

Abstract
The LytC lysozyme belongs to the autolytic system of Streptococcus pneumoniae and carries out a slow autolysis with optimum activity at 30 °C. Like all pneumococcal murein hydrolases, LytC is a modular enzyme. Its mature form comprises a catalytic module belonging to the GH25 family of glycosyl-hydrolases and a cell wall binding module (CBM), made of 11 sequence repeats, that is essential for activity and specifically targets choline residues present in pneumococcal lipoteichoic and teichoic acids. Here we show that the catalytic module is natively folded, and its thermal denaturation takes place at 45.4 °C. However, the CBM is intrinsically unstable, and the ultimate folding and stabilization of the active, monomeric form of LytC relies on choline binding. The complex formation proceeds in a rather slow way, and all sites (8.0 ± 0.5 sites/monomer) behave as equivalent (Kd = 2.7 ± 0.3 mm). The CBM stabilization is, nevertheless, marginal, and irreversible denaturation becomes measurable at 37 °C even at high choline concentration, compromising LytC activity. In contrast, the Cpl-1 lysozyme, a homologous endolysin encoded by pneumococcal Cp-1 bacteriophage, is natively folded in the absence of choline and has maximum activity at 37 °C. Choline binding is fast and promotes Cpl-1 dimerization. Coupling between choline binding and folding of the CBM of LytC indicates a high conformational plasticity that could correlate with the unusual alternation of short and long choline-binding repeats present in this enzyme. Moreover, it can contribute to regulate LytC activity by means of a tight, complementary binding to the pneumococcal envelope, a limited motility, and a moderate resistance to thermal denaturation that could also account for its activity versus temperature profile.
The integrity of the cell wall is essential for bacterial survival, and peptidoglycan-metabolizing enzymes have to work closely together to prevent the rupture of the cell wall and the cell lysis during cell growth and division (1). In particular, the potentially suicidal activity of autolytic enzymes should be strictly controlled (2), and tight binding to the cell envelope (3) seems to work as a mechanism for the regulation and control of many murein hydrolases from different bacterial species (4). In Streptococcus pneumoniae, the presence of phosphorylcholine residues on lipoteichoic and teichoic acids (5), a characteristic shared by other related microorganisms (6), determines the activity of murein hydrolases. In addition to the catalytic module (CM),3 these enzymes comprise a choline-binding module (CBM) that specifically recognizes and binds the choline residues on the cell wall (4). Moreover, it has been suggested that phosphorylcholine residues would be acting as a selective pressure factor to preserve the cell wall recognition module in all of the murolytic enzymes encoded by pneumococcus so far characterized. The CBM, usually located at the C terminus of choline-binding proteins, is made up of tandemly arranged homologous repeats of about 20 amino acids (Pfam accession code PF01473) (7). Each repeat forms a β-hairpin followed by a loop and a coiled region that tend to fold into a left-handed β-solenoid (8-10). Choline binds at the interface of two consecutive β-hairpins, in a cavity lined by three aromatic side chains (8-10), and the multivalent interacting surface presented by the CBM can provide a strong binding to the pneumococcal envelope. The same type of modular organization is found in the endolysins produced by most pneumococcal bacteriophages and suggests a genetic interchange during evolution between the host and the parasites (7).
Only two autolytic enzymes (autolysins) have been unequivocally identified so far in S. pneumoniae: the well known LytA amidase and the LytC lysozyme (11). LytC is an autolysin designed to remodel the cell wall, with maximum activity at 30 °C. This feature suggests that LytC could be more active in habitats like the upper, well ventilated respiratory tract (11). In fact, LytC plays a role in the colonization of the rat nasopharynx (12), where it could also contribute to DNA release in competent cells (13). LytC is directed to the outer surface by a leader peptide (33 residues), and it remains tightly bound to the cell wall in a mature, active form that comprises a CBM made of 11 (p1-p11) repeating units (264 residues) and a CM belonging to the GH-25 family of glycosyl hydrolases (204 residues). The unprocessed form is also detected in the cytoplasm (11).
The slow hydrolysis of pneumococcal cultures carried out by LytC contrasts with the fast, uncontrolled lysis of the host cell performed by Cpl-1, the lysozyme encoded by the pneumococcal phage Cp-1, in order to release the infective particles after the virion replication (11). Cpl-1 and LytC are built of homologous modules (Fig. 1) although assembled in opposite locations (7). In Cpl-1, whose three-dimensional structure has been solved (9), the catalytic module is localized at the N terminus (188 residues), and the CBM (139 residues) is made up of six repeating units (p1-p6) and a short terminal tail (7). However, only two choline-binding sites, located at the interfaces of the three first repeats (p1-p2 and p2-p3), seem to be functional, according to the crystal structure of Cpl-1 (9). In contrast, molecular modeling of LytC reveals a higher number of potential sites for choline. In addition, the alternation of shorter (17 residues) and longer (21-23 residues) repeats (see Fig. 1) in LytC leads to an uneven distribution of choline-binding sites along the CBM surface (14), previously unseen in other CBMs of known structure (8-10).
FIGURE 1.

Modular organization and sequence alignment of the CBM repeats of Cpl-1 and LytC. A, Cpl-1. B, LytC. CMs are shown in green, and CBMs are shown in orange. The first element in LytC (gray) is the signal peptide, and the last one of Cpl-1 (red and orange stripes) is the C-terminal tail. Numbers on the right indicate the number of residues in each repeat. Strictly conserved residues are marked with an asterisk, and colons indicate conservative substitutions. β-Strands appear as arrows. Orange, glycine; dark blue, hydrophobic; dark cyan, tyrosine; green, neutral polar; magenta, acidic; red, basic.
We have performed a comparative study of LytC and Cpl-1 lysozymes using different approaches. The differences found in structure, stability, conformational plasticity, choline-binding avidity, and complex dynamics contribute to determine the lytic profiles of these two lysozymes. Overall, LytC and Cpl-1 represent a good illustrative example of how natural selection can tailor at the genetic level different solutions using homologous building blocks and module shuffling.
EXPERIMENTAL PROCEDURES
Protein Purification and Chemicals—Protein expression and purification were performed as previously described (14). Before use, protein samples were extensively dialyzed at 4 °C against Pi buffer (20 mm phosphate buffer, pH 8.0) with or with-out choline, and centrifuged for 5 min at 11,600 × g. Protein concentrations were measured spectrophotometrically using molar absorption coefficients of 186,107 m-1 cm-1 (LytC; 55.210 kDa) and 97,114 m-1 cm-1 (Cpl-1; 39.249 kDa) (14). Choline concentration was measured by differential refractometry (15). All reagents (Sigma) were of analytical grade.
Enzymatic Activity—Thermal inactivation was determined by measuring cell wall lytic activities after 5 min of preincubation at selected temperatures. Activity essays were carried out in Pi buffer (pH 6.0), as described elsewhere (16), using optimal conditions (30 °C for LytC and 37 °C for Cpl-1).
Sedimentation Equilibrium—Sedimentation equilibrium experiments were performed by centrifugation of 80-μl samples of LytC (4 °C and 20,000 × g) or Cpl-1 (25 °C and 30,000 × g) in an Optima-XLA analytical ultracentrifuge (Beckman Instruments, Inc.), following previously reported protocols (17). Partial specific volumes of 0.72 ml/g (LytC) and 0.73 ml/g (Cpl-1) were calculated from the amino acid composition (18).
Size Exclusion Chromatography—Gel filtration was carried out at room temperature in a Superose 12 HR/10/30 column (Amersham Biosciences) equilibrated in Pi buffer, and, when stated, choline chloride or NaCl were added at the indicated concentration. Typically, 100-μl samples of protein or molecular weight markers (12-18 μm) were applied and eluted at a flow rate of 0.5 ml min-1.
Circular Dichroism—CD spectra (average of four scans) were recorded in a JASCO-J810 spectropolarimeter (Jasco Corp.) equipped with a Peltier type cell holder, using a scan rate of 20 nm min-1, a response time of 2 s, and a bandwidth of 1 nm. Typical protein concentrations were around 3-5 μm in the far UV region (1-mm path length) and 15-25 μm in the near UV region (1-cm path length). Buffer spectra were subtracted from the experimental data, and the corrected ellipticity was converted to mean residue ellipticity using average molecular masses per residue of 118 (LytC) and 116 (Cpl-1). Choline titration curves were obtained by measuring the far-UV CD spectra at varying choline concentrations and plotting the increase in ellipticity at 224-221 nm as a function of the ligand concentration. To minimize errors, titration of Cpl-1 with choline was carried out by serial addition of small volumes of concentrated choline stocks to the same protein sample (less than 10% total volume increase). Because choline binding to LytC was slow, the spectra at varying ligand concentrations were measured using samples (same protein stock) prepared by extensive dialysis against solutions containing the required concentration of choline in order to assure that the equilibrium was achieved. In all cases, data reported correspond to two independent series of measurements.
Thermal denaturation experiments were carried out by increasing the temperature at a heating rate of 20 °C/h and allowing the cell to equilibrate for 60 s before recording the ellipticity at the selected wavelength. Denaturation kinetics at a fixed temperature were monitored by following the time course of the protein ellipticity at 225 nm. Reactions were initiated by protein addition to the cell loaded with the selected buffer solution. When required, the experimental curves were analyzed using the least square fitting routine implemented in the Origin software.
Choline Binding Kinetics—Kinetics of choline binding to LytC were initially measured at 4 °C by following the change of the protein ellipticity at 225 and 285 nm in a Jasco-810 spectropolarimeter. Three independent measurements were carried out at each wavelength, and the resulting curves were fitted to a double exponential equation using the least square fitting routine implemented in the Origin software. Measurements at varying temperatures were also performed at 225 nm to analyze the influence of temperature and LytC denaturation on choline binding capacity.
Differential Scanning Calorimetry—Unless otherwise stated, differential scanning calorimetry (DSC) measurements were performed at a heating rate of 20 °C/h using an MCS microcalorimeter (Microcal, Inc.), under an extra constant pressure of 2 atm. Standard MCS and Microcal Origin DSC software were used for data acquisition and analysis. The excess heat capacity functions were obtained after subtraction of the buffer-buffer base line. The absence of a thermal effect on reheating previously scanned samples and the dependence of the heat capacity curves on the heating rate showed that thermal denaturations of LytC and Cpl-1 were irreversible and kinetically controlled (19).
Analysis of DSC Curves—The heat capacity function of
transitions described in terms of the two-state irreversible
model,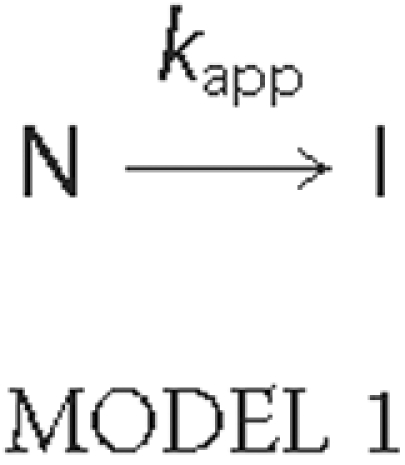
was analyzed using Equation 1, derived by Sánchez-Ruiz
(19) for processes following
first-order kinetics,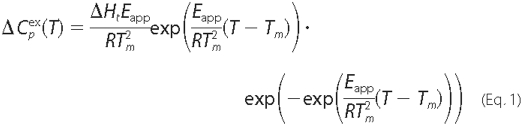
where R is the gas constant, and ΔHt,
Eapp, and Tm represent, respectively, the
total calorimetric enthalpy change, the activation energy of the denaturation
rate constant (kapp), and the temperature of the maximum
in the heat capacity transition. Denaturation processes evolving according to
the three-state irreversible denaturation
model,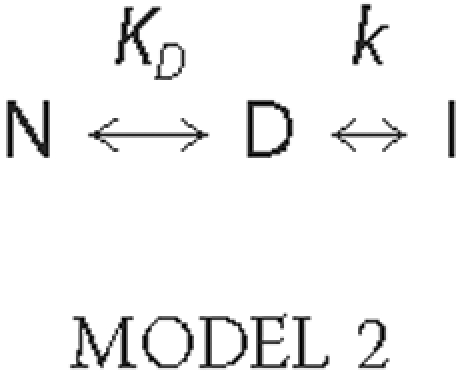
were analyzed using Equation 2
(20), as previously described
(17),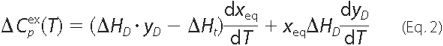
ΔHD and ΔHt represent, respectively, the calorimetric transition enthalpy for the equilibrium step, and the total enthalpy change on passing from N to I; xeq is the total fraction of forms in equilibrium, and yD is the fraction of the reversibly denatured state D.
Isothermal Titration Calorimetry—Choline binding to Cpl-1 was studied at 25 °C by isothermal titration calorimetry using an MCS microcalorimeter (Microcal, Inc.). Data acquisition and analysis were carried out using the MCS and Microcal Origin software. In a first assay, the total enthalpy of binding, ΔHapp, was measured by injecting 5 × 20 μl of 195 μm Cpl-1 into the calorimetric cell loaded with a saturating solution of choline in the same buffer. The value of ΔHapp was calculated as the heat evolved per mol of protein injected after correction of dilution effects (21). The choline titration curve was measured by injection of 100 mm choline, in small increments, over the sample cell loaded with a solution of around 160 μm Cpl-1 equilibrated in the same buffer (cell volume = 1.3537 ml). Data were acquired in triplicate. After peak area integration and subtraction of the dilution heat of the ligand, the titration curve, plotted as the total heat evolved at a given ligand concentration (22), was analyzed in terms of the reaction model shown in Scheme 1. The model assumes that Cpl-1 undergoes dimerization upon choline binding (see “Results”) and that each monomer contains two functional choline-binding sites (9).

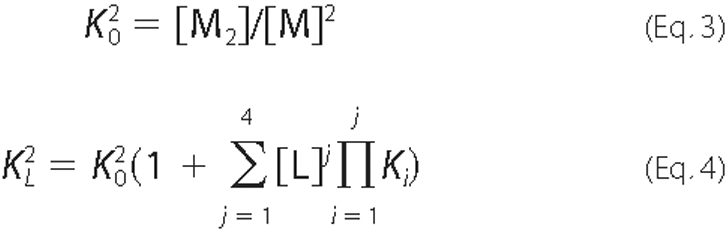
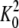 and
and 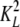 are,
respectively, the dimerization constants in the absence and in the presence of
a given concentration [L] of free choline, and Ki
(i = 1-4) is the binding constant to the Cpl-1 dimer for step
i. The following can be easily
derived,
are,
respectively, the dimerization constants in the absence and in the presence of
a given concentration [L] of free choline, and Ki
(i = 1-4) is the binding constant to the Cpl-1 dimer for step
i. The following can be easily
derived,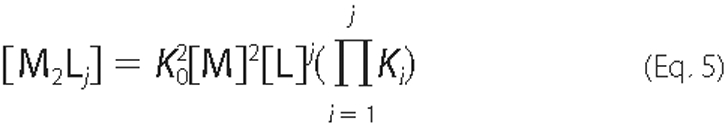
and for n equivalent and independent sites, the step binding constants can be expressed as follows,
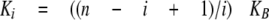 |
(Eq.6) |
where KB is the intrinsic binding constant to Cpl-1 dimer
(23). According to the CD
titration curves (see “Results”) and the three-dimensional
structure of the Cpl-1-choline complex
(9), the four choline-binding
sites found per Cpl-1 dimer can be considered equivalent. Hence, the
experimental enthalpy change at [L] can be expressed as
follows,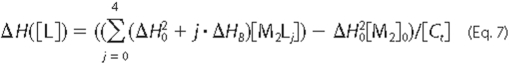
where 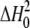 is the dimerization enthalpy
change of the free enzyme, ΔHB is the binding
enthalpy per mol of sites, and [M2]0 is the initial
dimer concentration in the absence of choline. Substitution of
[M2Lj] in terms of Equations 5 and 6 leads to
Equation 8,
is the dimerization enthalpy
change of the free enzyme, ΔHB is the binding
enthalpy per mol of sites, and [M2]0 is the initial
dimer concentration in the absence of choline. Substitution of
[M2Lj] in terms of Equations 5 and 6 leads to
Equation 8,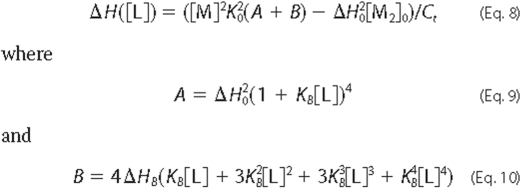
[M], the free monomer concentration, is given by Equation
11,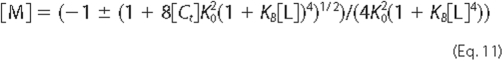
where [Ct] is the total molar protein concentration expressed as moles of monomer.
RESULTS
Differences in the Native Structures of LytC and Cpl-1 Lysozymes—The far UV region of the CD spectrum of murein hydrolases encoded by pneumococcus and its phages is characterized by the presence of a maximum in the region of 224-230 nm (15, 17, 24, 25), which is sensitive to the interaction of choline with the tryptophan residues present at the binding site (8-10). Interestingly, the presence of this band in the spectrum of LytC strictly depends on choline binding (Fig. 2A). Thus, attachment of LytC to choline moieties of lipoteichoic and teichoic acids seems to mediate a major reorganization of the polypeptide chain that strongly affects the environment of the aromatic residues present in its CBM. Indeed, the fine structure of the near UV spectra is strongly modified by the complex formation (Fig. 2B). In particular, the small positive bands between 277 and 292 nm vanish, and a well defined, negative band appears at 286 nm. In addition, the small minimum at 298 nm, due to tryptophan residues, blue-shifts by 2 nm, increasing its intensity by a factor of 3.5, and the broad maximum around 264 nm is also enhanced.
FIGURE 2.

CD spectra of lysozymes LytC and Cpl-1. Influence of choline binding. A and B depict, respectively, the far and near UV spectra of LytC (circles) and Cpl-1 (triangles) in the absence and in the presence of 140 mm choline (Pi buffer) recorded at 25 °C (Cpl-1) and 4 °C (LytC). C, the kinetics of choline binding to LytC following ellipticity variations at 225 nm (black trace) and 285 nm (gray trace; top scale). Measurements were made in Pi buffer at 4 °C and 140 mm choline.
The spectroscopic changes proceed in a rather slow way, taking more than 3 h to reach equilibrium upon in situ addition of 140 mm choline (pseudo-first order conditions). The time course of choline binding was followed by monitoring the ellipticity changes at 225 and 285 nm. Measurements were initially performed at 4 °C to prevent denaturation during the complex formation (see below). The process displayed a biphasic character (Fig. 2C), although ellipticity changes below 208 nm reached the equilibrium in a few min (not shown). This behavior points to the formation of an intermediate with essentially the same ellipticity than the final state in that region of the spectrum. Kinetics at 225 and 285 nm can be explained in terms of two-exponential processes (Fig. 2C), and the best fitting values for the rate constants and the phase amplitudes are summarized in Table 1. In contrast, the far UV CD spectrum of the Cpl-1 lysozyme (Fig. 2A) resembles those reported for other properly folded choline-binding proteins (15, 17, 24), and the CD changes promoted by choline binding take place within the dead time of mixing in both regions of the spectrum. In the near UV region, the interaction with choline primarily sharpened preexisting bands between 279 and 296 nm, increasing their intensities by about 4-fold. Such an effect suggests a reduction in the mobility of the aromatic residues that shape the choline-binding cavity upon the complex formation (9, 26).
TABLE 1.
Kinetic constants for binding of choline to LytC measured by spectropolarimetry
Measurements were at 4 °C in Pi buffer plus 140 mm choline.
|
λ (nm)
|
Fast phase
|
Slow phase
|
||
|---|---|---|---|---|
| k1 | ΔΘ1a | k2 | ΔΘ2a | |
| min−1 | % | min−1 | % | |
| 225 | 0.3 ± 0.1 | 34 ± 4 | 0.023 ± 0.003 | 66 ± 7 |
| 285 | 0.4 ± 0.1 | 26 ± 3 | 0.015 ± 0.002 | 74 ± 7 |
ΔΘ1 and ΔΘ2 are the relative ellipticity changes associated with the fast and the slow phase, respectively.
Preliminary sedimentation studies had showed that LytC forms large oligomers that dissociate upon choline binding (14). The regulation of oligomerization through choline-protein interactions was here first investigated by size exclusion chromatography. In the absence of choline, LytC elutes as a highly broad, asymmetric peak (supplemental Fig. 1A), whose position (15.7 min) is very close to the void volume of the column (15.3 min), as expected for an ensemble of high molecular weight aggregates. Protein equilibration in 140 mm choline increases the retention time up to 22.9 min (supplemental Fig. 1A). In contrast, there is only a negligible change in the elution profile in the presence of 140 mm NaCl (not shown), showing that LytC dissociation is not due to the increase of ionic strength induced by choline chloride addition. Choline-induced dissociation was further analyzed by sedimentation equilibrium experiments. In the absence of choline, LytC is fully displaced to the bottom of the cell in less than 5 min at sedimentation rates as low as 600 × g. This loss of absorbance decreases at increasing concentrations of choline, and around 50% of the protein becomes measurable at 2 mm choline, where the aggregates coexist with low molecular weight species. Dissociation is almost complete at 7 mm choline. At higher ligand concentration, data can be fitted to a single species model (supplemental Fig. 1, B and C) with an average molecular mass (Mw) of 56.8 ± 0.9 kDa, corresponding to a monomer (Mw/M0 = 1.03) (supplemental Fig. 1D). In contrast, Cpl-1 sediments as a monomer, but its Mw value increases in a highly cooperative way with choline concentration (K0.5 = 2 mm), reaching twice the molecular mass of the monomer under saturation conditions (supplemental Fig. 1D). Thus, choline regulates Cpl-1 self-association, inducing the dimerization of the phage-coded lysozyme, as it also does with amidases carrying CBMs with the same number of choline-binding repeats (17, 24, 27). Accordingly, on gel filtration chromatography, the free enzyme exhibits a single peak with an elution time of 27.7 min that shifts to 22.1 min upon Cpl-1-choline complex formation (supplemental Fig. 1A).
LytC and Cpl-1 Have Very Different Requirements on Choline Binding for Stability—The structural stability of the two lysozymes and the stabilization promoted by choline binding was analyzed by thermal denaturation studies. Fig. 3A shows the calorimetric profiles of LytC denaturation at increasing choline concentrations. In the absence of the ligand, the thermogram shows a single peak characterized by a Tm of 45.4 °C and a denaturation enthalpy of 59 kcal/mol, which is rather low, considering the molecular size of the pneumococcal lysozyme. Protein equilibration in choline-containing buffers induced the appearance of a new peak at a lower temperature, which is progressively up-shifted as choline concentration increases, with a concomitant rise of the total denaturation enthalpy (Table 2). At around 19 mm choline, the two peaks merge into an asymmetric endotherm that remains unresolved at 140 mm choline (Fig. 3, B and C). Although the thermograms obtained at low choline concentration varied somewhat between batches, probably due to the presence of heterogeneous oligomers in the samples, data at 3 and 7 mm were included (Table 2 and Fig. 3) to highlight the dramatic effect of choline binding on LytC stability.
FIGURE 3.

Thermal denaturation curves of the LytC lysozyme at increasing concentrations of choline; influence of the scan rate. A, influence of choline concentration on the thermal stability of LytC (36 μm) measured by DSC. The thermograms were recorded at 20 °C/h in Pi buffer, and ligand concentrations are specified in the curve labels. Dashed-dotted lines under the endotherms are the elementary transitions derived by deconvolution analysis as indicated under “Results”. Theoretical envelopes (continuous lines) superimposed the experimental curves. B, influence of the heating rate on the DSC profiles of LytC recorded in the presence of 140 mm choline. C, deconvolution of LytC endotherm (symbols) measured at 140 mm choline, assuming a two-state N → I irreversible denaturation model for each elementary transition under the endotherm. Dashed-dotted and continuous lines are as in A. For clarity, every fourth data point, on average, is plotted in B and C, although full data sets were used for fitting.
TABLE 2.
Influence of choline on thermal stability of LytC lysozyme
Measurements were in Pi buffer at a scan rate of 20.4 °C/h. ΔHcal is the calorimetric enthalpy change/mol of monomer obtained by integration of the endotherm. Tm1 and Tm2 are, respectively, the maximum temperatures for the CBB and the CM. Estimated errors are ±10% in ΔHcal and ±0.1-0.3 °C in Tm.
| Choline | Tm1 | Tm2 | ΔHcal |
|---|---|---|---|
| mm | °C | °C | kcal·mol−1 |
| 0 | 45.4 | 59 | |
| 3 | 26.2 | 37.5 | 144 |
| 7 | 33.4 | 43.2 | 173 |
| 19 | 39.2 | 45.8a | 243 |
| 40 | 43.3 | 47.4a | 264 |
| 60 | 45.9b | 266 | |
| 140 | 49.8b | 280 |
Shoulder.
Both transitions overlap.
Thermal denaturation of LytC is irreversible and kinetically controlled according to the effect of the scanning rate on the transitions (Fig. 3B). Nevertheless, the experimental curves recorded at choline concentrations where LytC is a monomer can be explained in terms of two transitions (Fig. 3, A and C). One of them, highly dependent on choline concentration, would account for the denaturation of the CBM, whereas the second one, strongly resembling the transition found in the absence of choline, would correspond to the denaturation of the CM. Up to 19 mm choline, both processes can be described in terms of the three-state N ↔ D → I model; however, at higher choline concentrations, the fraction of intermediates becomes rather low, and both transitions essentially follow a two-state irreversible model. The continuous lines in Fig. 3 correspond to the best theoretical fit to the experimental curves, using the parameters and models summarized in supplemental Table 1.
The linkage between folding and choline binding was also analyzed by monitoring the CD thermal transitions at 225 nm, both in the absence and in the presence of 140 mm choline (supplemental Fig. 2A). As mentioned above, this region, dominated by the chiral contributions of aromatic side chains, is a signature in spectra of native choline-binding proteins and provides a complementary probe of the CBM structure. In the absence of choline, Θ225 decreases in a rather progressive way (as expected for noncooperative structures), and variations start well below the beginning of the CM transition seen by DSC. However, upon choline binding, the cooperativity of the process is strongly enhanced, and the transition shifts to higher temperatures. These results clearly show that the CBM is only partially structured in the choline-free state and that the tertiary contacts characterizing folded native structures should be acquired upon attachment of LytC to the choline moieties of lipoteichoic and teichoic acids.
Thermal denaturation of Cpl-1 proceeds with an enthalpy change of 160 ± 10 kcal mol-1, and the DSC profile shows a major peak centered at 41.8 °C (transition A) and a broad, flat shoulder around 47 °C (transition B) (Fig. 4A). As choline concentration increases, the first peak is shifted upward and merges with the second one into a single, sharp, asymmetric peak at 19 mm choline. At higher ligand concentrations, transition A overpasses transition B, and they become completely resolved, again, around 140 mm choline (Fig. 4A). Table 3 summarizes the experimental data at the different conditions. The progressive stabilization of transition A upon choline binding allows its assignment to the Cpl-1 CBM. Hence, transition B would correspond to the denaturation of the catalytic module. These assignments are also supported by stability data obtained for the isolated CBM (28). Cpl-1 denaturation was irreversible, and experiments performed at different scan rates showed a clear up-shift of transition A at increasing heating rates and a moderate effect on transition B (supplemental Fig. 3). The shape of the endotherms and its dependence on the scan rate can be explained assuming that denaturation of each module proceeds in three steps according to the N ↔ D → I model, both in the absence and in the presence of 140 mm choline. The solid lines in Fig. 4, B and C, and supplemental Fig. 3 are the theoretical fits of the experimental envelopes calculated with the parameters given in supplemental Table 2. As shown in the insets (Fig. 4, B and C), the fraction of intermediate in equilibrium with the native state is significant for both modules. Denaturation proceeds in a different way at intermediate choline concentrations. Thus, at 19 mm choline, the endotherm can be explained, assuming that the whole polypeptide chain denatures simultaneously according to a three-state mechanism (data not shown), but data at 40 mm choline cannot be described in terms of simple models. Overall, variations in the shape of Cpl-1 endotherms with choline concentration and the effect of choline binding on the Tm value of both transitions correspond to the behavior expected for proteins with two strongly interacting modules (29, 30). Thus, the increase in the Tm of the CM observed at high choline concentrations reflects the stabilization of the catalytic barrel by the numerous contacts established with the CBM (9), whose denaturation occurs at higher temperatures. On the contrary, in the choline-free state, thermal unfolding of the CM essentially reflects its intrinsic thermal stability, since the process takes place when the CBM is almost denatured.
FIGURE 4.

Influence of choline concentration on the DSC transitions of Cpl-1.
A, influence of choline on the calorimetric profiles of Cpl-1
lysozyme (55 μm). Measurements were done in Pi buffer
at the ligand concentrations indicated in the curve labels. B and
C show, respectively, the analysis of Cpl-1 thermograms in the
absence and in the presence of 140 mm choline, assuming a
three-state N ↔ D → I denaturation model for each module (heating
rate 20.4 °C/h). Symbols correspond to the experimental curves,
and solid and dashed lines are, respectively, the best fits
of the curves and the individual transitions under the envelope calculated
using the parameters summarized in supplemental Table 2. Open circles
in B show the DSC transition evolved by a sample of Cpl-1 previously
heated up to 42.5 °C (blue triangles), corresponding to the CM
denaturation. For clarity, one data point of four is plotted on average,
although full data sets were used for fitting. Insets in B
and C show the population of the native, reversibly, and irreversibly
denatured states as a function of temperature. The red lines
correspond to the CM denaturation
( ,
,
 ,
,
 ), and the black
ones correspond to the CBM denaturation
(
), and the black
ones correspond to the CBM denaturation
( ,
,
 and
and
 ).
).
TABLE 3.
Influence of choline on thermal denaturation of Cpl-1 lysozyme
Measured in Pi buffer at a scan rate of 20.4 °C/h. ΔHcal is the calorimetric enthalpy change/mol of monomer obtained by integration of the whole endotherm. Tm1 and Tm2 are, respectively, the maximum temperatures for peak A (CBM) and peak B (CM). Experimental errors are ±7% in ΔHcal and ±0.1-0.3 °C in Tm.
| [Choline] | Tm1 | Tm2 | ΔHcal |
|---|---|---|---|
| mm | °C | °C | kcal·mol−1 |
| 0 | 41.8 | 47.2 | 160 |
| 19 | 52.4a | 210 | |
| 40 | 55.6 | 51.9 | 230 |
| 140 | 61.4 | 53.2 | 210 |
Both transitions merge into a single endotherm.
Cpl-1 denaturation was also followed by monitoring the ellipticity changes at 225 nm (supplemental Fig. 2B). The CD transition curves were highly cooperative both in the absence and in the presence of 140 mm choline and primarily reflect the denaturation of the Cpl-1 CBM. The apparent half-transition temperatures (T0.5 = 41.6 °C, unligated form; T0.5 = 59.5 °C, 140 mm choline) are in reasonable agreement with Tm values measured by DSC for transition A (Table 3), considering the asymmetry introduced by irreversibility in heat capacity transitions (Fig. 4).
Choline Binding Thermodynamics—Choline binding equilibria were first characterized measuring the changes induced in the CD spectra by choline binding (see “Experimental Procedures”). Fig. 5A shows the relative increase in ellipticity, ΔΘ/ΔΘmax, at 224 nm (LytC) and 221 nm (Cpl-1) as a function of choline concentration. The titration curves are monophasic and highly cooperative for both lysozymes, suggesting that all binding sites are approximately equivalent in each enzyme. The analysis of the binding curves in terms of the Hill equation (23) yielded apparent dissociation constants (Kd) of 2.7 ± 0.3 mm (LytC), and 3.6 ± 0.2 mm (Cpl-1) and cooperativity indexes (nH) of 2.4 ± 0.5 (LytC) and 3.0 ± 0.4 (Cpl-1). These results agree well with the half dissociation constant of Cpl-1 derived from ultracentrifugation data (K0.5 = 2 mm) and with the observation that choline-protein interactions are similar for all occupied sites, according to the choline-Cpl-1 complex structure (9).
FIGURE 5.

Choline binding to LytC and Cpl-1 lysozymes. A depicts the logarithmic plot of the CD titration curves of LytC (4 °C) and Cpl-1 (25 °C) measured by monitoring the ellipticity changes promoted by ligand binding. The solid lines are the best fits of the Hill equation, assuming a single set of binding sites. B shows the isothermal titration calorimetry titration curve of Cpl-1 with choline (25 °C) represented as the cumulative enthalpy of binding versus the total ligand concentration. The solid line represents the fit of the experimental data in terms of equilibria shown in Scheme 1 (see “Experimental Procedures”) and the thermodynamic parameters reported in Table 4. All measurements were in Pi buffer.
As shown above, the denaturation of the CBM of LytC above 19 mm choline can be essentially described in terms of an irreversible two-state N → I model. For this type of process, at a constant heating rate, the dependence of the transition temperature with the free ligand concentration follows Equation 12,
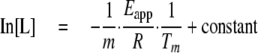 |
(Eq.12) |
where m is the complex stoichiometry, provided that [L] is well above protein concentration (19).
The stoichiometry of the LytC-choline complex was, thus, estimated to be 8.0 ± 0.5 from the slope of the plot of ln[L] versus 1/Tm (data not shown), using an Eapp of 320 ± 30 kcal/mol (average of eight independent experiments) for denaturation of the CBM of LytC.
The complexity of the processes associated to choline-LytC interactions
precludes its thermodynamic analysis by isothermal titration calorimetry. In
contrast, the association states of Cpl-1 are well defined both in the absence
and in the presence of choline, the complex formation is fast, and a number of
2 functional choline-binding sites/monomer was previously established
(9). In addition, according to
our experimental results, choline preferentially binds to Cpl-1 dimers, and
all binding sites behave as equivalent. The thermodynamics of choline-Cpl-1
complex formation was, therefore, characterized by isothermal titration
calorimetry and the titration curve analyzed in terms of the equilibria
described in Scheme 1 (see “Experimental Procedures”), consistent
with previous information. Fig.
5B shows the calorimetric titration curve expressed as
the cumulative enthalpy change as a function of choline concentration. For a
dimer with four equivalent sites of binding, the dependence of the total heat
evolved per mol of Cpl-1 monomer with the ligand concentration is given by
Equation 8 (see “Experimental Procedures”). The parameters
yielding the best fit of the model to the experimental data were
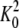 = 700 ± 100 m-1,
= 700 ± 100 m-1,
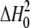 = -75 ± 10 kcal/mol of dimer,
KB = 520 ± 70 m-1, and
ΔHB = 4 ± 1 kcal/mol of site. The value
of-29.5 kcal calculated from these values for the contribution of each Cpl-1
monomer to the total enthalpy change is in good agreement with the average
value of-33 ± 2 kcal (mol of monomer)-1 independently
measured by injection of Cpl-1 in the cell loaded with a saturating solution
of choline (see “Experimental Procedures”).
= -75 ± 10 kcal/mol of dimer,
KB = 520 ± 70 m-1, and
ΔHB = 4 ± 1 kcal/mol of site. The value
of-29.5 kcal calculated from these values for the contribution of each Cpl-1
monomer to the total enthalpy change is in good agreement with the average
value of-33 ± 2 kcal (mol of monomer)-1 independently
measured by injection of Cpl-1 in the cell loaded with a saturating solution
of choline (see “Experimental Procedures”).
The thermodynamic parameters of binding reported in Table 4 showed that choline-Cpl-1 interactions are entropically driven, as expected for the hydrophobic contacts of the aromatic residues forming the binding site with the three choline methyl groups, and for the cation-π interaction established with the positive charge of choline (9). Cpl-1 dimerization contributes with-3.9 kcal·(mol of dimer)-1 to complex stabilization, a value comparable with the free energy change associated with the intrinsic protein-ligand interactions. The dimerization is enthalpically driven, suggesting that hydrogen bonds and van der Waals interactions should play a determinant role (31) in monomer-monomer interactions. Indeed, the dimerization surface, which primarily comprises the first repeat of the CBM and a few side chains in the coiled region of the second repeat (32), exposes numerous polar groups that might contribute to tightly pack the residues located in the monomer-monomer interface by hydrogen bond formation.
TABLE 4.
Thermodynamic parameters associated with choline binding to Cpl-1
Measurements were in Pi buffer at 25 °C. Dimerization parameters are expressed per mole of dimer, and the binding parameters are expressed per mole of binding site.
| ΔGa | ΔH | ΔSb | |
|---|---|---|---|
| kcal/mol | kcal/mol | cal/(mol.K) | |
| Dimerization | −3.9 ± 0.2 | −75 ± 10 | −240 ± 30 |
| Binding | −3.7 ± 0.2 | 4 ± 1 | 26 ± 4 |
ΔG = - RTlnK (K =
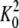 or KB); R is the
gas constant, T is the temperature in Kelvin, and
or KB); R is the
gas constant, T is the temperature in Kelvin, and
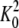 and KB are, respectively,
the dimerization constant and the intrinsic affinity constant for choline
binding (see Scheme 1).
and KB are, respectively,
the dimerization constant and the intrinsic affinity constant for choline
binding (see Scheme 1).
ΔS = (ΔH - ΔG)/T.
The Temperature-Activity Profile of LytC May Be Determined by the CBM Stability and Choline Binding Kinetics—LytC behaves as a choline-dependent autolytic enzyme at 30 °C, although cell lysis proceeds at slower rate than LytA-dependent autolysis (11). Moreover, M32 strain, a LytA-deficient mutant, suffers overnight autolysis when cultured at 30 °C but not at 37 °C. Thus, inactivation of the lytC gene makes the M32 double-deficient mutant resistant to autolysis at both temperatures (11), as it also happens when wild type strains are grown in media that replace choline by ethanolamine (33). Remarkably, cells of M32 grown at 37 °C and taken at different times of the early stationary phase of growth did autolyze when they were shifted to 30 °C (Fig. 6A). However, the recovery of the autolytic capacity was drastically reduced as the stationary phase went forward. On the contrary, when cells cultured at 30 °C were shifted to 37 °C during the stationary phase (Fig. 6B), the autolysis stopped at the levels expected for the cultures grown at this temperature.
FIGURE 6.

Thermal sensitivity of LytC and Cpl-1 lytic activities. A and B show the sensitization of lytA- M32 strain hydrolysis to different temperatures. Cultures were incubated at 37 °C (A) and 30 °C (B), and cellular density was followed by the optical density at 550 nm. After reaching the stationary phase, samples were withdrawn at different times (triangles, t1; squares, t2; diamonds,t3), and the autolytic capacity of the cells was monitored upon inversion of the incubation temperatures. C shows the percentage of activity retained by LytC and Cpl-1 lysozymes after preincubation for 5 min in Pi buffer (pH 6.0) with and without 40 mm choline at increasing temperatures. Activities were measured at 30 °C (LytC) and 37 °C (Cpl-1) by appropriate dilution of the enzyme in the cell wall-containing sample.
The drastic dependence of LytC CBM stability on choline binding, together with the slow character of this process and the fast degradation reported for marginally stable proteins (34), might compromise LytC activity at physiological temperatures, accounting for the autolysis versus temperature profile of the M32 strain. To further analyze this possibility, the influence of temperature on the kinetics of choline binding and CBM denaturation was characterized by following the ellipticity changes at 225 nm. The choline binding rate increased as the reaction temperature was augmented from 20 to 30 °C (Fig. 7A), although the signal increment was ∼25% reduced at this temperature. In both cases, a further signal increase was observed when the temperature was lowered to 4 °C, indicating a small percentage of reversibly unfolded forms. More importantly, at 37 °C, the ellipticity quickly decreased upon protein addition to the ligand solution, and the signal recovery by subsequent cooling to 4 °C was small, even for short reaction times. This demonstrates that at 37 °C choline binding is competing with the CBM denaturation, which becomes the dominant event. In fact, denaturation of the unbound species proceeded very quickly around 30 °C (Fig. 7B), and the structural recovery by subsequent cooling to 4 °C was rather small (not shown). As expected, the structure of the CBM is stabilized by choline binding. Nevertheless, at 37 °C, the complex denaturation proceeds in a slow, although measurable, rate even at 40 mm choline (Fig. 7B). At this ligand concentration, the full process can be described by the sum of two exponentials with very different half-life times (11.5 min and ∼7 h), the slowest part (∼90% total ellipticity decrease) corresponding to the irreversible denaturation of the CBM.
FIGURE 7.

Temperature dependence of choline binding and CBM thermal denaturation kinetics of LytC. Kinetics, monitored by measuring the protein ellipticity at 225 nm, were initiated by the addition of LytC (7.2 μm final concentration) to the cell loaded with Pi buffer (pH 6.0), supplemented, unless otherwise stated, with 40 mm choline. A shows the time course of choline binding at 20 °C (black trace), 30 °C (blue trace), and 37 °C (gray traces). At times indicated by arrows, the temperature was lowered to 4 °C, and the ellipticity changes were monitored again until re-equilibration was achieved. B shows the denaturation kinetics of the CBM of LytC in the free and choline-bound states. Measuring temperatures are indicated. The inset shows the fit of the denaturation kinetics at 40 mm choline and 37 °C in terms of two exponentials.
As expected from the CD results, the in vitro activity of LytC at 30 °C was drastically reduced upon preincubation for 5 min in Pi buffer at temperatures above 25 °C, whereas almost 60% of the activity was still preserved at 50 °C upon the addition of 40 mm choline to the preincubation buffer (Fig. 6C). After the same treatment, the choline-free form of Cpl-1 still retains about 80% activity upon preincubation at 55 °C (Fig. 6C), in agreement with the thermal stabilities displayed by its two modules and the higher reversibility of their denaturation processes.
Taken together, these results strongly indicate that the stabilization provided by the attachment of LytC to lipoteichoic and teichoic acids would preserve the folded, native form of the CBM, accounting for the autolysis suffered by M32 cells cultured at 30 °C, even when the temperature was increased up to 37 °C at the very late stages of the stationary phase. Moreover, the slow character of the irreversible denaturation suffered by the CBM at 37 °C after choline binding could also explain why M32 cells kept at 37 °C up to early stages of the stationary phase could recover the autolytic capacity when they were shifted to 30 °C as well as the in vitro protection of LytC against thermal inactivation. Nevertheless, a long exposure to higher temperatures when no new molecules of LytC are being synthesized would lead to an irreversible loss of the cell wall binding capacity with the subsequent blocking of the lytic activity, as we observed in M32 cultures (Fig. 6, A and B) (the actual rate being dependent on the local concentration of choline residues in the cell wall). The present results cannot rule out the possibility, however, that in addition to the role played by the CBM, other factors could also contribute to regulate the temperature-activity profile of LytC in vivo.
DISCUSSION
The murein hydrolases of the pneumococcal cell wall so far characterized present a modular organization that could be the result of an adaptive evolution directed by the presence of choline in the lipoteichoic and teichoic acids of the pneumococcal envelope. The acquisition of the cell wall binding module, which specifically targets the phosphorylcholine moieties of these teichoic acids, increases the catalytic efficiency on intact cell walls by several orders of magnitude (10, 11, 16). Such enhancement strongly suggests that choline binding does not merely increase the effective protein concentration in the cell wall (35, 36) but also favors the access of the muropeptide to the catalytic cavity (9, 10, 37, 38).
LytC behaves as an autolytic enzyme at 30 °C. The lysis of the LytA-deficient mutant M32 sharply decreases above this temperature, becoming completely resistant to autolysis at 37 °C (11). Here we show that the CBM module of LytC is intrinsically unstable at temperatures as low as 4 °C and that the ultimate folding and stabilization of the native, active form relies on choline binding. Indeed, the loss of the cell wall binding capacity, by either deletion of the CBM (C-LytC truncated protein, comprising the CM of LytC) or substitution of choline residues by its analog ethanolamine, reduces LytC activity by about 3 orders of magnitude (11), as similarly observed upon the thermal denaturation of the CBM. Besides, an unstable and partially folded CBM can also contribute to regulation of LytC activity, since the stability of tertiary structures is inversely related to degradation rates by intracellular proteases (34).
The conformational changes experienced by LytC upon choline binding directly affect the environment of the aromatic residues present in the β-hairpins of choline-binding repeats (14-17, 24, 39) and also promote LytC monomerization. When compared with other CBMs (8-10), the larger solvent-accessible surface and the reduced length of the β-hairpins forming the shorter choline-binding repeats of LytC, inferred from its three-dimensional model (14), may account for the dramatic instability of its CBM (17, 25, 40), whose structure is characterized, as in other β-solenoids, by the absence of long range contacts.
In contrast to LytC, the activity of Cpl-1 is optimal at 37 °C (11). DSC and CD denaturation data show that the phage lysozyme is folded at this temperature, regardless of whether is bound to choline or not. On the other hand, the intrinsic stability of the Cpl-1 CM is comparable with the LytC one. Again, this finding supports the notion that the extreme sensitivity of LytC against thermal inactivation primarily reflects the marginal stability of its CBM.
Although the mature form of LytC is tightly attached to the cell wall (11), where the folded native state would be favored by choline-protein interactions, the intrinsic instability of the CBM can also determine the specificity of binding and the complex dynamics. Physiologically unstructured regions whose folding is coupled to the interaction with the targeted molecules offer several important advantages in systems involved in cellular signaling and regulation: (i) they are inherently flexible and can be easily shaped by the environment; (ii) they can recognize a more extensive area than a compact folded state with restrained conformational flexibility (41); and (iii) tight binding would only occur when the complementarity with the target is maximal, since the free energy expended to bring about the induced folding transition would come at the cost of a reduction in the net free energy of complex formation. By this reason, although the apparent affinities of LytC and Cpl-1 for choline estimated from CD titration curves are similar (Kapp ∼ 3 mm), the favorable free energy change contributed by direct protein-ligand interactions, a measure of choline intrinsic affinity, should be higher for LytC than for Cpl-1. Six of the 11 choline-binding repeats of LytC have a tyrosine close to the fully conserved glycine (Fig. 1B). As proposed in the pneumococcal phosphorylcholine esterase Pce (10), such residues, not conserved in the Cpl-1 lysozyme, could provide an additional aromatic residue to certain choline-binding sites, increasing in this way the affinity of LytC for choline.
As mentioned above, CBMs enhance the activity of pneumococcal cell wall hydrolases by increasing the effective enzyme concentration in the proximity of the bond to be hydrolyzed and also by facilitating the access of the substrate into the catalytic cavity. Hence, the overall structure of the Cpl-1 dimer can be a determinant for the Cpl-1 lytic activity, as it has been previously shown to be for the pneumococcal LytA amidase (25). As in other polymeric systems, in vivo, the strength and selectivity of the binding relies on the ability of CBMs to form multiple, weak, noncovalent bonds with the choline moieties of the pneumococcal cell wall. Optimal ligand-protein interactions would therefore require a certain disposition of choline residues. In the Cpl-1 dimer, the choline-binding region presents a boomerang-like structure with the four functional sites localized around the dimerization interface and the CMs located at both extremes of the boomerang (9, 32). Thus, the dimerization of Cpl-1 doubles the potential cell wall binding capacity of the monomer but also imposes a spatial orientation of both the choline-binding sites and the active sites. In contrast, LytC is an elongated molecule whose CBM forms a triangular prism of ∼100 Å long with eight choline-binding sites distributed along its lateral faces and the CM at its C terminus. Since the strength of the interactions between CBMs and the cell wall results from the multidentate binding of phosphocholine moieties and choline-binding repeats, the higher binding capacity provided by the 11 repeats of the LytC CBM and the structural flexibility of this module could explain why the choline half-inhibitory concentration for lytic activity on cell walls is notably higher for LytC (IC50 = 30 mm) than for Cpl-1 (IC50 = 2 mm) (11).
In addition, a highly efficient enzyme should also require an easy transit across the glycopeptide network to reach new cleavable bonds. This would be impaired by a strong binding to the cell wall and also by slow rates of choline binding and dissociation, both conditions being displayed by LytC, whose activity in vivo might be thus controlled by reducing its mobility across the pneumococcal cell wall. Interestingly, deletion of the CBM of Cpl-1 decreases the activity of the truncated N-Cpl-1 protein (comprising the catalytic module of Cpl-1) on intact cell walls by 5 orders of magnitude, becoming comparable with the activity reported for the C-LytC protein, the isolated CM of the pneumococcal lysozyme (11, 16). These results further support the idea that the main differences between LytC and Cpl-1 activities, under optimal conditions, could be derived from the interaction of their CBMs with the cell wall and the restraints imposed by their modular structures in order to bind, simultaneously, the teichoic acids and the muropeptide moiety to be hydrolyzed.
In summary, pneumococcus has acquired a rather unstable and moderately efficient lysozyme, whose activity is under the control of the general metabolic regulatory system of pneumococcus (11). On the contrary, in the bacteriophage, a late gene codes for Cpl-1 lysozyme that acts in a coordinated way with the Cph-1 holin, which allows the lytic enzyme to access the pneumococcal peptidoglycan at the time of releasing the virus progeny (42). Current results are consistent with the hypothesis that regulation and control of the potentially suicidal activity of LytC could be achieved by a tight binding to the pneumococcal envelope, derived from the high choline avidity of its CBM, the slow dynamics of the complex, and, probably, a rapid protein turnover of the intracellular unprocessed form. In addition, thermal denaturation of its intrinsically unstable CBM above 30 °C compromises the lytic activity of LytC, which might be constrained to habitats like the upper, highly ventilated, respiratory tract, where LytC might also contribute to DNA liberation by competent cells (13). In contrast, the higher activity achieved by the bacteriophage-encoded enzyme could be linked, to a great extent, to a moderate capacity for interacting with teichoic acids, faster rates for choline binding and dissociation, choline-induced dimerization, and a well folded and stable CBM. Nevertheless, other factors directly related to substrate recognition by the CM cannot at present be ruled out.
Supplementary Material
This work was supported by Dirección General de Investigación Cientifica y Técnica Grants BIO2003-01952, BFU2006-10288, and SAF2006-00390. Additional funding was provided by the COMBACT program (S-BIO-0260/2006) of the Comunidad de Madrid, the CIBER of Respiratory Diseases (CIBERES), an initiative of the ISCIII, and the Glycodynamics Network (FP6-UE and MCTN-CT-2005-019561). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-3 and Tables 1 and 2.
Footnotes
The abbreviations used are: CM, catalytic module; CBM, pneumococcal choline-binding module; DSC, differential scanning calorimetry.
References
- 1.Höltje, J. V. (1998) Microbiol. Mol. Biol. 62 181-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Höltje, J. V., and Tuomanen, E. (1991) J. Gen. Microbiol. 137 441-454 [DOI] [PubMed] [Google Scholar]
- 3.Shockman, G. D., and Höltje, J. V. (1994) in The Bacterial Cell Wall (Ghuyssen, J. M., and Hakenbeck, R., eds) pp. 131-166, Elsevier, Amsterdam, Netherlands
- 4.López, R., García, E., García, P., and García, J. L. (1997) Microb. Drug Resist. 3 199-211 [DOI] [PubMed] [Google Scholar]
- 5.Tomasz, A. (1967) Science 157 694-697 [DOI] [PubMed] [Google Scholar]
- 6.García, J. L., Sánchez-Beato, A. R., Medrano, F. J., and López, R. (2000) in Streptococcus pneumoniae: Molecular Biology and Mechanisms of Disease (Tomasz, A., ed) pp. 231-244, Mary Ann Liebert, Inc., Larchmont, NY
- 7.López, R., and García, E. (2004) FEMS Microbiol. Rev. 28 553-580 [DOI] [PubMed] [Google Scholar]
- 8.Fernández-Tornero, C., López, R., García, E., Giménez-Gallego, G., and Romero, A. (2001) Nat. Struct. Biol. 8 1020-1024 [DOI] [PubMed] [Google Scholar]
- 9.Hermoso, J. A., Monterroso, B., Albert, A., Galán, B., Ahrazem, O., García, P., Martínez-Ripoll, M., García, J. L., and Menéndez, M. (2003) Structure 11 1239-1249 [DOI] [PubMed] [Google Scholar]
- 10.Hermoso, J. A., Lagartera, L., González, A., Stelter, M., García, P., Martínez-Ripoll, M., García, J. L., and Menéndez, M. (2005) Nat. Struct. Mol. Biol. 12 533-538 [DOI] [PubMed] [Google Scholar]
- 11.García, P., González, M. P., García, E., García, J. L., and López, R. (1999) Mol. Microbiol. 33 128-138 [DOI] [PubMed] [Google Scholar]
- 12.Gosink, K. K., Mann, E. R., Guglielmo, C., Tuomanen, E. I., and Masure, H. R. (2000) Infect. Immun. 68 5690-5695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claverys, J. P., and Håvarstein, L. S. (2007) Nat. Rev. Microbiol. 5 219-229 [DOI] [PubMed] [Google Scholar]
- 14.Monterroso, B., López-Zumel, C., García, J. L., Sáiz, J. L., García, P., Campillo, N. E., and Menéndez, M. (2005) Biochem. J. 391 41-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medrano, F. J., Gasset, M., López-Zumel, C., Usobiaga, P., García, J. L., and Menéndez, M. (1996) J. Biol. Chem. 271 29152-29161 [DOI] [PubMed] [Google Scholar]
- 16.Sanz, J. M., Díaz, E., and García, J. L. (1992) Mol. Microbiol. 6 921-931 [DOI] [PubMed] [Google Scholar]
- 17.Varea, J., Monterroso, B., Sáiz, J. L., López-Zumel, C., García, J. L., Laynez, J., García, P., and Menéndez, M. (2004) J. Biol. Chem. 279 43697-43707 [DOI] [PubMed] [Google Scholar]
- 18.Laue, T. M., Shah, B. D., Ridgeway, T. M., and Pelletier, S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding, S. E., Horton, H. C., and Rowe, A. J., eds) pp. 90-125, Royal Society of Chemistry, London, UK
- 19.Sánchez-Ruiz, J. M. (1992) Biophys. J. 61 921-935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freire, E., van Osdol, W. W., Mayorga, O. L., and Sánchez-Ruiz, J. M. (1990) Annu. Rev. Biophys. Biophys. Chem. 19 159-188 [DOI] [PubMed] [Google Scholar]
- 21.Velázquez-Campoy, A., Ohtaka, H., Nezami, A., Muzammil, S., and Freire, E. (2004) Curr. Protein Cell Biol. 23 17.8.1-17.8.24 [DOI] [PubMed] [Google Scholar]
- 22.Bains, G., Lee, R. T., Lee, Y. C., and Freire, E. (1992) Biochemistry 31 12624-12628 [DOI] [PubMed] [Google Scholar]
- 23.Gill, S. J., and Wyman, J. (1990) in Binding and Linkage (Kelly, A., ed) pp. 80-81, University Science Books, Mill Valley, CA
- 24.Sáiz, J. L., López-Zumel, C., Monterroso, B., Varea, J., Arrondo, J. L. R., Iloro, I., García, J. L., Laynez, J. L., and Menéndez, M. (2002) Protein Sci. 11 1788-1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varea, J., Sáiz, J. L., López-Zumel, C., Monterroso, B., Medrano, F. J., Arrondo, J. L. R., Iloro, I., Laynez, J., García, J. L., and Menéndez, M. (2000) J. Biol. Chem. 275 26842-26855 [DOI] [PubMed] [Google Scholar]
- 26.Strickland, E. H., Horwitz, J., and Billups, C. (1969) Biochemistry 8 3205-3213 [DOI] [PubMed] [Google Scholar]
- 27.Usobiaga, P., Medrano, F. J., Gasset, M., García, J. L., Sáiz, J. L., Rivas, G., Laynez, J., and Menéndez, M. (1996) J. Biol. Chem. 271 6832-6838 [DOI] [PubMed] [Google Scholar]
- 28.Sanz, J. M., García, J. L., Laynez, J., Usobiaga, P., and Menéndez, M. (1993) J. Biol. Chem. 268 6125-6130 [PubMed] [Google Scholar]
- 29.Brandts, J. F., Ju, C., Lin, L. N., and Mas, M. T. (1989) Biochemistry 28 8588-8596 [DOI] [PubMed] [Google Scholar]
- 30.Luque, I., Leavitt, S. A., and Freire, E. (2002) Annu. Rev. Biophys. Biomol. Struct. 31 235-256 [DOI] [PubMed] [Google Scholar]
- 31.Eftink, M., and Biltonen, R. (1980) in Biological Microcalorimetry (Beezer, A. E., ed) pp 343-412, Academic Press, London
- 32.Martínez-Buey, R., Monterroso, B., Menéndez, M. Diakun, G., Chacón, P., Hermoso, J. A., and Díaz, J. F. (2007) J. Mol. Biol. 365 411-424 [DOI] [PubMed] [Google Scholar]
- 33.Tomasz, A. (1968) Proc. Natl. Acad. Sci. U. S. A. 59 86-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parsell, D. A., and Sauer, R. T. (1989) J. Biol. Chem. 264 7590-7595 [PubMed] [Google Scholar]
- 35.Din, N., Damude, H. G., Gilkes, N. R., Miller, R. C., Warren, R. A. G., and Kilburn, D. G. (1994) Proc. Natl. Acad. Sci. U. S. A. 87 11383-11387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gill, J., Rixon, J. E., Bolam, D. N., McQueen-Mason, S., Simpson, P. J., Williamson, M. P., Hazlewood, G. P., and Gilbert, H. J. (1999) Biochem. J. 342 473-480 [PMC free article] [PubMed] [Google Scholar]
- 37.Severin, A., Horne, D., and Tomasz, A. (1997) Microb. Drug Resist. 3 391-400 [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Dorado, I., Campillo, N., Monterroso, B., Hesek, D., Lee, M., Páez, J. A., García, P., Martínez-Ripoll, M., García, J. L., Mobashery, S., Menéndez, M., and Hermoso, J. A. (2007) J. Biol. Chem. 282 24990-24999 [DOI] [PubMed] [Google Scholar]
- 39.Freskgård, P.-O., Mårtensson, L.-G., Jonasson, P., Jonsson, B.-H., and Carlsson, U. (1994) Biochemistry 33 14281-14288 [DOI] [PubMed] [Google Scholar]
- 40.Lagartera, L., González, A., Hermoso, J. A., Sáiz, J. L., García, P., García, J. L., and Menéndez, M. (2005) Protein Sci. 14 3013-3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dyson, H. J., and Wright, P. E. (2002) Curr. Opin. Struct. Biol. 12 54-60 [DOI] [PubMed] [Google Scholar]
- 42.Martín, A. C., López, R., and García, P. (1998) J. Bacteriol. 180 210-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.