

The interconversion of Ar—H and Ar—FG (FG = functional group) is an attractive process for the synthesis of functionalized aromatic rings and the development of transition-metal catalysts for achieving this transformation is currently an area of intense activity. Whilst a range of efficient catalyst systems have recently been reported, it is of note that nearly all of them require high temperatures and/or powerful oxidants.[1] Indeed, the development of an effective catalytic protocol for carbonylation using CO by palladium(II)-mediated C—H activation has long been recognized as being difficult and only recently has the first example been reported.[2] Herein we report a room-temperature, urea-directed, palladium-catalyzed carbonylation that provides access to biologically and synthetically useful anthranilic acid derivatives and heterocycles under mild conditions.
We recently reported the development of a palladium(II)-catalyzed 1,2-carboamination of dienes using aryl urea derivatives.[3] During these investigations, [Pd(OTs)2-(MeCN)2] (Ts = 4-toluenesulfonyl) was identified as a highly efficient precatalyst and we proposed that the reaction proceeded by a urea-directed C—H insertion to give a reactive Pd—Ar complex. In subsequent attempts to isolate Pd—Ar complexes, we found that reaction of the m-toluidine urea 1 (Scheme 1) with one equivalent of [Pd(OTs)2(MeCN)2] in anhydrous THF led to rapid precipitation of the ortho-palladate 2 as a grey amorphous solid, which was readily isolated in analytically pure form (79%). Crystallization of 2 from CH2Cl2/petroleum ether afforded the cationic hydrate complex 3 whose molecular structure was confirmed by X-ray crystallography (Figure 1).
Scheme 1.

Formation and reactions of PdII complex 2. NBS = N-bromosuccinimide, NCS = N-chlorosuccinimide.
Figure 1.
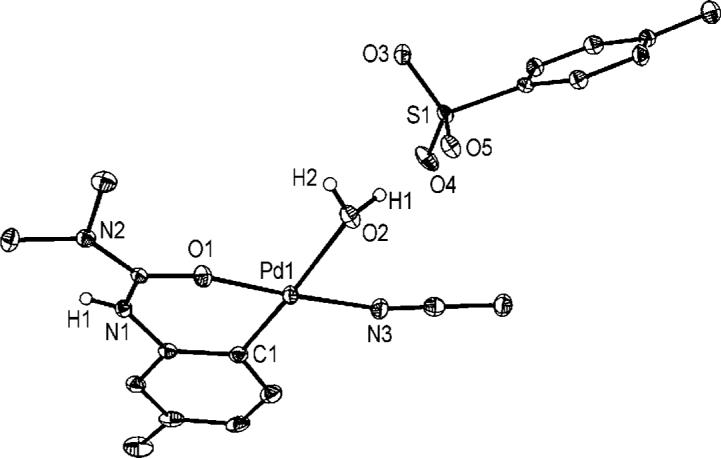
Molecular structure of 3 (thermal ellipsoids shown at 50% probability).
With a ready supply of 2 in hand we explored its reactivity towards a range of coupling partners and reagents (Scheme 1).[4,5] Methoxycarbonylation[6] (CO/MeOH) and arylation (Ph—M, M = B, Sn, etc.) were found to be effective for the formation of the anthranilic ester 4 and biaryl 5 respectively. Interestingly, carbonylation of 2 in the absence of MeOH led to the instantaneous formation of the cyclic imidate 6 by internal nucleophilic capture of the intermediate acyl palladate.[7]
The ability to selectively convert 2 into either anthranilate 4 or cyclic imidate 6 by stoichiometric reaction with either CO/MeOH or CO, led us to focus on developing these into catalytic processes. After a brief optimization study, it was found that a range of aryl urea derivatives (7) underwent rapid carbonylation ([Pd(OTs)2(MeCN)2], 5 mol%), benzoquinone (BQ, 2 equivalents), TsOH (1 equivalent), CO (1 atm), CH2Cl2, reaction time ≤ 5 hours) at ambient temperature (18 °C) to afford cyclic imidates[8] (8) in moderate to good yields (Table 1). In contrast to the reactions of aniline-derived urea derivatives with dienes,[3] this procedure did not require anhydrous conditions, although TsOH was essential for catalytic turnover. Prolonged reaction times resulted in lower yields, which are presumably a reflection of the acid sensitivity of the imidate products 8.
Table 1.
Room-temperature palladium(II)-catalyzed ortho-carbonylation of aryl urea derivatives.
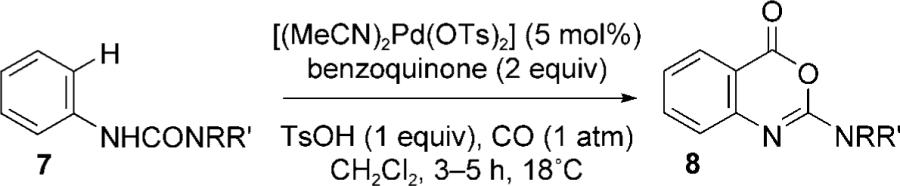 | |||||
|---|---|---|---|---|---|
| Entry | 7 | R | R′ | 8 | Yield [%] |
| 1 | 7a | Me | Me | 8a | 67 (75)[a] |
| 2 | 7b | Et | Et | 8b | 50 |
| 3 | 7c | iPr | iPr | 8c | 40 |
| 4 | 7d | morpholine | 8d | 49 | |
| 5 | 7e | Me | H | 8e | 34 |
| 6 | 7f | Et | H | 8f | 40 |
| 7 | 7g | iPr | H | 8g | 66 |
| 8 | 7h | tBu | H | 8h | 85 |
Reaction conducted with 10 mol% precatalyst.
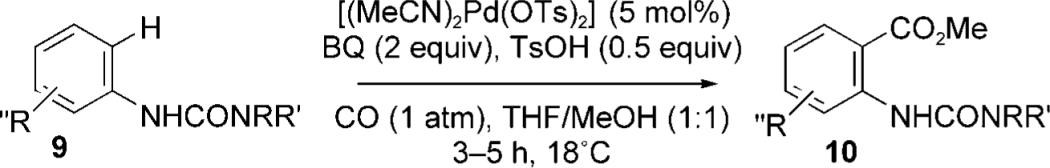
When these palladium(II)-catalyzed reactions were conducted in MeOH as solvent instead of CH2Cl2, methoxycarbonylation gave the desired anthranilate, but yields were compromised by contamination with significant quantities of an unidentified resinous material.[9] After screening a variety of cosolvents, we found that dilution of the MeOH with THF (1:1) gave excellent results with minimal by-product formation. It was also found that the amount of TsOH could be reduced to 0.5 equivalents. These conditions were then applied to a variety of substituted aniline derivatives (Table 2) and generally gave excellent yields of anthranilates 10. The reaction proceeded readily at room temperature with short reaction times and was unaffected by substitution on the non-aryl urea nitrogen atom. The reaction was generally tolerant of substitution in the aniline ring, although highly electron-withdrawing substituents were troublesome.[10] For example, the p-CF3-bearing substrate 9p gave only traces of anthranilate product (Table 2, entry 16) at ambient temperature, although a 30% yield of 10p could be obtained at 50 °C. The p-CO2Me analogue (9t) benefited similarly (Table 2, entry 20), which reinforced the conclusion that the palladium(II)-catalyzed C—H activation sequence is electrophilic in nature.[3] Interestingly, all three MeO isomers (Table 2, entries 13−15) reacted inefficiently at 18 °C but gave moderate to excellent yields of anthranilates at 50 °C. The 1-naphthylamine derivative (Table 2, entry 21) gave only cyclized imidate 11 (see below). Significantly, acetanilide failed to undergo reaction even at 50 °C. This latter result highlights the powerful activation effect of the urea moiety with this precatalyst.[3,11]
Table 2.
Room temperature palladium(II)-catalyzed methoxycarbonylation.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 9 | R | R′ | R″ | 10 | Yield [%] |
| 1 | 9a | Me | Me | H | 10a | 88 (97)[a] |
| 2 | 9b | Et | Et | H | 10b | 76 |
| 3 | 9c | iPr | iPr | H | 10c | 55 (75)[b] |
| 4 | 9d | morpholine | H | 10d | 75 | |
| 5 | 9e | Me | H | H | 10e | 61 |
| 6 | 9f | Et | H | H | 10f | 81 |
| 7 | 9g | iPr | H | H | 10g | 90 |
| 8 | 9h | tBu | H | H | 10h | 88 |
| 9 | 9i | Bn | H | H | 10i | 85 |
| 10 | 9j | Me | Me | o-Me | 10j | 88 |
| 11 | 9k | Me | Me | m-Me | 10k | 84 |
| 12 | 9l | Me | Me | p-Me | 10l | 78 |
| 13 | 9m | Me | Me | o-MeO | 10m | 15 (40)[c] |
| 14 | 9n | Me | Me | m-MeO | 10n | 26 (60)[c] |
| 15 | 9o | Me | Me | p-MeO | 10o | 31 (80)[c] |
| 16 | 9p | Me | Me | p-CF3 | 10p | 5 (30)[c] |
| 17 | 9q | Me | Me | m-Br | 10q | 70 |
| 18 | 9r | Me | Me | p-Br | 10r | 40 |
| 19 | 9 s | Me | Me | o,p-Me | 10 s | 56 |
| 20 | 9t | Me | Me | p-CO2Me | 10t | 36 (46)[c] |
| 21 |  |
54 | ||||
Reactions conducted with 10 mol% precatalyst.
Reaction conducted in 100% MeOH with 3 equiv BQ.
Reactions conducted at 50 °C.
In certain cases, the transient formation of cyclic imidates (8) was observed by TLC, which raised the question of whether ester formation (10) proceeds by direct solvolysis of an acyl palladate (12→10) or by a postcatalytic methanolysis of the imidate (13→10). Control experiments[12] demonstrated that the imidates are very labile[8] under the methanolic carbonylation conditions and that the likely mechanism for the formation of 10 from 9 is by a C—H insertion, carbonylation, cyclization, and solvolytic ring-opening sequence (Scheme 2).
Scheme 2.
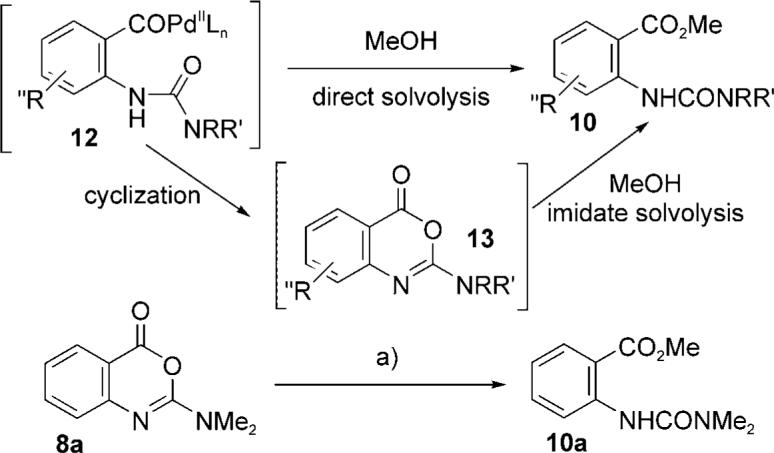
Mechanism of ester formation: direct solvolysis versus imidate formation. a) For reagents see Table 2, 1.5 h, 95%.
The synthetic utility of the urea anthranilates was further demonstrated by their cyclization to form quinazolinones, a key heterocyclic pharmacophore in numerous drug substances.[13] Indeed, carbonylation of 9i followed by the addition of potassium carbonate[14] and heating afforded the quinazolinone 14 in 80% overall yield in a one-pot sequence from 9i (Scheme 3). Both 9e and 9f reacted similarly to afford the corresponding quinazolinones in 57 and 70% yields, respectively.
Scheme 3.
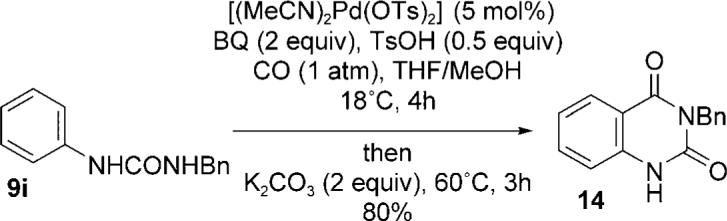
One-pot carbonylation—cyclization route to quinazolinones.
Finally, we investigated conditions for the selective cleavage of the urea moiety to provide convenient access to the methyl anthranilates and anthranilic acids. During this study we made the observation that aniline diisopropyl urea derivatives undergo an unprecedented facile solvolysis in methanol under neutral conditions.[15] Remarkably, we found that simply heating 10c in water gave methyl anthranilate 15 in 85% yield with no ester hydrolysis. The unusual reactivity of the diisopropyl urea moiety was further highlighted by the lack of reaction of 10a and 10b under identical conditions. Reaction of 10c with NaOH (1m) gave anthranilic acid 16 in excellent yield, which highlighted the diisopropyl urea moiety as an efficient “traceless” ortho-directing C—H activating group for anilines (Scheme 4).
Scheme 4.
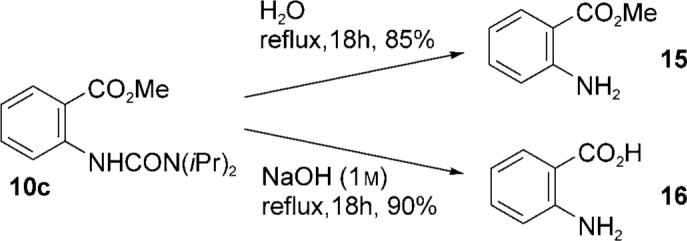
Facile hydrolysis of an aryl diisopropyl urea.
In summary, we have described a highly efficient, palladium(II)-catalyzed, and ortho-selective carbonylation sequence for aniline derivatives. The reaction proceeds efficiently under CO (1 atm), at room temperature (18 °C), and with 5% catalyst loadings. Key features of the reaction are the use of [Pd(OTs)2(MeCN)2] as precatalyst and the powerful activating effect of the aniline—urea moiety (compare Table 2, entry 1 (88%) versus acetanilide (0%)). The reaction conditions can be easily manipulated to produce either cyclic imidates (8) or methyl anthranilates (10). The use of N-aryl urea derivatives that bear a terminal N—H moiety allows the generation of quinazolinones by in situ base-mediated cyclization of the methoxycarbonylated products. Finally, we have demonstrated the unique ability of the diisopropyl urea moiety to function as a C—H-activating and -directing group that can be removed under neutral conditions, which represents the selective hydrolysis of a urea group in the presence of an ester group. We anticipate that these reactions will be of substantial utility for the preparation of anthranilic acids, and their derivatives, under mild conditions from the corresponding aniline.
Experimental Section
General procedure for methoxycarbonylation: A Radley's reduced-volume reaction vessel (10 mL) equipped with a stirring bar was charged with the desired urea derivative (1 mmol), benzoquinone (2 mmol), [Pd(OTs)2(MeCN)2] (5 mol%), tosic acid monohydrate (0.5 mmol), THF (0.5 mL), and methanol (0.5 mL). The vessel was connected to a Schlenk line and was briefly evacuated and stirred at room temperature (18 °C), followed by charging with CO to 1 atm. This cycle was repeated three times and the vessel left open to a dynamic atmosphere of CO (1 atm). The reaction mixture was then stirred at 18 °C and monitored by TLC. On completion (2−5 h), the reaction mixture was concentrated in vacuo, the residue dissolved in CH2Cl2 and washed with HCl (1m). The organic layer was then washed with water, dried over MgSO4, filtered and concentrated in vacuo. Purification by flash chromatography (10−40% EtOAc/petroleum ether afforded the pure products.
Footnotes
We thank the EPSRC (GR/R02382 and E061575) and AstraZeneca for support, the joint EPSRC/AstraZeneca/GlaxoSmithKline/Pfizer Organic Studentship Initiative, and Dr. M. Haddow for X-ray crystallography. G.C.L.-J. holds a Royal Society Wolfson Merit Award.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200805842.
Contributor Information
J. Gair Ford, AstraZeneca, Global Process R&D Silk Road, Macclesfield, Cheshire, SK10 2NA (UK).
Simon N. G. Tyler, AstraZeneca, Global Process R&D, Avlon Works Severn Road, Hallen, Bristol BS10 7ZE (UK)
Michel R. Gagné, Department of Chemistry, Caudill and Kenan Laboratories UNC-Chapel Hill, NC 27599−3290 (USA)
References
- 1.a Kakiuchi F, Kochi T. Synthesis. 2008:3013–3039. [Google Scholar]; b Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174–238. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; c Li BJ, Yang S-D, Shi ZJ. Synlett. 2008:949–957. [Google Scholar]; d Campeau L-C, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35. [Google Scholar]
- 2.During the preparation of this manuscript Yu and Giri reported the first palladium(II)-catalyzed ortho carbonylation of benzoic and phenylacetic acids by C-H insertion: Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:14082. doi: 10.1021/ja8063827..
- 3.Houlden CE, Bailey CD, Ford JG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. J. Am. Chem. Soc. 2008;130:10066. doi: 10.1021/ja803397y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For the stoichiometric reaction of aryl–PdII–amide complexes, see: Horino H, Inoue N. J. Org. Chem. 1981;46:4416..
- 5.For palladium-catalyzed C-H activation reactions with phenyl amides as directing groups, see: Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:2002. doi: 10.1021/ja0176907. (vinylation);Wang J-R, Yang C-T, Liu L, Guo Q-X. Tetrahedron Lett. 2007;48:5449.;Shi Z, Li B, Wan X, Cheng J, Fang Z, Cao B, Qin C, Wang Y. Angew. Chem. 2007;119:5650. doi: 10.1002/anie.200700590. (arylation with boronic acids);Angew. Chem. Int. Ed. 2007;46:5554.;Shi Z, Li B, Wan X, Cheng J, Fang Z, Cao B, Qin C, Wang Y. Angew. Chem. 2007;119:7874. doi: 10.1002/anie.200700590.;Angew. Chem. Int. Ed. 2007;46:7730–7730.;Brasche G, García-Fortanet J, Buchwald SL. Org. Lett. 2008;10:2207. doi: 10.1021/ol800619c. (arylation without boronic acids);Yang S, Li B, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:6066. doi: 10.1021/ja070767s.;Daugulis O, Zaitsev VG. Angew. Chem. 2005;117:4114. doi: 10.1002/anie.200500589.;Angew. Chem. Int. Ed. 2005;44:4046.;Li B-J, Tian S-L, Fang Z, Shi Z-J. Angew. Chem. 2008;120:1131–1134.;Angew. Chem. Int. Ed. 2008;47:1115–1118. doi: 10.1002/anie.200704092.;Yu W-Y, Sit WN, Lai K-M, Zhou Z, Chan ASC. J. Am. Chem. Soc. 2008;130:3304. doi: 10.1021/ja710555g. (ethoxycarbonylation);Wan X, Ma Z, Li B, Zhang K, Cao S, Zhang S, Shi Z. J. Am. Chem. Soc. 2006;128:7416. doi: 10.1021/ja060232j. (chlorination).
- 6.For CO insertion into palladacycles, see: Dupont J, Consorti C, Spencer J. Chem. Rev. 2005;105:2527. doi: 10.1021/cr030681r.;Ryabov AD. Synthesis. 1985:233..
- 7.For palladium-catalyzed urea ortho-carbonylation using thallium intermediates, see: Larock RC, Fellows CA. J. Am. Chem. Soc. 1982;104:1900.;Krantz A, Spencer RW, Tam TF, Liak T. US Patent 4,665,070. 1987;Krantz A, Spencer RW, Tam TF, Liak TJ, Copp LJ, Thomas EM, Rafferty SP. J. Med. Chem. 1990;33:464. doi: 10.1021/jm00164a002..
- 8.For the synthesis, biological activity, and reactions of 2-hetero-4H-3,1-benzoxazinones see: Coppola GM. J. Heterocycl. Chem. 2000;37:1369., and references therein.
- 9.PdII/CO has been used for the manufacture of polymers from diols. From NMR studies we suggest that polycarbonate oligomers are formed from hydroquinone repeating units and methanol termini: Okamoto M, Sugiyama J, Yamamoto T. J Appl. Polym. Sci. 2008;110:3902.;Okuyama K, Sugiyama J, Nagahata R, Asai M, Ueda M, Takeuchi K. Macromolecules. 2003;36:6953..
- 10.The p-NO2 derivative failed to react altogether.
- 11.We also found that methoxycarbonylation could be achieved with a) PdCl2 (10 mol %), BQ (3 equiv), CO, MeOH/THF (1:1) at 60 °C and b) Pd(OAc)2 (5 mol %), BQ (2 equiv), and TsOH (1 equiv) at 18 °C (see the Supporting Information for full details). However, significantly lower yields were obtained in these cases, which confirmed the superiority of the [Pd(OTs)2-(MeCN)2] precatalyst system for mild and selective C-H activation (Ref. [3]).
- 12.Pure 8a was subjected to the reaction conditions from Table 2. After 1.5 hours, all of 8a was consumed and the ester 10a was isolated in 95 % yield.
- 13.For recent reports on synthesis and biological activity of 2,4(1H,3H)-quinazolinediones see: Li Z, Huang HH, Sun H, Jiang H, Liu H. J. Comb. Chem. 2008;10:484. doi: 10.1021/cc800040z.;Larksarp C, Alper H. J. Org. Chem. 2000;65:2773. doi: 10.1021/jo991922r., and references therein.
- 14.For the formation of quinazolinediones from basic condensation of ortho-ester aryl urea groups, see: Papadopoulos EP, Torres CD. J. Heterocycl. Chem. 1982;19:269..
-
15.To the best of our knowledge, solvolysis of urea derivatives requires relatively harsh conditions, for example, high temperatures, strongly acidic or basic conditions, metal catalysis etc: Wang J, Li Q, Dong W, Kang M, Wang X, Peng S. Appl. Catal. 2004;261:191.;Kaminskaia NV, Kostíc NM. Inorg. Chem. 1998;37:4302. doi: 10.1021/ic980065r.;Ayyanger NR, Choudhary AR, Kalkote UR, Natu AA. Chem. Ind. 1998:599.. In contrast, we have found that aryl diisopropylamino ureas undergo solvolysis under particularly mild conditions. This is clearly a property imparted by the diisopropylamino group, and not a consequence of (contaminant) palladium catalysis as the following (palladium-free) example demonstrates: Capasso et al have demonstrated
that phenyldimethyl urea derivatives undergo hydrolysis under acidic or basic conditions via phenylisocyanate intermediates: Salvestrini S, DiCerbo P, Capasso S. J. Chem. Soc. Perkin Trans. 2. 2002:1889.. We propose that the ease with which phenyldiisopropyl urea derivatives undergo solvolysis may be a result of the leaving group ability of diisopropylamine being enhanced by the steric compression afforded by the isopropyl groups. Further mechanistic investigation is ongoing and will be reported in due course.