

Abstract
Rhinoviruses are the major cause of the common cold and acute exacerbations of asthma and chronic obstructive pulmonary disease. We previously reported rapid rhinovirus induction of intracellular superoxide anion, resulting in NF-κB activation and pro-inflammatory molecule production. The mechanisms of rhinovirus superoxide induction are poorly understood. Here we found that the proteolytic activation of the xanthine dehydrogenase/xanthine oxidase (XD/XO) system was required because pretreatment with serine protease inhibitors abolished rhinovirus-induced superoxide generation in primary bronchial and A549 respiratory epithelial cells. These findings were confirmed by Western blotting analysis and by silencing experiments. Rhinovirus infection induced intracellular depletion of reduced glutathione (GSH) that was abolished by pretreatment with either XO inhibitor oxypurinol or serine protease inhibitors. Increasing intracellular GSH with exogenous H2S or GSH prevented both rhinovirus-mediated intracellular GSH depletion and rhinovirus-induced superoxide production. We propose that rhinovirus infection proteolytically activates XO initiating a pro-inflammatory vicious circle driven by virus-induced depletion of intracellular reducing power. Inhibition of these pathways has therapeutic potential.
Rhinoviruses (RV)3 are the major cause of the commonest human acute infectious disease, the common cold (1). They are also associated with the majority of acute exacerbations of asthma (2, 3) and chronic obstructive pulmonary disease (COPD) (4, 5). No licensed effective antiviral is currently available for the treatment of the common cold (6, 7) and treatment of virus-induced asthma and COPD exacerbations is a major unmeet therapeutic need (8). Understanding the mechanisms of virus-induced exacerbation of airway diseases is required to identify molecular targets for therapeutic intervention.
The mechanisms underlying virus-induced exacerbations of airway diseases are poorly understood. However, rhinoviruses are believed to directly infect airway epithelium inducing pro-inflammatory cytokine production (9-11). This leads to recruitment and activation of inflammatory cells, resulting in airway inflammation (12, 13). We have recently demonstrated that bronchial epithelial cells from asthmatic subjects have a deficient innate immune response to rhinovirus infection, responsible for: (i) increased virus replication (14, 15) that could account for increased and more persistent inflammatory responses (12); (ii) increased severity and duration of lower respiratory tract symptoms and reductions in lung function (16) in rhinovirus-induced asthma exacerbations.
Increased oxidative stress is implicated in induction of the acute airway inflammation during exacerbations of asthma and COPD (17). Oxidants are directly involved in inflammatory responses via signaling mechanisms, including the redox-sensitive activation of transcription factors such as NF-κB (18, 19).
Recent data indicate that rhinovirus and other respiratory viruses can
alter cellular redox homeostatic balance toward a pro-oxidative condition
(20-22).
The molecular pathways responsible for such disequilibrium are virtually
unknown. A recent study suggested NADPH oxidase involvement in
rhinovirus-induced production of reactive oxygen species over a 6-h infection
(23). In a previous study we
documented that rhinovirus infection induces a rapid increase of intracellular
super-oxide anion (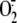 ),
which occurs within 15 min after infection. This early pro-oxidative response
was found to induce NF-κB activation and downstream pro-inflammatory
molecule production (24).
),
which occurs within 15 min after infection. This early pro-oxidative response
was found to induce NF-κB activation and downstream pro-inflammatory
molecule production (24).
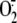 is a product of
cellular metabolism and mainly originates from the activity of two enzyme
systems: NADPH oxidase and xanthine dehydrogenase/xanthine oxidase (XD/XO)
(25). Here we studied the
molecular mechanisms by which rhinovirus induces rapid
is a product of
cellular metabolism and mainly originates from the activity of two enzyme
systems: NADPH oxidase and xanthine dehydrogenase/xanthine oxidase (XD/XO)
(25). Here we studied the
molecular mechanisms by which rhinovirus induces rapid
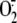 production in respiratory
epithelial cells. We also analyzed the mechanisms by which reducing agents can
abolish rhinovirus-induced
production in respiratory
epithelial cells. We also analyzed the mechanisms by which reducing agents can
abolish rhinovirus-induced
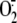 production and thus can
stabilize the intracellular redox state in respiratory epithelial cells
following infection. Finally, we demonstrated that blocking the activity of
the system responsible for rhinovirus-triggered
production and thus can
stabilize the intracellular redox state in respiratory epithelial cells
following infection. Finally, we demonstrated that blocking the activity of
the system responsible for rhinovirus-triggered
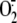 generation inhibited
rhinovirus-induced inflammatory mediator production in respiratory epithelial
cells.
generation inhibited
rhinovirus-induced inflammatory mediator production in respiratory epithelial
cells.
EXPERIMENTAL PROCEDURES
Cell Culture
Ohio HeLa cells were obtained from the MRC Common Cold Unit, Salisbury, UK, and A549 cells, a type II respiratory cell line, were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Primary human bronchial epithelial cells (HBEC) were obtained by bronchial brushing from healthy volunteers, and cultured as previously described (14, 24, 26).
Virus Stocks
Rhinovirus type 16 (RV16, a major group rhinovirus) was obtained from the MRC Common Cold Unit. Viral stocks were prepared by infection of sensitive cell monolayers (Ohio HeLa, HeLa) as described elsewhere (24, 26). TCID50/ml values were determined and the rhinovirus serotype was confirmed by neutralization with serotype-specific antibodies (ATCC) (27). For selected experiments rhinovirus type 1B (RV1B, minor group), obtained from the MRC Common Cold Unit, was used to evaluate whether the results were group/receptor restricted. For selected experiments filtration of the virus from inoculum, to remove viral particles, was performed as previously described (24, 26). Filtered virus stocks were used as negative control. Virus at a multiplicity of infection of 1 was used for all the experiments.
Infections, Harvesting of Cells, Preparation of Cell Homogenates, and Preparation of Membrane and Cytosolic Fractions
Confluent A549 or HBEC cells were exposed to rhinovirus, medium alone, or filtered virus (f-RV) inoculum for different time intervals (20 min to 8 h). Cell layers were thereafter washed three times in cold phosphate-buffered saline (PBS) before harvesting by scraping. Harvested cells were centrifuged and the cell pellet was resuspended in phosphate buffer (10 mm, pH 7.2). Cell lysis was obtained by repeated (three times) freezing and thawing. For preparation of cytosolic fractions, the cell homogenate was then ultracentrifuged at 20,000 × g for 30 min, the cell fragments pelleted, and the supernatant (cytosol) collected. Where indicated, to obtain the membrane fraction, the cell homogenates were centrifuged at 800 × g for 10 min to separate nuclei from cell membranes. Supernatants were harvested and again centrifuged at 2,000 × g, supernatants discarded, and membrane pellets diluted in 0.1 m sucrose solution. A final centrifugation at 11,000 × g for 20 min was performed at 4 °C. Pellets containing membranes were diluted in 100 μl of PBS buffer. Protein content was determined photometrically using the Bio-Rad protein assay (Bio-Rad).
Protease and XO Inhibition
In selected experiments cells were pretreated, before infection, as follows: 12 h (0.25 to 10 mm) GSH (Sigma), or 12 h (0.25 to 2.5 mm)H2S (Acqua Breta, Riolo Terme SpA, Ravenna, Italy) or 4 h (20 μm) oxypurinol (4, 6-dihydroxyprazol (3, 4-d)pyrimidine, Sigma), a permanent inactivator of xanthine oxidase (28). Where indicated, a 4-h pretreatment with antiproteases (0.625 μm) serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF) or 1.25 μm serine and cysteine protease inhibitor leupeptin (Leu), or 1.25 μm aspartic protease inhibitor pepstatin (Pep), or 1.25 μm serine protease inhibitor aprotinin (Apr), or 1.25 μm metalloprotease inhibitor phenanthroline (Phe) or cysteine protease inhibitor E-64, all from Sigma), or diluent alone was performed (29-34). Diluent was made of phosphate buffer, pH 7.2, with a maximal final concentration of 0.4%. PMSF only was previously resuspended in ethanol before dilution in PBS. Antiproteases were removed immediately before infection.
Cytochrome c Reduction Kinetics
The intracellular production of
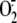 was
spectrophotometrically evaluated by superoxide dismutase (SOD)-inhibitable
cytochrome c reduction kinetics, as previously described
(24,
35). Kinetics were carried out
in 2-ml quartz cuvettes at 37 °C for 20 min in a Uvikon 860 (Kontron
Instruments) spectrophotometer in the presence or absence of SOD (500 IU/ml,
Sigma). Concentration of cytochrome c from beef heart (Sigma) was
10-5 m. Absorbance readings were taken at 550 nm (peak
of reduced cytochrome). Newly generated
was
spectrophotometrically evaluated by superoxide dismutase (SOD)-inhibitable
cytochrome c reduction kinetics, as previously described
(24,
35). Kinetics were carried out
in 2-ml quartz cuvettes at 37 °C for 20 min in a Uvikon 860 (Kontron
Instruments) spectrophotometer in the presence or absence of SOD (500 IU/ml,
Sigma). Concentration of cytochrome c from beef heart (Sigma) was
10-5 m. Absorbance readings were taken at 550 nm (peak
of reduced cytochrome). Newly generated
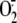 was measured in each
sample and expressed as micromolar, according to standardized procedures
(25). Measurements were based
on absorbance differences in the presence or absence of SOD, after 5 min of
kinetics, when the kinetic slope of cytochrome c reduction was
steepest. Data were normalized per mg of protein.
was measured in each
sample and expressed as micromolar, according to standardized procedures
(25). Measurements were based
on absorbance differences in the presence or absence of SOD, after 5 min of
kinetics, when the kinetic slope of cytochrome c reduction was
steepest. Data were normalized per mg of protein.
Uric Acid Kinetics
Uric acid kinetics were performed at 1 h infection to evaluate the
involvement of XD/XO in rhinovirus-induced
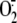 generation, as uric acid
represents the other end product of xanthine degradation by XO. Uric acid
kinetics were spectrophotometrically monitored at 293 nm in a UviKon
spectrophotometer (Kontron), according to standard procedures
(36,
37), on 500 μl of
supernatant collected in PBS, pH 7.4, after addition of xanthine (0.1
mm, Sigma). After 15 min of kinetics, uricase (1.0 units/ml) was
added to evaluate the amount of uric acid produced.
generation, as uric acid
represents the other end product of xanthine degradation by XO. Uric acid
kinetics were spectrophotometrically monitored at 293 nm in a UviKon
spectrophotometer (Kontron), according to standard procedures
(36,
37), on 500 μl of
supernatant collected in PBS, pH 7.4, after addition of xanthine (0.1
mm, Sigma). After 15 min of kinetics, uricase (1.0 units/ml) was
added to evaluate the amount of uric acid produced.
NADPH Oxidase Assay
NADPH oxidase assay was performed at different time intervals (20 min to 3
h) to evaluate the involvement of this system in rhinovirus-induced
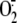 generation. Cell
homogenates were centrifuged as described above to separate nuclei from cell
membranes. To reconstitute NADPH oxidase, supernatants containing membranes
were centrifuged again at 40,000 × g. The reaction mixture
contained 200 μl of supernatant and 50 μl of diluted membrane pellet.
After 2 min, 200 μm NADPH and 5 mm MgCl2
were added in the presence or absence of the specific inhibitor of NADPH
oxidase diphenylene iodonium chloride (0.92 μg/ml, Sigma)
(38). Cytochrome c
was added to a concentration of 0.1 mm and PBS, pH 7.2, to a final
volume of 0.5 ml, and the reduction kinetics were monitored for 15 min at 37
°C as previously described.
generation. Cell
homogenates were centrifuged as described above to separate nuclei from cell
membranes. To reconstitute NADPH oxidase, supernatants containing membranes
were centrifuged again at 40,000 × g. The reaction mixture
contained 200 μl of supernatant and 50 μl of diluted membrane pellet.
After 2 min, 200 μm NADPH and 5 mm MgCl2
were added in the presence or absence of the specific inhibitor of NADPH
oxidase diphenylene iodonium chloride (0.92 μg/ml, Sigma)
(38). Cytochrome c
was added to a concentration of 0.1 mm and PBS, pH 7.2, to a final
volume of 0.5 ml, and the reduction kinetics were monitored for 15 min at 37
°C as previously described.
Western Blot Analysis for Xanthine Dehydrogenase/Oxidase
Whole cell proteins were extracted from A549 cells as previously described (39). At least 50 mg/lane of whole cell proteins were subjected to a 4-12% Tris glycine gel electrophoresis, and transferred to nitrocellulose filters by blotting. Filters were blocked for 45 min at room temperature in Tris-buffered saline (TBS), 0.05% Tween 20, 5% nonfat dry milk. The filters were then incubated with rabbit anti-human XD/XO (LS-C26419; from LifeSpan Biosciences) for 1 h at room temperature in TBS, 0.05% Tween 20, 5% nonfat dry milk at dilution of 1:500. Filters were washed three times in TBS, 0.5% Tween 20 and after being incubated for 45 min at room temperature with goat anti-rabbit antibody conjugated to horseradish peroxidase (Dako) in TBS, 0.05% Tween 20, 5% nonfat dry milk, at a dilution of 1:4000. After three further washes in TBS, 0.05% Tween 20 visualization of the immunocomplexes was performed using ECL as recommended by the manufacturer (Amersham Biosciences). As an internal control we reprobed each filter with an anti-human actin antibody (Santa Cruz Biotechnology). The 145- and 85-kDa bands of the XD/XO system ((full-length XD and the post-cleavage fragment containing the active site, respectively (40, 41)) and the 43-kDa (actin) band were quantified using densitometry with Vision Works® LS software (UVP) and expressed as the ratio with the corresponding actin optical density value of the same lane.
Knockdown of Xanthine Dehydrogenase Expression
RNA interference was used to specifically suppress expression of XD in A549 cells. Cells were transfected in 6-well plates with small interfering RNA (siRNA) using siPORT™ NeoFX™ Transfection Agent (Applied Biosystem), as described by the manufacturer. The following siRNA (all from Ambion) were used: siRNAs for XD (s14918; target sequence: sense, GCAUCGUCAUGAGUAUGUAtt; antisense, UUUAUAGCAUCCUCAAUUGtg), siRNA for GAPDH (4390849) and nonsilencing siRNA (4390843). Total mRNA was extracted by using the RiboPure™ kit (Ambion) as per the manufacturer's instructions. 1 μg of mRNA was used to perform the reverse transcription assay with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystem). XD mRNA expression was monitored by Real Time RT-PCR using the TaqMan® Gene Expression Assay (Applied Biosystem) specific for XD (catalog number Hs00166010_m1) following the manufacturer's recommendations. The reaction was carried out in a Rotor-Gene™ 6000 instrument (Corbett Life Science). Results were normalized to 18S rRNA (sense, 5′-CGC CGC TAG AGG TGA AAT TCT-3′; antisense, 5′-CAT TCT TGG CAA ATG CTT TCG-3′ 300 nm each, probe, 5′-FAM ACC GGC GCA AGA CGG ACC AGA TAMRA-3′, 175 nm) and expressed as XD mRNA relative levels as compared with nonsilencing siRNA-transfected cells by using the RotorGene software (Corbett Research) and the two standard curve methods for relative quantitation (42).
High Performance Liquid Chromatography (HPLC) Analysis of Intracellular GSH
A549 cells were cultured at 85% confluence, incubated with RV16, medium alone, or f-RV for different periods (20 min to 1 h), then trypsinized, collected, and harvested in cryovials with 1.2 ml of 3% metaphosphoric acid in sterile conditions to avoid GSH oxidation and finally frozen in liquid nitrogen until used. Cell homogenate was obtained and protein content was determined as previously described. Intracellular GSH concentration was evaluated by HPLC in a Kontron Instruments apparatus (Milan, Italy) equipped with a C18 hydrophobic column (5 μm particle size, 4.6 × 250 mm), a 420 pump (range 0.005-10 ml/min), a 425 gradient former, and an injection valve with a 20-μl sampling loop. Elution was carried out at room temperature in isocratic gradient (75% methanol and KH2PO4 buffer, pH 3, 1 ml/min speed). GSH was analyzed at 200 nm by a 432 UV detector (Kontron) with an IBM integrated software PC Pack. Homogenates of cells were centrifuged at 40,000 × g for 20 min at 4 °C and the supernatant collected and concentrated on Amicon Ultra 10,000 centrifugal filter devices (Millipore, Bedford, MA) to a final volume of about 300 μl. Samples were analyzed without derivatization against standards of pure lyophilized GSH (Biomedica Foscama), diluted in 1 ml of normal saline solution. The final concentration was obtained by serial dilutions of the lyophilized product. In selected experiments cells were pretreated, before infection, as previously described, for 12 h with GSH, H2S, (0.25 to 2.5 mm), or 4 h with oxypurinol (20 μm). Where indicated, a 4-h pretreatment with protease inhibitors (PMSF, 0.625 μm; Leu, 1.25 μm; Pep, 1.25 μm; Apr, 1.25 μm; Phe, 1.25 μm; E-64, 1.25 μm) or diluent was performed. All protease inhibitors were removed immediately before infection.
Rhinovirus Replication
Titration Assay in a Sensitive Cell Line—Rhinovirus replication was evaluated by titration assay in a sensitive cell line (HeLa) (9). Cells were seeded in a 96-well plate. Where indicated, subconfluent cells were treated with the highest concentration used in the study for each of the tested compounds for the time intervals previously specified (4 h for Leu, Pep, PMSF, Apr, E-64, oxypurinol; 12 h for H2S and GSH) or diluent alone before the infection. Cells were exposed for 1 h to 10-fold serial dilution of RV16, from not diluted down to 10-8 (4 wells per condition). After a 1-h infection, virus unbound to cultured cells was removed and fresh medium added. The cells were incubated in 4% minimal essential medium (Invitrogen) at 37 °C for 5 days, fixed in methanol, and stained with 0.1% crystal violet. The cytopathic effect was evaluated by visual assessment and assessment of the continuity of the monolayer. For each experiment TCID50/ml values were calculated (27).
TaqMan® Real-time PCR—A549 cells were seeded in 6-well plates at 1.7 × 105 cells/ml. Where indicated, subconfluent cells were treated with the highest concentration used in the study for each of the tested compounds for the time intervals previously specified (4 h for Leu, Pep, PMSF, Apr, E-64, and oxypurinol; 12 h for H2S and GSH) or diluent alone before the infection. Cell lysates were harvested at 4 and 8 h following the infection. Total RNA was extracted from cell lysates by using a commercially available kit (RNeasy Kit, Qiagen) following the manufacturer's recommendations. Viral RNA in cell lysates was measured by TaqMan RT-PCR. For this purpose 2 μg of total RNA were used for cDNA synthesis (Omniscript RT kit, Qiagen). TaqMan quantitative PCR was carried out using primers and probe for rhinovirus (sense, 5′-GTG AAG AGC CSC RTG TGC T-3′ 50 nm; antisense, 5′-GCT SCA GGG TTA AGG TTA GCC-3′ 300 nm; probe, 5′-FAM-TGA GTC CTC CGG CCC CTG AAT G-TAMRA-3′, 175 nm) and 18S rRNA (see above) (15). Reactions consisted of 2 μl of cDNA (cDNA for 18S was diluted 1:100), 12.5 μl of 2× QuantiTect Probe PCR Master Mix (Qiagen), primers, and probes at the final concentrations listed above and RNase-free water to a total volume of 25 μl. Reactions were performed on a Rotor-Gene™ 6000 instrument (Corbett Life Science). Viral RNA expressions were normalized to 18S rRNA and compared with standard curves and expressed as copies per μg of RNA.
Enzyme-linked Immunosorbent Assays for Chemokines
Subconfluent A549 cells were pretreated for 4 h with oxypurinol (20 μm) before RV16 or f-RV inoculum or medium alone treatment. After a 1-h infection unbounded virus was removed and fresh medium added. Supernatants were harvested at 4 h and levels of IL-8 and GRO-α were assessed using commercially available enzyme-linked immunosorbent assay kits (R&D System) following the manufacturer's instructions. Detection limits for IL-8 and GRO-α enzyme-linked immunosorbent assay were ∼10 and 15 pg/ml, respectively.
NF-κB Transcription Factor Activation
Nuclear extracts were prepared from A549 cells using the Nuclear Extract Kit (Active Motif). NF-κB activation was assessed in A549 cell nuclear extracts using the TransAM™ p65 Transcription Factor Assay Kit (Active Motif) following the manufacturer's recommendations. Nuclear extract of Jurkat cells provided by the manufacturer (Active Motif) were used as positive controls.
Statistical Analysis
Group data were expressed as mean ± S.E. Analysis of variance was used to determine differences between groups. Paired or unpaired Student's t tests were performed after the analysis of variance when appropriate. All experiments were carried out at least 5 times. Bonferroni adjustment was applied where indicated. A probability value of <0.05 was considered significant.
RESULTS
Rhinovirus-induced 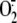 Production in Respiratory Epithelial Cells Is Cytosolic Not Membrane
Associated—
Production in Respiratory Epithelial Cells Is Cytosolic Not Membrane
Associated—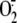 production was evaluated by SOD-inhibitable cytochrome c reduction
kinetics. In our previous study we found that RV16 infection rapidly induced
intracellular
production was evaluated by SOD-inhibitable cytochrome c reduction
kinetics. In our previous study we found that RV16 infection rapidly induced
intracellular 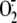 production,
which was maximal at 1 h in A549 cells, a type II respiratory epithelial cell
line (24,
35). That study evaluated
intracellular
production,
which was maximal at 1 h in A549 cells, a type II respiratory epithelial cell
line (24,
35). That study evaluated
intracellular 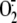 generation,
i.e. cell membranes were precipitated before SOD-inhibitable
cytochrome c reduction assay was performed. Because other workers had
implicated NADPH oxidase in RV induction of reactive oxygen species
(23) and because NADPH oxidase
is a membrane bound system, we first sought to identify the cellular site of
generation,
i.e. cell membranes were precipitated before SOD-inhibitable
cytochrome c reduction assay was performed. Because other workers had
implicated NADPH oxidase in RV induction of reactive oxygen species
(23) and because NADPH oxidase
is a membrane bound system, we first sought to identify the cellular site of
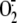 production. In the search
of cellular sources of RV16-induced
production. In the search
of cellular sources of RV16-induced
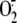 production, we first
confirmed our previous findings of rapid induction of
production, we first
confirmed our previous findings of rapid induction of
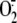 by RV16 in membrane-free
cytosolic fractions (Fig.
1A). We next investigated total cell homogenates, which
included cell membranes (Fig.
1B) and found that
by RV16 in membrane-free
cytosolic fractions (Fig.
1A). We next investigated total cell homogenates, which
included cell membranes (Fig.
1B) and found that
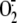 production was again
rapidly increased after RV16 infection, detectable at 20-120 min in A549 total
cell homogenates. Because
production was again
rapidly increased after RV16 infection, detectable at 20-120 min in A549 total
cell homogenates. Because 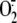 production (normalized per mg of protein) was almost doubled in the absence of
cell membranes (see y axes of Fig.
1, A and B), whereas protein quantities were
halved (14.8 ± 1.9 mg/experiment versus 32.1 ± 3.1
mg/experiment, in the absence and presence of cell membrane, respectively), we
reasoned that the vast majority, if not all of RV16 early production of
production (normalized per mg of protein) was almost doubled in the absence of
cell membranes (see y axes of Fig.
1, A and B), whereas protein quantities were
halved (14.8 ± 1.9 mg/experiment versus 32.1 ± 3.1
mg/experiment, in the absence and presence of cell membrane, respectively), we
reasoned that the vast majority, if not all of RV16 early production of
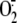 came from the cytosolic
fraction. To investigate membrane production, direct
came from the cytosolic
fraction. To investigate membrane production, direct
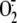 production was evaluated
in cell membranes obtained from A549 epithelial cells infected with RV16. No
induction was observed (p = 0.82 versus unstimulated
controls) at any time point (20, 60, 120, 180, 240, 300, 360, 420, and 480
min), indicating that the cytosol, not the membrane, is the site of RV16
induced
production was evaluated
in cell membranes obtained from A549 epithelial cells infected with RV16. No
induction was observed (p = 0.82 versus unstimulated
controls) at any time point (20, 60, 120, 180, 240, 300, 360, 420, and 480
min), indicating that the cytosol, not the membrane, is the site of RV16
induced 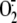 production.
production.
FIGURE 1.

Rhinoviruses-induced production of superoxide anion
(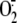 ) in A549 respiratory
epithelial cells and HBEC. The production is independent of the NADPH
oxidase system. In panels A, B, and E, confluent cells were
exposed to live RV16 (closed circles), medium alone (open
circles), or RV16 physically removed by filtration (f-RV, dash line,
diamonds) for different time intervals (20 min to 8 h).
) in A549 respiratory
epithelial cells and HBEC. The production is independent of the NADPH
oxidase system. In panels A, B, and E, confluent cells were
exposed to live RV16 (closed circles), medium alone (open
circles), or RV16 physically removed by filtration (f-RV, dash line,
diamonds) for different time intervals (20 min to 8 h).
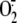 production was evaluated
in cytosolic fractions of A549 cells (A, n = 7) and HBEC (E,
n = 5) and in total A549 cell homogenates (B, n = 7) by
SOD-inhibitable cytochrome c reduction kinetics.
C,
production was evaluated
in cytosolic fractions of A549 cells (A, n = 7) and HBEC (E,
n = 5) and in total A549 cell homogenates (B, n = 7) by
SOD-inhibitable cytochrome c reduction kinetics.
C,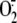 production by
a mixture of cytosolic and membrane fraction from A549 cells was evaluated at
different intervals following exposure to live RV16 (black circles),
RV16 physically removed by filtration (open circles), medium alone
(diamonds), without (continuous line) or with (dashed
line) NADPH added to the reaction mixture. The addition of NADPH did not
affect RV16-induced cytosol
production by
a mixture of cytosolic and membrane fraction from A549 cells was evaluated at
different intervals following exposure to live RV16 (black circles),
RV16 physically removed by filtration (open circles), medium alone
(diamonds), without (continuous line) or with (dashed
line) NADPH added to the reaction mixture. The addition of NADPH did not
affect RV16-induced cytosol
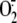 production (n =
5). D,
production (n =
5). D, 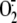 production was not changed when a specific inhibitor of NADPH oxidase
(diphenylene iodonium chloride) was added to the reaction mixture (RV16
physically removed by filtration (open circles), medium alone
(diamonds) or live RV16 infection (black circles), without
(continuous line) or with (dashed line) NADPH) confirming
lack of involvement of NADPH oxidase (n = 5). F and
G,
production was not changed when a specific inhibitor of NADPH oxidase
(diphenylene iodonium chloride) was added to the reaction mixture (RV16
physically removed by filtration (open circles), medium alone
(diamonds) or live RV16 infection (black circles), without
(continuous line) or with (dashed line) NADPH) confirming
lack of involvement of NADPH oxidase (n = 5). F and
G, 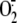 production
evaluated in the cytosolic fraction of HBEC (F, n = 5)) and A549
cells (G, n = 7) exposed to live RV1B (closed circles),
medium alone (open circles), or RV1B physically removed by filtration
(dash line, diamonds). Newly generated
production
evaluated in the cytosolic fraction of HBEC (F, n = 5)) and A549
cells (G, n = 7) exposed to live RV1B (closed circles),
medium alone (open circles), or RV1B physically removed by filtration
(dash line, diamonds). Newly generated
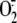 was measured in each
sample and expressed as micromolar and normalized per mg of protein
(***, p < 0.001; *, p < 0.05
compared with medium alone treated cells and f-RV inoculated cells).
was measured in each
sample and expressed as micromolar and normalized per mg of protein
(***, p < 0.001; *, p < 0.05
compared with medium alone treated cells and f-RV inoculated cells).
Rhinovirus Induction of
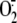 Is Independent of the
NADPH Oxidase System—The fact that
Is Independent of the
NADPH Oxidase System—The fact that
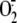 production occurs in
cytosolic fractions and not cell membranes suggests that a membrane-bound
enzyme system such as NADPH oxidase is not involved, however, this finding
does not rule out such a possibility. We therefore investigated whether
rhinovirus-induced
production occurs in
cytosolic fractions and not cell membranes suggests that a membrane-bound
enzyme system such as NADPH oxidase is not involved, however, this finding
does not rule out such a possibility. We therefore investigated whether
rhinovirus-induced 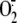 production could derive from the activity of NADPH oxidase in experimental
conditions where both the cytosolic and membrane fractions of the homogenates
were pooled together. The membrane fractions were added, together with the
substrate of NADPH oxidase, NADPH, in the presence and absence of an inhibitor
of the NADPH enzyme system, diphenylene iodonium chloride
(38). NADPH addition would
increase
production could derive from the activity of NADPH oxidase in experimental
conditions where both the cytosolic and membrane fractions of the homogenates
were pooled together. The membrane fractions were added, together with the
substrate of NADPH oxidase, NADPH, in the presence and absence of an inhibitor
of the NADPH enzyme system, diphenylene iodonium chloride
(38). NADPH addition would
increase 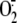 production if
production is NADPH oxidase-dependent. We found that both the addition of
NADPH (Fig. 1C) and
diphenylene iodonium chloride (Fig.
1D) to the reaction mixture did not change
production if
production is NADPH oxidase-dependent. We found that both the addition of
NADPH (Fig. 1C) and
diphenylene iodonium chloride (Fig.
1D) to the reaction mixture did not change
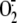 production induced by
RV16 infection, indicating that the NADPH oxidase system is not relevant to
early
production induced by
RV16 infection, indicating that the NADPH oxidase system is not relevant to
early 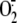 generation induced
by RV16. Thus, from now on, intracellular
generation induced
by RV16. Thus, from now on, intracellular
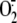 production and
intracellular redox equilibrium will be solely evaluated in cytosolic
fractions.
production and
intracellular redox equilibrium will be solely evaluated in cytosolic
fractions.
Rhinovirus Induction of
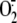 in Primary Bronchial
Epithelial Cells Is Virus-specific and Serotype/Receptor
Independent—We previously demonstrated RV induction of
in Primary Bronchial
Epithelial Cells Is Virus-specific and Serotype/Receptor
Independent—We previously demonstrated RV induction of
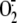 in A549 cells was
virus-specific (24) and
confirmed this in the present study, as an inoculum from which virus had been
removed by molecular weight filtration (f-RV16) did not induce any
in A549 cells was
virus-specific (24) and
confirmed this in the present study, as an inoculum from which virus had been
removed by molecular weight filtration (f-RV16) did not induce any
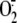 production
(Fig. 1, A and
B). We next wished to investigate RV induction of
production
(Fig. 1, A and
B). We next wished to investigate RV induction of
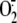 in primary bronchial
epithelial cells, as we had only previously demonstrated this at a single time
point of 20 min. Similar results were found in primary HBEC obtained by
bronchial brushing from healthy volunteers, with induction being significant
at 20 and 120 min and peaking at 60 min
(Fig. 1E). To
investigate whether these results were confined to the major RV group, to
which RV16 belongs, which binds ICAM-1 as surface receptor
(43,
44), experiments were
performed using RV1B of the minor RV group, which uses members of the low
density lipoprotein receptor family. The fact that following RV1B infection
the kinetics of
in primary bronchial
epithelial cells, as we had only previously demonstrated this at a single time
point of 20 min. Similar results were found in primary HBEC obtained by
bronchial brushing from healthy volunteers, with induction being significant
at 20 and 120 min and peaking at 60 min
(Fig. 1E). To
investigate whether these results were confined to the major RV group, to
which RV16 belongs, which binds ICAM-1 as surface receptor
(43,
44), experiments were
performed using RV1B of the minor RV group, which uses members of the low
density lipoprotein receptor family. The fact that following RV1B infection
the kinetics of 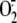 production both in the A549 cell line (Fig.
1F) and HBEC (Fig.
1G) were not different to those observed with RV16
indicates that the effect is receptor independent.
production both in the A549 cell line (Fig.
1F) and HBEC (Fig.
1G) were not different to those observed with RV16
indicates that the effect is receptor independent.
Rhinovirus Induces 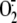 Production via the XD/XO Enzyme System—To confirm that
Production via the XD/XO Enzyme System—To confirm that
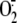 is generated via the
XD/XO enzyme system, we next investigated RV induction of uric acid, the other
product, besides
is generated via the
XD/XO enzyme system, we next investigated RV induction of uric acid, the other
product, besides 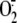 , of XO
degradation of the purine base xanthine. These experiments were conducted at 1
h after infection, i.e. at the peak of
, of XO
degradation of the purine base xanthine. These experiments were conducted at 1
h after infection, i.e. at the peak of
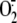 generation.
Fig. 2A shows the
kinetics of uric acid production, after addition of substrate xanthine, in
cytosolic fractions of A549 cell homogenates after exposure for 1 h to RV16,
f-RV16, or medium alone. Uric acid was detected only in RV16-infected samples.
Its identification was confirmed by addition, after 15 min of the kinetic
assay, of uricase, which degrades uric acid to allantoin
(36,
37). As expected, uric acid
was rapidly degraded by uricase confirming that the XD/XO system was activated
by rhinovirus infection (Fig.
2A). Similar findings were observed in homogenate samples
obtained from HBEC, in the same experimental conditions
(Fig. 2B), confirming
rhinovirus activation of XD/XO in primary cells. The fact that following RV1B
infection the kinetics of uric acid production were identical
(Fig. 2C) to those
observed with RV16 (Fig.
2A) indicates that the effect is receptor independent. To
further confirm the role of the XD/XO enzyme system in
generation.
Fig. 2A shows the
kinetics of uric acid production, after addition of substrate xanthine, in
cytosolic fractions of A549 cell homogenates after exposure for 1 h to RV16,
f-RV16, or medium alone. Uric acid was detected only in RV16-infected samples.
Its identification was confirmed by addition, after 15 min of the kinetic
assay, of uricase, which degrades uric acid to allantoin
(36,
37). As expected, uric acid
was rapidly degraded by uricase confirming that the XD/XO system was activated
by rhinovirus infection (Fig.
2A). Similar findings were observed in homogenate samples
obtained from HBEC, in the same experimental conditions
(Fig. 2B), confirming
rhinovirus activation of XD/XO in primary cells. The fact that following RV1B
infection the kinetics of uric acid production were identical
(Fig. 2C) to those
observed with RV16 (Fig.
2A) indicates that the effect is receptor independent. To
further confirm the role of the XD/XO enzyme system in
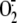 production we performed
experiments in the presence of oxypurinol, a permanent inactivator of the
oxidase form of the enzyme (XO), the only form of the enzyme able to produce
production we performed
experiments in the presence of oxypurinol, a permanent inactivator of the
oxidase form of the enzyme (XO), the only form of the enzyme able to produce
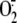 (28). At 1 h infection
RV16-induced cytosol
(28). At 1 h infection
RV16-induced cytosol 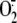 production was completely quenched when A549 cells were pretreated with
oxypurinol (Fig. 2D).
Similar findings were observed in homogenate samples obtained from HBEC in the
same experimental conditions (Fig.
2E), as in both cases oxypurinol reduced
production was completely quenched when A549 cells were pretreated with
oxypurinol (Fig. 2D).
Similar findings were observed in homogenate samples obtained from HBEC in the
same experimental conditions (Fig.
2E), as in both cases oxypurinol reduced
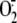 production to levels
observed with medium or diluent alone or inactivated virus.
production to levels
observed with medium or diluent alone or inactivated virus.
FIGURE 2.

XO involvement in rhinovirus-induced cytosolic superoxide anion
(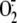 ) production.
A (n = 5) and B (n = 5), uric acid
production kinetics in confluent A549 cells and HBEC, respectively. Cells were
exposed to live RV16 (squares), RV16 physically removed by filtration
(triangles), or medium alone (open circles). The
arrow indicates when uricase was added to the system. Newly generated
uric acid was measured spectophotometrically in each sample and expressed as
micromolar and normalized per mg of protein. C, uric acid production
in A549 exposed to minor group rhinovirus RV1B (live RV1B (squares),
RV1B physically removed by filtration (triangles), or medium alone
(open circles)) (n = 5). D (n = 5) and
E (n = 5), A549 cells (D) and HBEC (E)
were exposed for 1 h to live RV16 (RV16), RV16 physically removed by
filtration (f-RV16), medium alone (Medium), or diluent alone
(Diluent). Where indicated cells were pretreated 4 h before the
infection with oxypurinol and than exposed for 1 h to medium alone
(Oxy) or RV16 (Oxy+RV16) (***,
p < 0.001 versus medium, diluent, f-RV16, Oxy-treated
cells and versus RV16-infected cells pretreated with Oxy).
) production.
A (n = 5) and B (n = 5), uric acid
production kinetics in confluent A549 cells and HBEC, respectively. Cells were
exposed to live RV16 (squares), RV16 physically removed by filtration
(triangles), or medium alone (open circles). The
arrow indicates when uricase was added to the system. Newly generated
uric acid was measured spectophotometrically in each sample and expressed as
micromolar and normalized per mg of protein. C, uric acid production
in A549 exposed to minor group rhinovirus RV1B (live RV1B (squares),
RV1B physically removed by filtration (triangles), or medium alone
(open circles)) (n = 5). D (n = 5) and
E (n = 5), A549 cells (D) and HBEC (E)
were exposed for 1 h to live RV16 (RV16), RV16 physically removed by
filtration (f-RV16), medium alone (Medium), or diluent alone
(Diluent). Where indicated cells were pretreated 4 h before the
infection with oxypurinol and than exposed for 1 h to medium alone
(Oxy) or RV16 (Oxy+RV16) (***,
p < 0.001 versus medium, diluent, f-RV16, Oxy-treated
cells and versus RV16-infected cells pretreated with Oxy).
Rhinovirus Induces Proteolytic Activation of XD/XO Enzymatic
System—In other experimental systems, the XD/XO enzymatic system is
able to produce 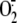 when it
is converted to the oxidase form by proteolytic activity, exerted by a serine
protease, which partially hydrolyzes the enzyme to its active form
(40,
45). To investigate the
mechanisms of rhinovirus induction of XD/XO, cells were next infected in the
presence or absence of serine protease inhibitors, or with cysteine or
metalloprotease inhibitors as controls. RV16 infection (20 min to 1 h) failed
to generate
when it
is converted to the oxidase form by proteolytic activity, exerted by a serine
protease, which partially hydrolyzes the enzyme to its active form
(40,
45). To investigate the
mechanisms of rhinovirus induction of XD/XO, cells were next infected in the
presence or absence of serine protease inhibitors, or with cysteine or
metalloprotease inhibitors as controls. RV16 infection (20 min to 1 h) failed
to generate 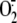 in the
cytosol when epithelial cells were pretreated for 4 h with protease inhibitors
PMSF, leupeptin (Leu), pepstatin (Pep), or aprotinin (Apr), which all act as
serine protease inhibitors (Fig.
3A). Serine protease involvement was confirmed as neither
the metalloprotease inhibitor phenanthroline (Phe,
Fig. 3B) nor the
cysteine protease inhibitor E-64 (Fig.
3C)
(29-34)
had any effect on rhinovirus induction of
in the
cytosol when epithelial cells were pretreated for 4 h with protease inhibitors
PMSF, leupeptin (Leu), pepstatin (Pep), or aprotinin (Apr), which all act as
serine protease inhibitors (Fig.
3A). Serine protease involvement was confirmed as neither
the metalloprotease inhibitor phenanthroline (Phe,
Fig. 3B) nor the
cysteine protease inhibitor E-64 (Fig.
3C)
(29-34)
had any effect on rhinovirus induction of
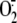 . This data confirm the
involvement of specific proteolytic mechanisms mediated by serine proteases in
rhinovirus-induced activation of XO. To directly evaluate the effects of RV
infection on XD/XO proteins, we performed Western blotting analysis using an
antibody able to recognize XD/XO expression. The 145-kDa band corresponding to
full-length XD was significantly reduced at 30 min (2-fold; p <
0.05) and at 1 h (4-fold; p < 0.01) after RV16 infection as
compared with uninfected control cells (p < 0.05). Such a
reduction was paralleled by 2-fold increased expression of the 85-kDa band,
containing the active site, at 1 h infection (p < 0.05)
(Fig. 4, A and
B). By measuring the XD/XO ratio we found a progressive
significant reduction at 30 min and 1 h after RV16 infection as compared with
uninfected control cells (3-fold; p < 0.05 and 10-fold; p
< 0.01, respectively) (Fig.
4C). Moreover 4-h serine-protease inhibitor Apr, but not
4-h cysteine-inhibitor E64 pretreatment, abolished XD cleavage
(Fig. 4, A-C).
. This data confirm the
involvement of specific proteolytic mechanisms mediated by serine proteases in
rhinovirus-induced activation of XO. To directly evaluate the effects of RV
infection on XD/XO proteins, we performed Western blotting analysis using an
antibody able to recognize XD/XO expression. The 145-kDa band corresponding to
full-length XD was significantly reduced at 30 min (2-fold; p <
0.05) and at 1 h (4-fold; p < 0.01) after RV16 infection as
compared with uninfected control cells (p < 0.05). Such a
reduction was paralleled by 2-fold increased expression of the 85-kDa band,
containing the active site, at 1 h infection (p < 0.05)
(Fig. 4, A and
B). By measuring the XD/XO ratio we found a progressive
significant reduction at 30 min and 1 h after RV16 infection as compared with
uninfected control cells (3-fold; p < 0.05 and 10-fold; p
< 0.01, respectively) (Fig.
4C). Moreover 4-h serine-protease inhibitor Apr, but not
4-h cysteine-inhibitor E64 pretreatment, abolished XD cleavage
(Fig. 4, A-C).
FIGURE 3.

Effects of protease inhibitors on rhinovirus-induced cytosolic
superoxide anion (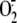 )
production. A, effects of serine protease inhibitors PMSF,
leupeptin (Leu), pepstatin (Pep), aprotinin (Apr)
on RV16-induced cytosolic
)
production. A, effects of serine protease inhibitors PMSF,
leupeptin (Leu), pepstatin (Pep), aprotinin (Apr)
on RV16-induced cytosolic 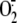 production in A549 cells (n = 5). B (n = 7) and
C (n = 6), effects of metalloprotease inhibitor
phenanthroline (Phe)(B) or cysteine protease inhibitor E-64
(C) on RV16-induced cytosolic
production in A549 cells (n = 5). B (n = 7) and
C (n = 6), effects of metalloprotease inhibitor
phenanthroline (Phe)(B) or cysteine protease inhibitor E-64
(C) on RV16-induced cytosolic
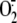 production in A549 cells.
In A-C, where indicated (+) cells were pretreated for 4 h with
protease inhibitors, then exposed, for 20 min (empty bars) or 1 h
(filled bars) to RV16 or medium alone (in A, ***
p < 0.001 versus all other conditions; B and
C, *** p < 0.001 versus medium alone
treated cells with or without inhibitor pre-treatment).
production in A549 cells.
In A-C, where indicated (+) cells were pretreated for 4 h with
protease inhibitors, then exposed, for 20 min (empty bars) or 1 h
(filled bars) to RV16 or medium alone (in A, ***
p < 0.001 versus all other conditions; B and
C, *** p < 0.001 versus medium alone
treated cells with or without inhibitor pre-treatment).
FIGURE 4.

Effect of rhinovirus on the XD/XO system and effect of XD knock-down on rhinovirus-induced superoxide anion production. A-C, Western blotting analysis of the XD/XO system. Where indicated (+) A549 cells were pretreated 4 h with protease inhibitors (cysteine protease inhibitor E-64 or the serine protease inhibitor aprotinin (Apr)), then exposed, for 30 min or 1 h to RV16 or medium alone. A, full-length XD (145 kDa band) and the 85-kDa band containing the C-terminal active site of the enzyme are shown on a representative film. Actin is shown as loading control. B and C, densitometric analyses of Western blotting assays are represented (n = 3, *, p < 0.05 versus medium alone treated cells). D, XD mRNA expression in A549 cells transfected with medium (white bars), GAPDH siRNA (gray bars), and XD siRNA (black bars)(n = 3, *, p < 0.05 versus all corresponding conditions). E, superoxide anion production following a 1-h RV16 infection in A549 cells transfected with medium alone (white bar), nonsilencing siRNA (bright gray bar), GAPDH siRNA (dark gray bar), and XD siRNA (black bar)(n = 3, *, p < 0.05 versus RV16-infected cells in all other experimental conditions).
XD Knockdown Reduces Rhinovirus-induced
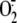 Production—To
further confirm the role of the XD/XO system in rhinovirus-induced
Production—To
further confirm the role of the XD/XO system in rhinovirus-induced
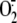 production, we performed
experiments in which XD expression in A549 cells was knocked down by siRNA.
Transfection of XD siRNAs resulted in marked suppression of XD mRNA expression
as compared with scrambled siRNA-transfected cells
(Fig. 4D), with a peak
of inhibition at 24 h. At this time point, XD knockdown suppressed
RV16-induced
production, we performed
experiments in which XD expression in A549 cells was knocked down by siRNA.
Transfection of XD siRNAs resulted in marked suppression of XD mRNA expression
as compared with scrambled siRNA-transfected cells
(Fig. 4D), with a peak
of inhibition at 24 h. At this time point, XD knockdown suppressed
RV16-induced 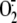 production
spectrophotometrically measured by SOD-inhibitable cytochrome c
reduction kinetics (Fig.
4E). Control siRNAs had no effect either on XD mRNA
expression (Fig. 4D)
or on RV16-induced
production
spectrophotometrically measured by SOD-inhibitable cytochrome c
reduction kinetics (Fig.
4E). Control siRNAs had no effect either on XD mRNA
expression (Fig. 4D)
or on RV16-induced 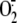 production (Fig.
4E).
production (Fig.
4E).
Rhinovirus Depletes Intracellular Reduced GSH—To investigate
the consequences of RV-induced
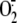 production on
intracellular redox equilibrium, we evaluated whether the concentration of the
intracellular reducing agent GSH is modified by RV16 infection. In A549 cells,
as the duration of RV16 infection increased, endogenous stores of GSH were
progressively reduced and complete depletion was observed at 1 h after
infection (Fig. 5A).
Similar results were found with RV1B (Fig.
5B). The HPLC absorbance peak for GSH is representatively
shown for medium-treated cells in Fig.
5C and depletion of the HLPC peak in RV16-infected cells
is representatively shown in Fig.
5D. As observed with
production on
intracellular redox equilibrium, we evaluated whether the concentration of the
intracellular reducing agent GSH is modified by RV16 infection. In A549 cells,
as the duration of RV16 infection increased, endogenous stores of GSH were
progressively reduced and complete depletion was observed at 1 h after
infection (Fig. 5A).
Similar results were found with RV1B (Fig.
5B). The HPLC absorbance peak for GSH is representatively
shown for medium-treated cells in Fig.
5C and depletion of the HLPC peak in RV16-infected cells
is representatively shown in Fig.
5D. As observed with
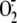 induction, RV16-induced
depletion of intracellular GSH at 1 h after infection was completely inhibited
when A549 cells were pretreated with either oxypurinol or serine protease
inhibitors, but not with metalloprotease inhibitor phenanthroline (Phe) or
cysteine protease inhibitor (E-64) (Fig.
5E). Thus, RV16-induced depletion of intracellular GSH
occurs via XO activation and subsequent cytosol
induction, RV16-induced
depletion of intracellular GSH at 1 h after infection was completely inhibited
when A549 cells were pretreated with either oxypurinol or serine protease
inhibitors, but not with metalloprotease inhibitor phenanthroline (Phe) or
cysteine protease inhibitor (E-64) (Fig.
5E). Thus, RV16-induced depletion of intracellular GSH
occurs via XO activation and subsequent cytosol
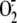 production. The same
findings were also observed when A549 cells were infected with minor group
RV1B (Fig. 5F), to
indicate that the effect was receptor independent.
production. The same
findings were also observed when A549 cells were infected with minor group
RV1B (Fig. 5F), to
indicate that the effect was receptor independent.
FIGURE 5.

Effect of RV infection on intracellular reduced GSH concentration in A549 and modulation by oxypurinol or protease inhibitors. Intracellular GSH was evaluated by HPLC in A549 cells incubated with RV16 (A, continuous line), RV1B (B, continuous line), or medium alone (A and B, dashed line) for 20 min to 1 h (n = 5, ***, p < 0.001 versus control samples). C, representative HPLC profiles of intracellular GSH detection in A549 cells treated for 1 h with medium alone. D, representative HPLC profiles of intracellular GSH detection in A549 cells treated for 1 h with RV16. In panels C and D an arrow indicates the position of the GSH peak. E (n = 5), HPLC evaluation of intracellular GSH in A549 cells exposed for 1 h to medium alone (white bar), live RV16 (RV16), or RV16 physically removed by filtration (f-RV16, light gray bar). Where indicated A549 cells were pretreated for 4 h with oxypurinol (Oxy) or protease inhibitors PMSF, Leu, Pep, Apr, Phe, and E-64, then exposed for 1 h to medium alone (gray bars) or live RV16 (black bars). In panel F (n = 5) A549 cells were exposed to RV1B (***, p < 0.001).
Increasing Intracellular GSH Inhibits Rhinovirus-induced Intracellular
GSH Depletion and 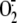 Production—Because reduction of intracellular reducing power is
“per se” a known mechanism of activation of XO
(46,
47), we next investigated
whether by increasing intracellular GSH with the reducing agents
H2S or exogenous GSH, we could block the activation of XO induced
by RV infection. We first showed that pretreatment of A549 cells with
H2S and exogenous GSH increased intracellular GSH levels in a
dose-dependent manner (Fig.
6A). A549 cells were then pretreated for 12 h with 2
mm H2S to enhance intracellular GSH
(Fig. 6B) and then
infected for 1 h with RV16. No reduction of endogenous GSH was observed after
a 1-h RV16 infection (Fig.
6C), confirming that enhancing intracellular GSH
protected cells against virus-induced GSH depletion. Similar protection was
observed with 10 mm exogenous GSH (data not shown) treatment before
the infection. Pretreatment with either 2 mm H2S or 10
mm exogenous GSH, not only increased intracellular GSH levels but
also completely inhibited RV16 induced
Production—Because reduction of intracellular reducing power is
“per se” a known mechanism of activation of XO
(46,
47), we next investigated
whether by increasing intracellular GSH with the reducing agents
H2S or exogenous GSH, we could block the activation of XO induced
by RV infection. We first showed that pretreatment of A549 cells with
H2S and exogenous GSH increased intracellular GSH levels in a
dose-dependent manner (Fig.
6A). A549 cells were then pretreated for 12 h with 2
mm H2S to enhance intracellular GSH
(Fig. 6B) and then
infected for 1 h with RV16. No reduction of endogenous GSH was observed after
a 1-h RV16 infection (Fig.
6C), confirming that enhancing intracellular GSH
protected cells against virus-induced GSH depletion. Similar protection was
observed with 10 mm exogenous GSH (data not shown) treatment before
the infection. Pretreatment with either 2 mm H2S or 10
mm exogenous GSH, not only increased intracellular GSH levels but
also completely inhibited RV16 induced
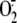 production at 1 h after
infection, as assessed by SOD-inhibitable cytochrome c reduction
assay (Fig. 6D). In
these experiments uric acid production was similarly suppressed
(Fig. 6E). Thus, the
results indicated that increasing GSH intracellular storage with exogenous GSH
or H2S completely inhibited rhinovirus-induced XO activation and
production at 1 h after
infection, as assessed by SOD-inhibitable cytochrome c reduction
assay (Fig. 6D). In
these experiments uric acid production was similarly suppressed
(Fig. 6E). Thus, the
results indicated that increasing GSH intracellular storage with exogenous GSH
or H2S completely inhibited rhinovirus-induced XO activation and
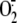 production.
production.
FIGURE 6.

Effects of exogenous H2S and GSH pre-treatment on endogenous GSH concentration and on RV16 superoxide anion and uric acid production. A, effects of increasing doses of reducing agents (H2S (closed circles) or GSH (open circles)) on intracellular concentration of GSH in A549 cells (n = 5). B, representative HPLC profiles of intracellular GSH detection in control uninfected A549 cells pretreated with 2 mm exogenous H2S. C, representative HPLC profile of intracellular GSH detection in A549 cells pretreated with 2 mm exogenous H2S and infected with RV16. In panels B and C an arrow indicates the GSH peak. D, A549 cells were infected for 1 h with RV16 or with or without 12 h pretreatment with 2 mm H2S(H2S) or 10 mm exogenous GSH (GSH). Superoxide anion production was assessed with SOD-inhibitable cytochrome c reduction assay (n = 5, ***, p < 0.001). E, uric acid production kinetics in confluent A549 cells exposed to RV16 infection with diluent (squares), 2 mm H2S(open circles), or 10 mm GSH (triangles) with a 12-h pre-treatment. The arrow indicates when uricase was added to the system. Newly generated uric acid was measured spectrophotometrically in each sample and expressed as micromolar and normalized per mg of protein (n = 5).
XO Inhibition Reduces Rhinovirus-induced Chemokine
Production—RV infection of bronchial epithelial cells induces the
expression of several proinflammatory cytokines, including many that are
involved in neutrophil chemoattraction and activation (i.e. IL-8 and
Gro-α). Neutrophil inducing cytokines are not effectively suppressed by
the currently available asthma therapies, i.e. steroids or long
acting β agonists (10).
Because the induction of these mediators occurs through oxidative sensitive
pathways (e.g. NF-κB signaling activation
(17)), we evaluated whether
oxypurinol inhibition of XO-mediated
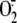 generation affects
rhinovirus-induced IL-8 and Gro-α production in A549 respiratory
epithelial cells. Significant induction of IL-8 and Gro-α was apparent
at 4 h post-RV16 infection (Fig. 6,
A and B). Oxypurinol pre-treatment significantly
reduced both IL-8 (Fig.
7A) and Gro-α RV16-induced production
(Fig. 7B), whereas
diluent had no effect. These data confirmed that inhibition of XO was
effective in suppressing rhinovirus-induced neutrophil chemokine
production.
generation affects
rhinovirus-induced IL-8 and Gro-α production in A549 respiratory
epithelial cells. Significant induction of IL-8 and Gro-α was apparent
at 4 h post-RV16 infection (Fig. 6,
A and B). Oxypurinol pre-treatment significantly
reduced both IL-8 (Fig.
7A) and Gro-α RV16-induced production
(Fig. 7B), whereas
diluent had no effect. These data confirmed that inhibition of XO was
effective in suppressing rhinovirus-induced neutrophil chemokine
production.
FIGURE 7.

Effect of oxypurinol pre-treatment on RV16-induced chemokine (IL-8 and GRO-α) production and on NF-κB transcription factor activation. A (n = 5) and B (n = 5), A549 cells were exposed for 1 h to medium alone (Medium), RV16 physically removed by filtration (f-RV16) or live RV16 (RV16). Where indicated cells were pretreated for 4 h with oxypurinol (Oxy+RV16) or diluent (Diluent+RV16) before the infection. Supernatants were harvested at 4 h and levels of IL-8 (panel A) and GRO-α (panel B) were assessed (***, p < 0.001). C, A549 cells were exposed for 20 min to RV16 with (dark gray bar) or without (black bar) 20 μm oxypurinol (Oxy) after a 4-h pretreatment and NF-κB activation was assessed in nuclear extracts (n = 3, *, p < 0.05 versus medium alone).
Effect of Rhinovirus Infection on NF-κB Activation,
Modulation by Oxypurinol—To assess whether rhinovirus infection
activates NF-κB and whether this is mediated by
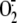 production, we measured
activated NF-κB in nuclear extracts in A549 cells following RV16
infection with or without a 4-h oxypurinol pretreatment. p65 nuclear
concentration was 40.7 ± 6.3 pg/μlin unstimulated conditions. In
accordance with previous data
(24,
26) we found that 30 min RV16
infection significantly induced p65 nuclear translocation (2-fold
versus unstimulated; p < 0.05). Four h pre-treatment with
20 μm oxypurinol significantly inhibited RV16-induced
NF-κB activation (p < 0.05 versus RV16-infected
cells not pretreated) (Fig.
7C). These data indicate that rhinovirus-induced
production, we measured
activated NF-κB in nuclear extracts in A549 cells following RV16
infection with or without a 4-h oxypurinol pretreatment. p65 nuclear
concentration was 40.7 ± 6.3 pg/μlin unstimulated conditions. In
accordance with previous data
(24,
26) we found that 30 min RV16
infection significantly induced p65 nuclear translocation (2-fold
versus unstimulated; p < 0.05). Four h pre-treatment with
20 μm oxypurinol significantly inhibited RV16-induced
NF-κB activation (p < 0.05 versus RV16-infected
cells not pretreated) (Fig.
7C). These data indicate that rhinovirus-induced
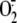 is involved in the
activation of the NF-κB-dependent pro-inflammatory pathways.
is involved in the
activation of the NF-κB-dependent pro-inflammatory pathways.
Effect of Tested Compounds on Rhinovirus Replication and
Infectivity—Control experiments were performed to assess whether
any of the antioxidant approaches used above had any antiviral activity that
could provide an alternative explanation for our findings. HeLa cells were
infected with RV16 for 1 h, i.e. until the peak of
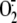 generation was reached
and intracellular GSH fully depleted, in the presence or absence of
antiproteases, oxypurinol or GSH, or H2S pretreatment for the time
intervals previously specified for each compound. The chosen concentrations
for each compound were the highest utilized in the present study. No
difference was found in the progression of RV16-induced cytopathic effects
measured on a daily basis by visual assessment (data not shown). At 5 days
there was no difference in virus yields expressed as mean TCID50/ml
values between treated and control samples for all tested compounds
(Fig. 8A). These
results were confirmed by TaqMan PCR assay showing progressive rhinovirus
replication (i.e. increased rhinovirus RNA), which was not affected
by antiprotease pretreatment (data not shown at 4 h infection; data at 8 h
represented in Fig.
8B).
generation was reached
and intracellular GSH fully depleted, in the presence or absence of
antiproteases, oxypurinol or GSH, or H2S pretreatment for the time
intervals previously specified for each compound. The chosen concentrations
for each compound were the highest utilized in the present study. No
difference was found in the progression of RV16-induced cytopathic effects
measured on a daily basis by visual assessment (data not shown). At 5 days
there was no difference in virus yields expressed as mean TCID50/ml
values between treated and control samples for all tested compounds
(Fig. 8A). These
results were confirmed by TaqMan PCR assay showing progressive rhinovirus
replication (i.e. increased rhinovirus RNA), which was not affected
by antiprotease pretreatment (data not shown at 4 h infection; data at 8 h
represented in Fig.
8B).
FIGURE 8.

Effect of tested compounds on rhinovirus replication and infectivity. Where indicated cells were treated for 4 h (oxypurinol (Oxy), serine protease inhibitor PMSF, serine and cysteine protease inhibitor Leu, aspartic protease inhibitor Pep, serine protease inhibitor Apr, metalloprotease inhibitor Phe, cysteine protease inhibitor E-64 (E64), diluent (Diluent)) or 12 h (H2S, GSH) with the highest concentration used in the study for each of the tested compounds before the infection. Rhinovirus infectivity and replication were evaluated by performing titration assays (A, n = 3) in a sensitive cell line (HeLa) and (B, n = 3) by TaqMan RT-PCR for rhinovirus RNA at 8 h after the infection.
DISCUSSION
In this study we found that
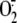 generation induced by
rhinovirus infection is initiated by proteolytic activation of the XD/XO
enzyme system. Consequently, newly generated
generation induced by
rhinovirus infection is initiated by proteolytic activation of the XD/XO
enzyme system. Consequently, newly generated
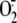 leads to progressive
depletion of intracellular GSH storage, a condition that can further activate
the XD/XO system. Inhibition of XO activity completely abolished
rhinovirus-induced
leads to progressive
depletion of intracellular GSH storage, a condition that can further activate
the XD/XO system. Inhibition of XO activity completely abolished
rhinovirus-induced 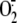 production and intracellular GSH depletion, as did a variety of serine
protease inhibitors. We also found that by enhancing intracellular GSH storage
with exogenous H2S or GSH rhinovirus infection was rendered unable
to induce
production and intracellular GSH depletion, as did a variety of serine
protease inhibitors. We also found that by enhancing intracellular GSH storage
with exogenous H2S or GSH rhinovirus infection was rendered unable
to induce 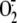 production and
to affect intracellular GSH levels.
production and
to affect intracellular GSH levels.
Several studies have described oxidant generation following respiratory virus infection both in vitro and in vivo (20, 48). In some studies a role for virus-induced oxidants in the production of inflammatory responses/mediators has been identified (49). However, the molecular mechanisms regulating generation of oxidant species by viruses in biological systems have never been fully investigated.
The findings of the present study indicate a complex mechanism of oxidant
induction following activation of XO induced by rhinovirus infection
(Fig. 9). A “vicious
circle” would represent the final scenario where activation of XO,
initiated immediately after infection via proteolysis of XD to XO, is
thereafter implemented via a non-proteolytic mechanism mediated by oxidative
consumption of intracellular reducing capacity via depletion of GSH stores.
Depletion of intracellular reducing agents is a known mechanism of activation
of XO and 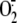 production
(46,
47). The involvement of
rhinovirus-induced oxidants in GSH consumption was confirmed by the finding
that, when XO activation was inhibited, rhinovirus infection did not result in
GSH depletion. The sequence of events represented in
Fig. 9 is supported by the
timing of the different steps involved, with
production
(46,
47). The involvement of
rhinovirus-induced oxidants in GSH consumption was confirmed by the finding
that, when XO activation was inhibited, rhinovirus infection did not result in
GSH depletion. The sequence of events represented in
Fig. 9 is supported by the
timing of the different steps involved, with
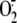 production being rapidly
induced 20 min after infection (Fig.
1), whereas GSH depletion is undetectable 20 min after infection
and thereafter progressively increases for 40 min, being complete at 60 min
after infection (Fig. 5, A and
B). The fact that in a reducing environment no uric acid
was produced and that the intracellular concentration of GSH was unchanged
after rhinovirus infection confirms the inverse relationship between GSH
intracellular concentration and XO activation. Previous studies have
documented GSH depletion following viral infections
(50), however, the underlying
mechanisms were not described.
production being rapidly
induced 20 min after infection (Fig.
1), whereas GSH depletion is undetectable 20 min after infection
and thereafter progressively increases for 40 min, being complete at 60 min
after infection (Fig. 5, A and
B). The fact that in a reducing environment no uric acid
was produced and that the intracellular concentration of GSH was unchanged
after rhinovirus infection confirms the inverse relationship between GSH
intracellular concentration and XO activation. Previous studies have
documented GSH depletion following viral infections
(50), however, the underlying
mechanisms were not described.
FIGURE 9.

Proposed mechanisms of rhinovirus-induced cytosolic super-oxide anion
(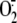 ) production.
Following rhinovirus infection xanthine oxidase is activated via proteolysis
of xanthine dehydrogenase (1).
) production.
Following rhinovirus infection xanthine oxidase is activated via proteolysis
of xanthine dehydrogenase (1).
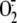 and uric acid are
thereafter produced from the enzyme substrate xanthine (2).
Intracellular oxidant production results in the depletion of intracellular
reducing capacity (including reduced glutathione depletion) (3),
which in turns induces xanthine oxidase activation via a non-proteolytic
mechanism (4). Antiprotease shield and exogenously induced increased
intracellular reducing capacity (green pathways), respectively, block the two
distinct mechanisms of xanthine oxidase activation initiated by rhinovirus
infection (
and uric acid are
thereafter produced from the enzyme substrate xanthine (2).
Intracellular oxidant production results in the depletion of intracellular
reducing capacity (including reduced glutathione depletion) (3),
which in turns induces xanthine oxidase activation via a non-proteolytic
mechanism (4). Antiprotease shield and exogenously induced increased
intracellular reducing capacity (green pathways), respectively, block the two
distinct mechanisms of xanthine oxidase activation initiated by rhinovirus
infection (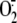 , superoxide
anion; XA, xanthine).
, superoxide
anion; XA, xanthine).
The involvement of a NADPH oxidase-like enzyme in rhinovirus-induced
oxidative stress has been previously described
(23). In contrast to the study
by Kaul and colleagues (23),
our study focuses strictly on the early oxidative events occurring within
cells immediately after rhinovirus infection (with a peak at 1 h after
infection). Also, by using a method able to specifically detect newly
generated 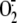 , i.e.
the SOD-inhibitable cytochrome c reduction assay
(51), we directly evaluated
, i.e.
the SOD-inhibitable cytochrome c reduction assay
(51), we directly evaluated
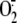 intracellular production,
and not oxidative stress in general. Moreover, at variance with Kaul and
colleagues (23), we employed
oxypurinol, which completely inactivates XO by direct binding to the enzyme
active site (28), and not
allopurinol, which only partially inhibits the enzyme. Treatment with
oxypurinol completely abolished rhinovirus-induced
intracellular production,
and not oxidative stress in general. Moreover, at variance with Kaul and
colleagues (23), we employed
oxypurinol, which completely inactivates XO by direct binding to the enzyme
active site (28), and not
allopurinol, which only partially inhibits the enzyme. Treatment with
oxypurinol completely abolished rhinovirus-induced
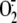 generation, thus
confirming the specificity of our findings on XO activation. These and other
differences between the two studies, in particular different samples for
analyses (intracellular versus extracellular compartments) and
different timing are likely explanations for the different results reported.
In the experimental setting evaluating the functional effect of oxypurinol on
cytokine production, to exclude nonspecific interference, we tested the effect
of oxypurinol on IL-1β-induced IL-6 production. This is a pathway for
which mechanisms other than oxidant generation are considered relevant
(52,
53). We found that IL-1β
significantly induced IL-6 in a dose-dependent manner and that oxypurinol does
not affect IL-6 production induced by 4-h IL-1β stimulation (data not
shown).
generation, thus
confirming the specificity of our findings on XO activation. These and other
differences between the two studies, in particular different samples for
analyses (intracellular versus extracellular compartments) and
different timing are likely explanations for the different results reported.
In the experimental setting evaluating the functional effect of oxypurinol on
cytokine production, to exclude nonspecific interference, we tested the effect
of oxypurinol on IL-1β-induced IL-6 production. This is a pathway for
which mechanisms other than oxidant generation are considered relevant
(52,
53). We found that IL-1β
significantly induced IL-6 in a dose-dependent manner and that oxypurinol does
not affect IL-6 production induced by 4-h IL-1β stimulation (data not
shown).
Pretreatment with all four serine antiproteases investigated, but not the
cysteine and metalloprotease inhibitors phenanthroline and E-64, also
completely prevented 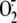 production in response to rhinovirus infection. Previous studies have
documented that limited proteolysis of XD with serine proteases converts the
enzyme to the active XO form
(40). With the exception of
phenanthroline and E-64, all anti-proteases here utilized can act as serine
protease inhibitors, or serine-cysteine protease inhibitors
(29,
30,
32). Pepstatin, an inhibitor
of aspartate proteases, may also act on serine proteases because of active
site target similarities (31).
The fact that phenanthroline and E-64, two protease inhibitors for which no
serine protease inhibitory activity is documented, do not prevent
rhinovirus-induced
production in response to rhinovirus infection. Previous studies have
documented that limited proteolysis of XD with serine proteases converts the
enzyme to the active XO form
(40). With the exception of
phenanthroline and E-64, all anti-proteases here utilized can act as serine
protease inhibitors, or serine-cysteine protease inhibitors
(29,
30,
32). Pepstatin, an inhibitor
of aspartate proteases, may also act on serine proteases because of active
site target similarities (31).
The fact that phenanthroline and E-64, two protease inhibitors for which no
serine protease inhibitory activity is documented, do not prevent
rhinovirus-induced 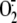 production and intracellular GSH depletion supports the serine specificity of
the pathway of activation of XD to XO following rhinovirus infection.
production and intracellular GSH depletion supports the serine specificity of
the pathway of activation of XD to XO following rhinovirus infection.
Not only activity but also the amount of XD protein was found to be
decreased after rhinovirus infection with a parallel increase of the
proteolytic fragment containing the active site of the enzyme. Further
confirmation comes from silencing experiments showing that XD knockdown
suppresses rhinovirus-induced
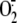 production.
production.
Control experiments were performed to evaluate the effects of the compounds used in the study on rhinovirus replication, to assess whether they have antiviral activities that could provide an alternative explanation for our findings. Virus binding to host cells, virus cell entry, and infectivity would be the most relevant events in our experimental conditions, where the mechanisms analyzed begin a few minutes after the infection has started (24). By using HeLa cells we were able to examine the effects of the tested compounds independently from the up-regulatory effect of rhinovirus on its own receptor (ICAM-1) observed in respiratory epithelial cells (26). Indeed, ICAM-1 surface expression decreases on rhinovirus-infected HeLa cells in parallel with the appearance and severity of cytopathic effects (data not shown). Moreover, we further exclude any direct anti-RV16 effect of the proteases used in the present study by assessing viral replication by TaqMan Real Time PCR (Fig. 8B). Taken together these experiments demonstrated that the tested compounds at the highest concentrations used in our experimental setting had no effect on rhinovirus replication and infectivity.
Although we cannot exclude the involvement of rhinovirus 3C proteolytic enzyme in XD/XO activation, we believe this possibility is unlikely to be relevant in our experimental conditions where oxidant activation was already detectable at 20 min and peaked at 1 h after infection, whereas 2-4 h infection is required for rhinovirus 3C protease to be produced (54).
Despite the effort and resources expended, no antiviral drugs are currently
marketed for the prevention or treatment of rhinovirus infection
(6). In the absence of
effective anti-viral therapies, development of therapies that blocked the
inflammatory responses to infection would be a major advance. Our
demonstration of the molecular mechanisms by which rhinovirus activates
oxidant generation, a crucial step in the complex inflammatory response to
infection (23,
24) could open new
possibilities in the search for therapeutic targets for future intervention.
In particular, the documentation that: (a) exogenous interventions
able to increase intracellular reducing agent storage can block
rhinovirus-mediated activation of the vicious circle that leads to sustained
production of 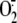 , and
(b) inhibition of XO, with protease inhibitors or specific inhibitors
(oxypurinol) inhibits
, and
(b) inhibition of XO, with protease inhibitors or specific inhibitors
(oxypurinol) inhibits 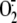 and
pro-inflammatory mediator production, indicate promising options for the
development of treatment for rhinovirus-induced diseases including the common
cold and exacerbations of asthma and COPD.
and
pro-inflammatory mediator production, indicate promising options for the
development of treatment for rhinovirus-induced diseases including the common
cold and exacerbations of asthma and COPD.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: RV, rhinoviruses; f-RV, filtered virus; COPD,
chronic obstructive pulmonary disease; HBEC, human bronchial epithelial cell;
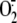 , superoxide anion; XD,
xanthine dehydrogenase; XO, xanthine oxidase; PMSF, phenylmethylsulfonyl
fluoride; Leu, serine and cysteine protease inhibitor leupeptin; Pep, aspartic
protease inhibitor pepstatin; Apr, serine protease inhibitor aprotinin; Phe,
metalloprotease inhibitor phenanthroline; Oxy, oxypurinol; SOD, superoxide
dismutase; HPLC, high performance liquid chromatography; siRNA, small
interfering RNA; RT, reverse transcriptase; PBS, phosphate-buffered saline;
TBS, Tris-buffered saline; H2S, sulfidric acid; IL, interleukin;
ICAM-1, intercellular adhesion molecule 1.
, superoxide anion; XD,
xanthine dehydrogenase; XO, xanthine oxidase; PMSF, phenylmethylsulfonyl
fluoride; Leu, serine and cysteine protease inhibitor leupeptin; Pep, aspartic
protease inhibitor pepstatin; Apr, serine protease inhibitor aprotinin; Phe,
metalloprotease inhibitor phenanthroline; Oxy, oxypurinol; SOD, superoxide
dismutase; HPLC, high performance liquid chromatography; siRNA, small
interfering RNA; RT, reverse transcriptase; PBS, phosphate-buffered saline;
TBS, Tris-buffered saline; H2S, sulfidric acid; IL, interleukin;
ICAM-1, intercellular adhesion molecule 1.
References
- 1.Stanway, G. (1994) in Encyclopedia of Virology (Webster, R., and Granoff, A., eds) pp. 1253-1259, Academic Press, London
- 2.Johnston, S. L., Pattemore, P. K., Sanderson, G., Smith, S., Lampe, F., Josephs, L., Symington, P., O'Toole, S., Myint, S. H., Tyrrell, D. A., and Holgate, S. T. (1995) Br. Med. J. 310 1225-1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grissell, T. V., Powell, H., Shafren, D. R., Boyle, M. J., Hensley, M. J., Jones, P. D., Whitehead, B. F., and Gibson, P. G. (2005) Am. J. Respir. Crit. Care Med. 172 433-439 [DOI] [PubMed] [Google Scholar]
- 4.Seemungal, T., Harper-Owen, R., Bhowmik, A., Moric, I., Sanderson, G., Message, S., Maccallum, P., Meade, T. W., Jeffries, D. J., Johnston, S. L., and Wedzicha, J. A. (2001) Am. J. Respir. Crit. Care Med. 164 1618-1623 [DOI] [PubMed] [Google Scholar]
- 5.Papi, A., Bellettato, C. M., Braccioni, F., Romagnoli, M., Casolari, P., Caramori, G., Fabbri, L. M., and Johnston, S. L. (2006) Am. J. Respir. Crit. Care Med. 173 1114-1121 [DOI] [PubMed] [Google Scholar]
- 6.Jefferson, T. O., and Tyrrell, D. (2007) Cochrane Database Syst. Rev. 18 CD002743. [DOI] [PubMed] [Google Scholar]
- 7.Heikkinen, T., and Jarvinen, A. (2003) The Lancet 361 51-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallia, P., Contoli, M., Caramori, G., Pandit, A., Johnston, S. L., and Papi, A. (2007) Curr. Pharm. Des. 13 73-97 [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulos, N. G., Bates, P. J., Bardin, P. G., Papi, A., Leir, S. H., Fraenkel, D. J., Meyer, J., Lackie, P. M., Sanderson, G., Holgate, S. T., and Johnston, S. L. (2000) J. Infect. Dis. 181 1875-1884 [DOI] [PubMed] [Google Scholar]
- 10.Edwards, M. R., Haas, J., Panettieri, R. A., Jr., Johnson, M., and Johnston, S. L. (2007) J. Biol. Chem. 282 15366-15375 [DOI] [PubMed] [Google Scholar]
- 11.Edwards, M. R., Hewson, C. A., Laza-Stanca, V., Lau, H.-T. H., Mukaida, N., Hershenson, M. B., and Johnston, S. L. (2007) Mol. Immunol. 44 1587-1597 [DOI] [PubMed] [Google Scholar]
- 12.Fraenkel, D. J., Bardin, P. G., Sanderson, G., Lampe, F., Johnston, S. L., and Holgate, S. T. (1995) Am. J. Respir. Crit. Care Med. 151 879-886 [DOI] [PubMed] [Google Scholar]
- 13.Calhoun, W. J., Dick, E. C., Schwartz, L. B., and Busse, W. W. (1994) J. Clin. Investig. 94 2200-2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wark, P. A., Johnston, S. L., Bucchieri, F., Powell, R., Puddicombe, S., Laza-Stanca, V., Holgate, S. T., and Davies, D. E. (2005) J. Exp. Med. 201 937-947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Contoli, M., Message, S. D., Laza-Stanca, V., Edwards, M. R., Wark, P. A., Bartlett, N. W., Kebadze, T., Mallia, P., Stanciu, L. A., Parker, H. L., Slater, L., Lewis-Antes, A., Kon, O. M., Holgate, S. T., Davies, D. E., Kotenko, S. V., Papi, A., and Johnston, S. L. (2006) Nat. Med. 12 1023-1026 [DOI] [PubMed] [Google Scholar]
- 16.Corne, J. M., Marshall, C., Smith, S., Schreiber, J., Sanderson, G., Holgate, S. T., and Johnston, S. L. (2002) Lancet 359 831-834 [DOI] [PubMed] [Google Scholar]
- 17.Caramori, G., and Papi, A. (2004) Thorax 59 170-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, N., and Karin, M. (1999) FASEB J. 13 1137-1143 [PubMed] [Google Scholar]
- 19.Janssen-Heininger, Y. M., Poynter, M. E., and Baeuerle, P. A. (2000) Free Radic. Biol. Med. 28 1317-1327 [DOI] [PubMed] [Google Scholar]
- 20.Peterhans, E. (1997) J. Nutr. 127 Suppl. 5, 962S-965S [DOI] [PubMed] [Google Scholar]
- 21.Casola, A., Burger, N., Liu, T., Jamaluddin, M., Brasier, A. R., and Garofalo, R. P. (2001) J. Biol. Chem. 276 19715-19722 [DOI] [PubMed] [Google Scholar]
- 22.Zsengeller, Z. K., Ross, G. F., Trapnell, B. C., Szabo, C., and Whitsett, J. A. (2001) Am. J. Physiol. 280 L503-L511 [DOI] [PubMed] [Google Scholar]
- 23.Kaul, P., Biagioli, M. C., Singh, I., and Turner, R. B. (2000) J. Infect. Dis. 181 1885-1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papi, A., Papadopoulos, N. G., Stanciu, L. A., Bellettato, C. M., Pinamonti, S., Degitz, K., Holgate, S. T., and Johnston, S. L. (2002) FASEB J. 16 1934-1936 [DOI] [PubMed] [Google Scholar]
- 25.Halliwell, B., and Gutteridge, J. (1999) Free Radical Biology & Medicine, Oxford, University Pess, Oxford, UK
- 26.Papi, A., and Johnston, S. L. (1999) J. Biol. Chem. 274 9707-9720 [DOI] [PubMed] [Google Scholar]
- 27.Johnston, S. L., and Tyrrell, D. A. J. (1997) in Diagnostic Procedures of Viral, Rickettsial and Clamydial Infections (Lennette, E. H., and Schmidt, N. J., eds) pp. 553-563, American Public Health Association, Washington, D. C.
- 28.Spector, T. (1988) Biochem. Pharmacol. 37 349-352 [DOI] [PubMed] [Google Scholar]
- 29.Stracher, A. (1999) Ann. N. Y. Acad. Sci. 884 52-59 [DOI] [PubMed] [Google Scholar]
- 30.Bhattacharya, S., Ghosh, S., Chakraborty, S., Bera, A. K., Mukhopadhayay, B. P., Dey, I., and Banerjee, A. (2001) BMC Struct. Biol. 1 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kageyama, T. (2002) Cell Mol. Life Sci. 59 288-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wegner, J. (2003) J. Extra Corpor. Technol. 35 326-338 [PubMed] [Google Scholar]
- 33.Yeh, C. T., Lai, H. Y., Chu, S. P., and Tseng, I. C. (2004) Biochem. Biophys. Res. Commun. 323 32-37 [DOI] [PubMed] [Google Scholar]
- 34.Tamai, M., Matsumoto, K., Omura, S., Koyama, I., Ozawa, Y., and Hanada, K. (1986) J. Pharmacobio-Dyn. 9 672-677 [DOI] [PubMed] [Google Scholar]
- 35.Pinamonti, S., Muzzoli, M., Chicca, M. C., Papi, A., Ravenna, F., Fabbri, L. M., and Ciaccia, A. (1996) Free Radic. Biol. Med. 21 147-155 [DOI] [PubMed] [Google Scholar]
- 36.Priest, D. G., and Pitts, O. M. (1972) Anal. Biochem. 50 195-205 [DOI] [PubMed] [Google Scholar]
- 37.Bartl, K., Brandhuber, M., and Ziegenhorn, J. (1979) Clin. Chem. 25 619-621 [PubMed] [Google Scholar]
- 38.Morre, D. J. (2002) Antioxid. Redox Signal. 4 207-212 [DOI] [PubMed] [Google Scholar]
- 39.Varani, K., Caramori, G., Vincenzi, F., Adcock, I., Casolari, P., Leung, E., Maclennan, S., Gessi, S., Morello, S., Barnes, P. J., Ito, K., Chung, K. F., Cavallesco, G., Azzena, G., Papi, A., and Borea, P. A. (2006) Am. J. Respir. Crit. Care Med. 173 398-406 [DOI] [PubMed] [Google Scholar]
- 40.Hille, R., and Nishino, T. (1995) FASEB J. 9 995-1003 [PubMed] [Google Scholar]
- 41.Harrison, R. (2002) Free Radic. Biol. Med. 33 774-797 [DOI] [PubMed] [Google Scholar]
- 42.Bustin, S. A. (2000) J. Mol. Endocrinol. 25 169-193 [DOI] [PubMed] [Google Scholar]
- 43.Greve, J. M., Davis, G., Meyer, A. M., Forte, C. P., Yost, S. C., Marlor, C. W., Kamarck, M. E., and McClelland, A. (1989) Cell 56 839-847 [DOI] [PubMed] [Google Scholar]
- 44.Uncapher, C. R., DeWitt, C. M., and Colonno, R. J. (1991) Virology 180 814-817 [DOI] [PubMed] [Google Scholar]
- 45.Enroth, C., Eger, B. T., Okamoto, K., Nishino, T., and Pai, E. F. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 10723-10728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassoun, P. M., Yu, F. S., Zulueta, J. J., White, A. C., and Lanzillo, J. J. (1995) Am. J. Physiol. 268 L809-L817 [DOI] [PubMed] [Google Scholar]
- 47.Lee, M. M., Green, F. H., Schurch, S., Cheng, S., Bjarnason, S. G., Leonard, S., Wallace, W., Possmayer, F., and Vallyathan, V. (2004) Mol. Cell Biochem. 259 15-22 [DOI] [PubMed] [Google Scholar]
- 48.Jobsis, R. Q., Schellekens, S. L., Fakkel-Kroesbergen, A., Raatgeep, R. H., and de Jongste, J. C. (2001) Mediators Inflamm. 10 351-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Psarras, S., Caramori, G., Contoli, M., Papadopoulos, N., and Papi, A. (2005) Curr. Pharm. Des. 11 2053-2062 [DOI] [PubMed] [Google Scholar]
- 50.Ciriolo, M. R., Palamara, A. T., Incerpi, S., Lafavia, E., Bue, M. C., De Vito, P., Garaci, E., and Rotilio, G. (1997) J. Biol. Chem. 272 2700-2708 [DOI] [PubMed] [Google Scholar]
- 51.McCord, J. M., and Fridovich, I. (1973) Photochem. Photobiol. 17 115-121 [DOI] [PubMed] [Google Scholar]
- 52.Cahill, C. M., and Rogers, J. T. (May 30, 2008) J. Biol. Chem. 283 25900-25912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stylianou, E., O'Neill, L. A., Rawlinson, L., Edbrooke, M. R., Woo, P., and Saklatvala, J. (1992) J. Biol. Chem. 267 15836-15841 [PubMed] [Google Scholar]
- 54.Amineva, S. P., Aminev, A. G., Palmenberg, A. C., and Gern, J. E. (2004) J. Gen. Virol. 85 2969-2979 [DOI] [PubMed] [Google Scholar]