PubMed] [Google Scholar]
- 45.Thorup-Andersen, M., Brondsted, L., Pearson, B. M., Mulholland, F., Parker, M., Pin, C., Wells, J. M., and Ingmer, H. (2005) Microbiology 151 905-915 [DOI] [PubMed] [Google Scholar]
- 46.Volker, U., Engelmann, S., Maul, B., Riethdorf, S., Volker, A., Schmid, R., Mach, H., and Hecker, M. (1994) Microbiology 140 741-752 [DOI] [PubMed] [Google Scholar]
- 47.Schmalisch, M., Langbein, I., and Stulke, J. (2002) J. Mol. Microbiol. Biotechnol. 4 495-501 [PubMed] [Google Scholar]
- 48.van Vliet, A. H. M., Baillon, M. L. A., Penn, C. W., and Ketley, J. M. (1999) J. Bacteriol. 181 6371-6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D'Autreaux, B., Touati, D., Bersch, B., Latour, J. M., and Michaud-Soret, I. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 16619-16624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Vliet, A. H. M., Ketley, J. M., Park, S. F., and Penn, C. W. (2002) FEMS Microbiol. Rev. 26 173-186 [DOI] [PubMed] [Google Scholar]
- 51.Janvier, B., Constantinidou, C., Aucher, P., Marshall, Z. V., Penn, C. W., and Fauchere, J. L. (1998) Res. Microbiol. 149 95-107 [DOI] [PubMed] [Google Scholar]
- 52.Guerry, P., PerezCasal, J., Yao, R. J., McVeigh, A., and Trust, T. J. (1997) J. Bacteriol. 179 3997-4002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raphael, B. H., and Joens, L. A. (2003) Can. J. Microbiol. 49 727-731 [DOI] [PubMed] [Google Scholar]
- 54.Moore, C. M., Nakano, M. M., Wang, T., Ye, R. W., and Helmann, J. D. (2004) J. Bacteriol. 186 4655-4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richardson, A. R., Dunman, P. M., and Fang, F. C. (2006) Mol. Microbiol. 61 927-939 [DOI] [PubMed] [Google Scholar]
- 56.Duetz, W. A., Ruedi, L., Hermann, R., O'Connor, K., Buchs, J., and Witholt, B. (2000) Appl. Environ. Microbiol. 66 2641-2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Vliet, A. H. M., Rock, J. D., Madeleine, L. N., and Ketley, J. M. (2000) FEMS Microbiol. Lett. 188 115-118 [DOI] [PubMed] [Google Scholar]
- 58.Goodhew, C. F., Elkurdi, A. B., and Pettigrew, G. W. (1988) Biochim. Biophys. Acta 933 114-123 [DOI] [PubMed] [Google Scholar]
- 59.Yu, H., Sato, E. F., Nagata, K., Nishikawa, M., Kashiba, M., Arakawa, T., Kobayashi, K., Tamura, T., and Inoue, M. (1997) FEBS Lett. 409 161-165 [DOI] [PubMed] [Google Scholar]
- 60.Mason, M. G., Nicholls, P., Wilson, M. T., and Cooper, C. E. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 708-713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roos, V., and Klemm, P. (2006) Infect. Immun. 74 3565-3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sebbane, F., Lemaitre, N., Sturdevant, D. E., Rebeil, R., Virtaneva, K., Porcella, S. F., and Hinnebusch, B. J. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 11766-11771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Velayudhan, J., Jones, M. A., Barrow, P. A., and Kelly, D. J. (2004) Infect. Immun. 72 260-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hughes, M. N., and Poole, R. K. (1991) J. Gen. Microbiol. 137 725-734 [Google Scholar]
- 65.Narberhaus, F. (1999) Mol. Microbiol. 31 1-8 [DOI] [PubMed] [Google Scholar]
- 66.Homuth, G., Domm, S., Kleiner, D., and Schumann, W. (2000) J. Bacteriol. 182 4257-4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allan, E., Clayton, C. L., McLaren, A., Wallace, D. M., and Wren, B. W. (2001) Microbiology 147 2285-2292 [DOI] [PubMed] [Google Scholar]
- 68.Stintzi, A. (2003) J. Bacteriol. 185 2009-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Konkel, M. E., Kim, B. J., Klena, J. D., Young, C. R., and Ziprin, R. (1998) Infect. Immun. 66 3666-3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lai, Y., Du, L., Dunsmore, K. E., Jenkins, L. W., Wong, H. R., and Clark, R. S. B. (2005) J. Neurochem. 94 360-366 [DOI] [PubMed] [Google Scholar]
- 71.Weaver, J., Porasuphatana, S., Tsai, P., Pou, S., Roman, L. J., and Rosen, G. M. (2005) Biochim. Biophys. Acta 1726 302-308 [DOI] [PubMed] [Google Scholar]
- 72.Stehr, M., Schneider, G., Aslund, F., Holmgren, A., and Lindqvist, Y. (2001) J. Biol. Chem. 276 35836-35841 [DOI] [PubMed] [Google Scholar]
- 73.Baillon, M. L. A., vanVliet, A. H. M., Ketley, J. M., Constantinidou, C., and Penn, C. W. (1999) J. Bacteriol. 181 4798-4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baker, L. M. S., Raudonikiene, A., Hoffman, P. S., and Poole, L. B. (2001) J. Bacteriol. 183 1961-1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Comtois, S. L., Gidley, M. D., and Kelly, D. J. (2003) Microbiology 149 121-129 [DOI] [PubMed] [Google Scholar]
- 76.Bryk, R., Griffin, P., and Nathan, C. (2000) Nature 407 211-215 [DOI] [PubMed] [Google Scholar]
- 77.Wan, X. Y., Zhou, Y., Yan, Z. Y., Wang, H. L., Hou, Y. D., and Jin, D. Y. (1997) FEBS Lett. 407 32-36 [DOI] [PubMed] [Google Scholar]
- 78.Olczak, A. A., Seyler, R. W., Olson, J. W., and Maier, R. J. (2003) Infect. Immun. 71 580-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reid, A. N., Pandey, R., Palyada, K., Whitworth, L., Doukhanine, E., and Stintzi, A. (2008) Appl. Environ. Microbiol. 74 1598-1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding, H. G., and Demple, B. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 5146-5150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakano, M. M., Geng, H., Nakano, S., and Kobayashi, K. (2006) J. Bacteriol. 188 5878-5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.D'Autreaux, B., Tucker, N. P., Dixon, R., and Spiro, S. (2005) Nature 437 769-772 [DOI] [PubMed] [Google Scholar]
- 83.DelgadoNixon, V. M., Gonzalez, G., and GillesGonzalez, M. A. (2000) Biochemistry 39 2685-2691 [DOI] [PubMed] [Google Scholar]
- 84.Nioche, P., Berka, V., Vipond, J., Minton, N., Tsai, A. L., and Raman, C. S. (2004) Science 306 1550-1553 [DOI] [PubMed] [Google Scholar]
- 85.Clark, R. W., Lanz, N. D., Lee, A. J., Kerby, R. L., Roberts, G. P., and Burstyn, J. N. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 891-896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moncada, S., and Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell Biol. 3 214-220 [DOI] [PubMed] [Google Scholar]
- 87.Hagen, T., Taylor, C. T., Lam, F., and Moncada, S. (2003) Science 302 1975-1978 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.