

Abstract
Cation transmission/electron transfer reagent anion storage mode electron transfer ion/ion reactions and beam-type collisional activation of the polypeptide ions are performed in rapid succession in the high pressure collision cell (Q2) of a quadrupole/time-of-flight tandem mass spectrometer (QqTOF), where the electron transfer reagent anions are accumulated. Duty cycles for both electron transfer dissociation (ETD) and collision-induced dissociation (CID) experiments are improved relative to ion trapping approaches since there are no discrete ion storage and reaction steps for ETD experiments and no discrete ion storage step and frequency tuning for CID experiments. For this technique, moderately high resolution and mass accuracy are also obtained due to mass analysis via the TOF analyzer. This relatively simple approach has been demonstrated with a triply charged tryptic peptide, a triply charged tryptic phosphopeptide, and a triply charged tryptic N-linked glycopeptide. For the tryptic peptide, the sequence is identified with more certainty than would be available from a single method alone due to the complementary information provided by these two dissociation methods. Because of the complementary information derived from both ETD and CID dissociation methods, peptide sequence and post-translational modification (PTM) sites for the phosphopeptide are identified. This combined ETD and CID approach is particularly useful for characterizing glycopeptides because ETD generates information about both peptide sequence and locations of the glycosylation sites while CID provides information about the glycan structure.
Introduction
Tandem mass spectrometry (MS) is a powerful tool for obtaining primary structure information from polypeptides. Indeed, it has become central to modern proteomics, particularly in the identification of a priori unknown proteins via protein digestion and subsequent structural characterization of the resulting peptides. The extent and quality of information that can be obtained from a tandem MS experiment is dependent both upon the chemical reaction that takes place between stages of MS and the figures of merit of the mass spectrometer used to analyze the products.1
Over the past two decades, much attention has been directed to gas-phase reactions that provide peptide structural information, with most success being obtained via peptide fragmentation.2 For this reason, the dissociation chemistries of various ionic forms of peptides (e.g., protonated species, metal-cationized species, radical cations, etc.) have been extensively explored,3-6 as well as the means for inducing fragmentation7. Much emphasis has been placed on multiply-protonated peptides because such ions are readily formed via electrospray ionization, which, in turn, is readily adapted to on-line separations, such as high performance liquid chromatography (HPLC). By far, collisional activation has been the method most widely applied to induce fragmentation of multiply protonated peptides formed via spray ionization. This technique, often referred to also as collision-induced dissociation (CID),8 has been sufficiently successful to warrant the widespread adoption of tandem mass spectrometry as a core tool in modern proteomics research. However, no form of ion activation has yet proved to be without limitations as a structural probe. Collisional activation, as it is implemented in most tandem mass spectrometers, for example, often fails to provide information regarding the sites of post-translational modifications (PTMs). Other activation methods, however, have been shown to be complementary to collisional activation. Prominent among these are electron-capture dissociation (ECD)9, 10 and electron transfer dissociation (ETD)11, 12, which have been shown to be particularly useful in locating PTM sites13-16. ETD and ECD have largely been implemented in so-called “tandem-in-time” instruments, which typically require separate ion accumulation, ion isolation, ion activation/dissociation, and mass analysis steps.1, 10, 12 CID has been implemented in both “tandem-in-time” and “tandem-in-space” instruments.1 In the latter type of instrument, ionization, ion isolation, ion/activation, and mass analysis steps occur more-or-less simultaneously.
Given the often complementary nature of the information provided by CID and ETD, it is desirable to be able to generate information from both techniques. Ideally, this would be done without significant compromise in the performance of either approach or with significant costs in time, duty cycle, etc. The recent implementation of transmission mode electron transfer ion/ion reactions on a hybrid linear ion trap allows for an increase in duty cycle since no discrete ion/ion mutual storage step is needed.17 In addition, one advantage associated with this transmission mode ETD method is that it doesn't require instrument modifications to superpose radio frequency (RF) on the containment lenses of the linear ion trap, as is required for reactions that employ mutual storage mode.
In this work, we demonstrate an approach that allows for the acquisition of both CID and ETD data in rapid succession in a hybrid quadrupole/time-of-flight tandem mass spectrometer. The approach involves a cation transmission/ETD reagent anion storage mode experiment via an ion/ion electron transfer with time-of-flight (TOF) acquisition of the product ion spectrum followed by a beam-type CID experiment with TOF data acquisition. Both the ETD and CID experiments are conducted in transmission mode, which obviates a discrete ion storage step and frequency tuning for CID. In this sense, both experiments are conducted in “tandem-in-space” fashion. The only interruption in data acquisition occurs when there is a periodic break to allow for ETD reagent ion accumulation in the collision cell. This step is mandated by the geometry of the instrument used here and introduced an element of “tandem-in-time” into the overall process. In any case, the resulting approach constitutes a relatively simple method for obtaining the structural information afforded by ETD and CID in rapid succession. This method is demonstrated by performing rapidly alternating ETD and CID experiments of a triply charged tryptic peptide from mygolobin, a triply charged tryptic phosphopeptide from α-casein and a triply charged tryptic glycopeptide from Erythrina cristagalli lectin.
Experimental
Materials
Methanol, acetonitrile and glacial acetic acid were purchased from Mallinckrodt (Phillipsburg, NJ). Horse myoglobin, bovine α-casein, lectin from Erythrina cristagalli (Cockspur Coral Tree), TPCK-treated trypsin, ammonium bicarbonate and azobenzene were obtained from Sigma-Aldrich (St. Louis, MO). Trifluoroacetic acid (TFA) was purchased from Pierce (Rockford, IL). All materials were used without further purification.
Trypsin Digestion
Myoglobin (1 mg) was dissolved in 0.5 mL of aqueous 200 mM ammonium bicarbonate. TPCK-treated trypsin (20 μL of a 1 mg/mL aqueous solution) was added to the protein solution to effect the digestion. The protein solution was incubated at 38°C in a water bath for ∼12 h followed by separation on a reverse-phase HPLC (Agilent 1100, Palo Alto, CA) using an Aquapore RP-300 (7 μm particle size, 100 × 4.6 mm i.d.) column (Perkin-Elmer, Wellesley, MA) operated at 1 mL/min. A linear 60 min gradient from 0 to 100% buffer B was used, where buffer A was 0.1% (v) TFA aqueous solution and buffer B was 59.5/39.6/0.09 (v/v/v) acetonitrile/water/TFA. The fractions were dried in vacuum and then dissolved in 200 μL of 49.5/49.5/1 (v/v/v) methanol/water/acetic acid for positive nano-ESI. E. cristagalli lectin and α-casein were also trypsin digested, then separated by reverse-phase HPLC using a similar procedure as that outlined for myoglobin. The only difference is the digestion time, ∼4 h for E. cristagalli lectin and ∼24 h for alpha-casein.
Mass Spectrometry
All experiments were performed using a QqTOF tandem mass spectrometer (QSTAR XL, Applied Biosystems/MDS Sciex, Concord, ON, Canada) modified for ion/ion reaction experiments.18, 19 A home-built pulsed dual nano-ESI/atmosphere pressure chemical ionization (APCI) source,20 as shown schematically in Scheme 1, was coupled directly to the interface of the QSTAR mass spectrometer to generate multiply charged peptide cations and azobenzene radical anions, respectively. Both positive ion transmission mode ETD (passing the analyte cations while the reagent radical anions are trapped) and beam type CID were effected in the Q2 linear ion trap (LIT), controlled by Daetalyst 3.14, a research version of software provided by MDS Sciex. Daetalyst 3.14 enabled both ETD and CID experiments in a single scan function. The collision energy (CE) of ions under the beam-type CID is defined by the product of ion charge and the DC potential difference between Q2 and Q0 quadrupole arrays. That is, CE = (Q0-Q2)*Z, where Z represents the charge of the ions. For different polypeptide ions, all the potentials applied to the ion optical elements are the same, except the potentials applied from Q0 through Q2 in the beam-type CID step. The potential difference, Q0-Q2, might need to be optimized to generate high quality CID spectra. However, the time required to adjust the Q0-Q2 potential difference in the beam-type CID step is much less compared to the time needed for the frequency and amplitude tuning during ion trap CID. The pressure in the Q2 LIT was optimized at ∼5 mTorr with nitrogen as the bath gas.
Scheme 1.
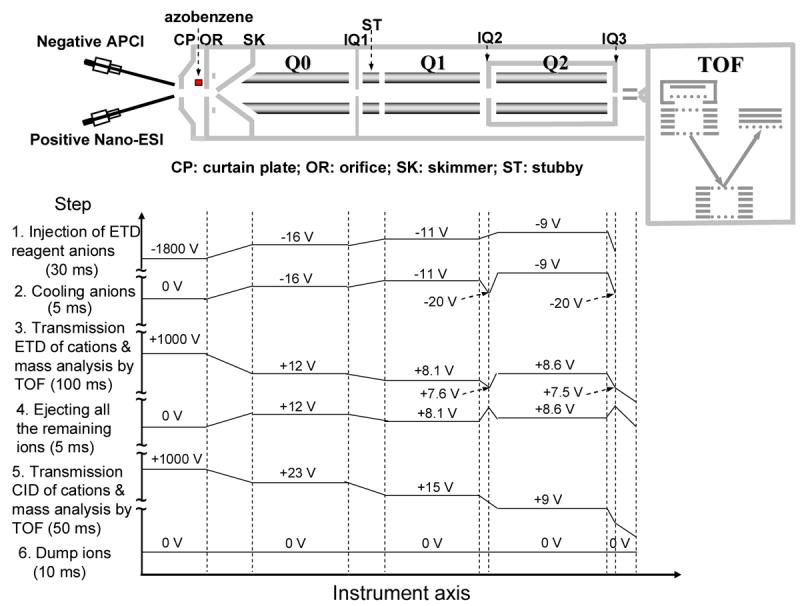
Schematic of the QSTAR mass spectrometer equipped with a home-built pulsed dual nano-ESI/APCI source. The plots below show the potentials along the instrument axis at different steps for alternating ETD and CID experiments of triply charged tryptic peptide HGTVVLTALGGILK from myoglobin.
A typical scan function for alternating cation transmission mode ETD and beam type CID consists of the following steps: (1) pulsing the high voltage (−2 kV) applied to the APCI needle and injecting the azobenzene radical anions, isolated by Q1 in the mass-resolving mode, into the Q2 LIT where ions are trapped by DC (30 ms); (2) switching off the high voltage on the APCI needle while the anions were cooled in Q2 for 5 ms; (3) switching on the high voltage (+1.0-1.5 kV) applied to the nano-ESI emitter, and passing the Q1 isolated analyte ions through the Q2 LIT to effect ETD and acquisition of product ions by reflectron TOF mass analyzer (100 ms); (4) ejecting all the remaining anions from Q2 (5 ms); (5) performing beam type CID with TOF data acquisition of product ions (50 ms); (6) dump any remaining ions that might be present in Q0-Q2 (10 ms). The injection q value (Mathieu dimensionless parameter)21 for the azobenzene reagent anions was ∼0.39 and the DC trapping voltage applied to IQ2 and IQ3, the containment lenses of Q2, was 1 V relative to the Q2 DC offset. For the tryptic glycopeptide from E. cristagalli lectin, an auxiliary AC in resonance with the secular frequency of the doubly charged electron transfer without dissociation (ET, no D) product ions was applied to Q2 in step 3 to enhance ETD.22 This collisional activation approach in the radial direction during the transmission mode electron transfer ion/ion reaction is demonstrated in detail elsewhere.23 The potentials along the instrument axis at different steps plotted in Scheme 1 were typical for the ETD experiments, while the potentials applied from Q0 to Q2 were optimized for the CID of each of the different analyte ions. Scheme 1 summarizes the potentials applied in the rapidly alternating ETD and CID experiment of the triply charged peptide HGTVVLTALGGILK from trypsin digestion of myoglobin. The TOF mass spectra shown herein were typically the averages of 100 individual scans.
Results and Discussion
Duty cycles of ETD and CID experiments performed in rapid succession
The fractional duty cycles for ETD and CID experiments in this study are defined as following:
| (1) |
| (2) |
The duty cycle of the orthogonal acceleration TOF is m/z-dependent and typically ranges, in fractional terms, from 0.05 to 0.324. Recently, alternating ETD and CID have been implemented on quadrupole linear ion traps (LITs) and quadrupole ion traps (QITs) and used for data-dependent analyses.12, 25-27 In these ion trap instruments, ETD and CID experiments are normally performed by switching between them automatically under software control in the data-dependent mode. Duty cycles for the ETD and CID experiments are comparable. For typical combined ETD and CID experiments performed on a LIT instrument, ∼20 ms cation injection time and ∼300 ms total cycle time are commonly reported in the literature for both ETD and CID experiments.12 Therefore, duty cycles for ETD and CID experiments are the same at and would each be if ETD and CID experiments were performed sequentially on the same analyte ion. For the alternating ETD and CID experiments performed in this study, time spent in every step was the same for all three peptides studied here. ETD and CID spectra acquired in a single rapidly alternating scan function for the triply charged tryptic peptide with single letter sequence HGTVVLTALGGILK from myoglobin are shown in Figure 1. The injection and trapping q values for the triply charged HGTVVLTALGGILK in both ETD and CID steps were 0.21. In this experiment, the cation transmission time for ETD is the same as the electron transfer ion/ion reaction time, which is 100 ms, since the reaction was performed in the cation transmission mode. The cation transmission time for CID is the same as the collisional activation time, 50 ms, because beam-type CID was performed via the passing of the precursor ions through the Q2 LIT. Therefore, and . The overall analyte duty cycle (i.e., the sum of the duty cycles for ETD and CID) for the rapidly alternating ETD and CID experiment on the QqTOF instrument is 0.75 multiplied by the time-of-flight duty cycle. Assuming the latter to be in the range of 0.05 to 0.3, the ETD and CID duty cycles associated with the alternating experiments on the QqTOF instrument are comparable to those associated with the LIT experiment outlined above and provide superior mass measurement accuracy and resolving power. The overall cycle time (200 ms) of the QqTOF experiment is also shorter than the LIT experiment due to the parallel nature of the transmission mode approach. Furthermore, this approach is relatively simple because beam-type CID does not require the frequency tuning associated with resonance excitation ion trap CID experiments. As shown in Figure 1, ETD and CID of the triply charged tryptic peptide generate different sets of fragment ions. These two sets of fragment ions can be used together to identify the peptide with more certainty. 13 out of 13 peptide backbone N-Cα cleavages are observed in the ETD spectrum while 12 out of 13 peptide backbone amide bonds are cleaved by CID. Therefore, in this particular case, the ETD data alone could be used to completely sequence the peptide while either dissociation method can provide rich information for protein identification purposes.
Figure 1.
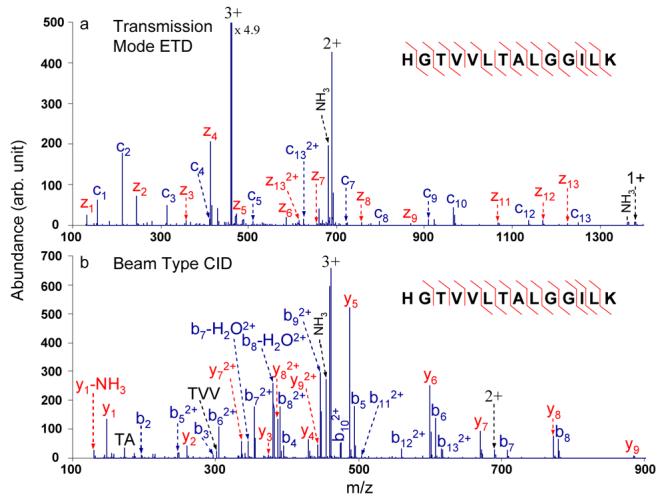
MS/MS spectra of the tryptic peptide HGTVVLTALGGILK from myoglobin: (a) transmission mode ETD of the triply charged peptide with azobenzene radical anions; (b) beam type CID of the triply charged peptide (CE = 42 eV).
Alternating ETD and CID experiments of tryptic peptides with PTMs
Phosphopeptide
The rapid alternating ETD and CID approach was applied to the polypeptide ions with PTMs in order to characterize them through the complementary information afforded by ETD and CID MS/MS analyses. CID of phosphopeptide ions often leads to loss of phosphoric acid (H3PO4), which can prevent localization of the modification site. The resulting CID spectra may also lack extensive structural information from peptide backbone cleavages, which is necessary to sequence the phosphopeptides and locate the phosphorylation sites. As shown in Figure 2b, the loss of H3PO4 from the peptide precursor ion and y-type fragment ions indicate that the peptide is phosphorylated. A neutral-loss triggered scan has been used to identify phosphopeptides.28, 29 However, more information is needed in order to characterize the sequence of the phosphopeptide and the phosphorylation sites. Figure 2a shows the ETD spectrum of the triply charged phosphopeptide with single letter sequence VPQLEIVPNpSAEER. The injection and trapping q values for the triply charged phosphorylated precursor ions in both ETD and CID steps were 0.19. Preservation of the phosphorylation site in the peptide precursor ions and fragment ions provides complementary information for this phosphopeptide, which is necessary to identify the modification site. Besides the location of the phosporylation site, 22 out of 26 possible c- and z-type fragment ions are observed in the ETD spectrum (85% sequence coverage) compared to only 10 out of 26 possible b- and y-type fragment ions observed in the CID spectrum (62% sequence coverage), where sequence coverage is defined as a percentage of possible backbone bond cleavages. Therefore, the ETD spectrum of this triply charged phosphopeptide generates more sequence information than the corresponding CID spectrum. The b7 and y7 species formed in the CID process provide complementary information compared to that derived from the ETD data. By combining the information derived from both ETD and CID data acquired in a single scan function, greater sequence coverage (92%) and the localization of the phosphorylation site are obtained.
Figure 2.
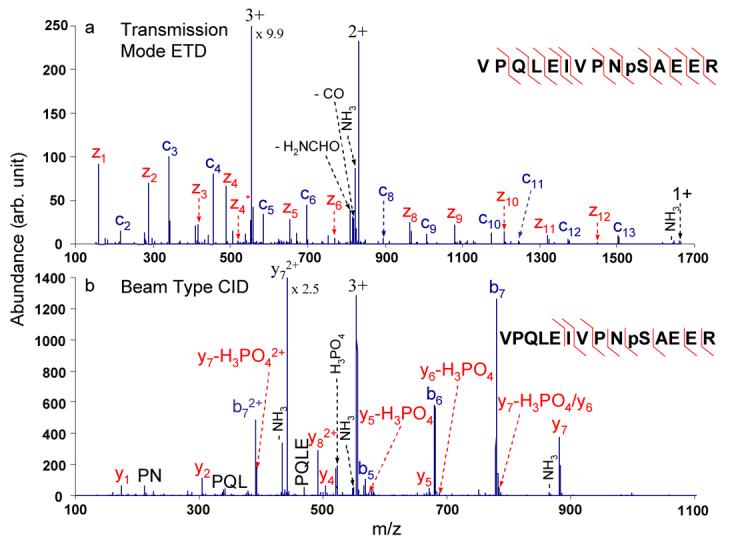
MS/MS spectra of the tryptic phosphopeptide VPQLEIVPNpSAEER from alpha-casein: (a) transmission mode ETD of the triply charged peptide with azobenzene radical anions; (b) beam type CID of the triply charged peptide (CE = 54 eV). z* denotes the O2 adduct of z ions.30
Glycopeptide
Glycosylation is one of the most common protein PTMs, and serves a variety of important functions in biological systems. CID of glycopeptides preferentially cleaves the glycosidic bonds with few or no peptide backbone fragment ions produced, which provides the structural information for the glycan exclusively. The N-glycosylated peptide used in this experiment was studied before by combining ECD/infrared multiphoton dissociation (IRMPD) and ETD/CID dissociation methods, which both provided information for the peptide sequence and the glycan structure.16, 31 The glycan structure and the glycopeptide sequence are known and shown in Figure 3. Beam-type CID spectrum of the triply charged glycopeptide is shown in Figure 3b, in which only the fragment ions corresponding to the cleavages of the glycosidic bonds are observed with no fragmentation of the peptide backbone. The glycan sequence can be determined by the product ions generated upon CID, although the exact linkage types of individual sugars and the locations of Xylose (Xyl) and Fucose (Fuc) cannot be inferred. Peptide sequence information and location of the glycosylation site could not be obtained from the CID experiment.
Figure 3.
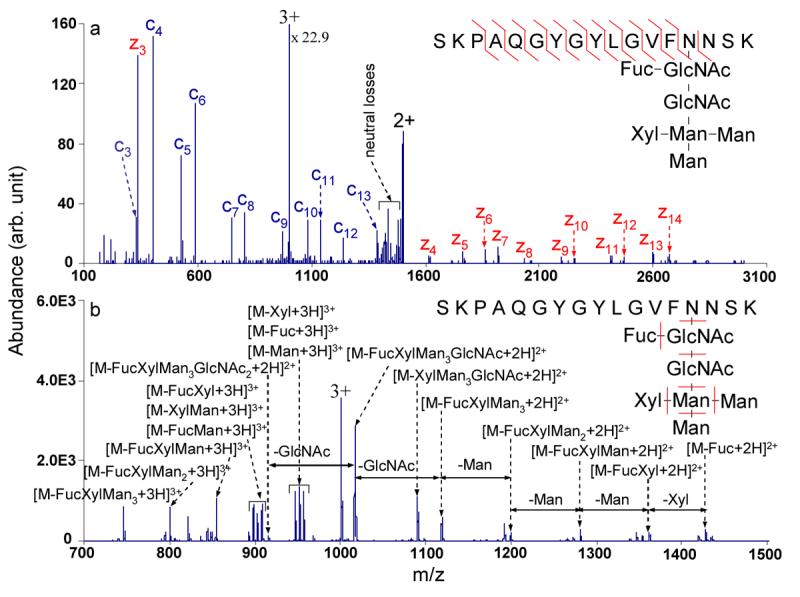
MS/MS spectra of the tryptic glycopeptide from lectin: (a) transmission mode ETD of the triply charged peptide with azobenzene radical anions (activation on doubly charged intact electron transfer species during the reaction: 48.60 kHz, 1.0 Vpp); (b) beam type CID of the triply charged peptide (CE = 93 eV).
The spectrum acquired for cation transmission mode electron transfer ion/ion reaction of the triply charged glycopeptide with azobenzene molecular anions is shown in Figure 3a. The injection q values for the triply charged glycosylated peptide ion in transmission mode ETD and CID steps were 0.12 and 0.10, respectively. During the transmission mode electron transfer reaction, an auxiliary AC in resonance with the doubly charged ET, no D product was applied to effect the radial activation on the non-dissociated electron transfer product to yield more c and z-type ETD fragment ions. The amplitude was finely tuned to fragment the electron transfer without dissociation product with few or no glycosidic bond cleavages. The electron transfer ion/ion reaction of the triply charged glycopeptide with azobenzene radical anions did not produce any c and z-type fragment ions when no radical activation was performed on the doubly charged ET, no D product, only neutral-loss peaks were observed (data not shown). The low ETD efficiency of the triply charged glycopeptide may be due to intramolecular interactions that prevent fragments from separating. With radial activation of the doubly charged intact electron transfer species, binding of the c/z-type fragments afforded by the non-covalent interactions can be broken to reveal c and z-type fragment ions. 12 out of 16 peptide backbone bonds are cleaved with 75% sequence coverage obtained and 23 out of 32 possible c/z-type fragment ions are generated upon activation and observed in Figure 3a. The odd-electron c-type ions and the even-electron z-type ions are present in the spectrum, which is consistent with the observations reported from CID of the intact charge reduced species from electron capture32 and electron transfer22, 30. Few observable glycosidic bond cleavages are seen under collisional activation in the radial direction. Most of the glycopeptide sequence information is derived from the backbone cleavages after radial activation on the electron transfer without dissociation product. The glycosylation site is determined to be the fourth amino acid from the C-terminus, the asparagine residue, by comparing the z3 and z4 fragment ions. Therefore, information for the peptide sequence, the glycosylation site and the glycan structure is obtainable by analyzing the data from rapid alternating ETD and CID experiments. This alternating ETD/CID approach appears to be particularly suitable for characterizing the glycosylated peptides due to the complementary feature of ETD and CID dissociation methods.
Conclusions
Transmission mode ETD and CID experiments of polypeptide ions are performed in rapid succession on a QqTOF instrument and controlled by Daetalyst 3.14 software in a single data collection cycle. Compared to ion traps that can perform alternating ion trapping ETD/CID experiments with intervening mass analysis steps, the QqTOF approach illustrated here has comparable duty cycles, higher repetition rate, and superior mass resolving power and mass measurement accuracy. Since both the CID and ETD experiments are performed in transmission mode, whereby ionization, mass-selection, dissociation, and mass analysis occur in parallel, the present approach also has significant advantages in repetition rate and duty cycle over analogous ion trapping CID and ETD experiments performed on the same platform. In principle, the approach described herein should be particularly well-suited for coupling with on-line liquid chromatography (LC), which places a premium on high spectral acquisition rates.
Acknowledgments
This research was supported by the National Institute of General Medical Sciences under Grant GM 45372 and MDS Sciex, an Industrial Associate of the Department of Chemistry.
References
- 1.McLuckey SA, Wells JM. Chem. Rev. 2001;101:571–606. doi: 10.1021/cr990087a. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R, Goodlett DR. Chem. Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 3.Anbalagan V, Patel JN, Niyakorn G, Van Stipdonk MJ. Rapid Commun. Mass Spectrom. 2003;17:291–300. doi: 10.1002/rcm.912. [DOI] [PubMed] [Google Scholar]
- 4.Han H, Xia Y, McLuckey SA. J. Proteome Res. 2007;6:3062–3069. doi: 10.1021/pr070177t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karnezis A, Barlow CK, O'Hair RA, McFadyen WD. Rapid Commun. Mass Spectrom. 2006;20:2865–2870. doi: 10.1002/rcm.2671. [DOI] [PubMed] [Google Scholar]
- 6.Lam CN, Ruan ED, Ma CY, Chu IK. J. Mass Spectrom. 2006;41:931–938. doi: 10.1002/jms.1052. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura T. Farumashia. 2005;41:1071–1075. [Google Scholar]
- 8.Wells JM, McLuckey SA. Methods Enzymol. 2005;402:148–185. doi: 10.1016/S0076-6879(05)02005-7. [DOI] [PubMed] [Google Scholar]
- 9.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Anal. Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 10.Zubarev RA, Kelleher NL, McLafferty FW. J. Am. Chem. Soc. 1998;120:3265–3266. [Google Scholar]
- 11.Coon JJ, Syka JEP, Shabanowitz J, Hunt DF. Int. J. Mass Spectrom. 2004;236:33–42. [Google Scholar]
- 12.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc. Nat. Acad. Sci. U.S.A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirgorodskaya E, Roepstorff P, Zubarev RA. Anal. Chem. 1999;71:4431–4436. doi: 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- 14.Hakansson K, Cooper HJ, Emmett MR, Costello CE, Marshall AG, Nilsson CL. Anal. Chem. 2001;73:4530–4536. doi: 10.1021/ac0103470. [DOI] [PubMed] [Google Scholar]
- 15.Shi SD, Hemling ME, Carr SA, Horn DM, Lindh I, McLafferty FW. Anal. Chem. 2001;73:19–22. doi: 10.1021/ac000703z. [DOI] [PubMed] [Google Scholar]
- 16.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. J. Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang X, Hager JW, McLuckey SA. Anal. Chem. 2007;79:3363–3370. doi: 10.1021/ac062295q. [DOI] [PubMed] [Google Scholar]
- 18.Shevchenko A, Chernushevich I, Ens W, Standing KG, Thomson B, Wilm M, Mann M. Rapid Commun. Mass Spectrom. 1997;11:1015–1024. doi: 10.1002/(SICI)1097-0231(19970615)11:9<1015::AID-RCM958>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 19.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang X, Londry FA, Yang MJ, McLuckey SA. Anal. Chem. 2006;78:4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang X, Xia Y, McLuckey SA. Anal. Chem. 2006;78:3208–3212. doi: 10.1021/ac052288m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.March RE. J. Mass Spectrom. 1997;32:351–369. [Google Scholar]
- 22.Swaney DL, McAlister GC, Wirtala M, Schwartz JC, Syka JEP, Coon JJ. Anal. Chem. 2007;79:477–485. doi: 10.1021/ac061457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia Y, Han H, McLuckey SA. Anal. Chem. 2008;80 doi: 10.1021/ac702188q. DOI 10.1021/ac702188q. [DOI] [PubMed] [Google Scholar]
- 24.Chernushevich IV, Loboda AV, Thomson BA. J. Mass Spectrom. 2001;36:849–865. doi: 10.1002/jms.207. [DOI] [PubMed] [Google Scholar]
- 25.Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2199–2204. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Frolov A, Tang N, Hoffmann R, van de Goor T, Metz TO, Smith RD. Rapid Commun. Mass Spectrom. 2007;21:661–666. doi: 10.1002/rcm.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Good DM, Wirtala M, McAlister GC, Coon JJ. Mol. Cell. Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Wagner V, Gessner G, Heiland I, Kaminski M, Hawat S, Scheffler K, Mittag M. Eukaryot. Cell. 2006;5:457–468. doi: 10.1128/EC.5.3.457-468.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casado-Vela J, Ruiz EJ, Nebreda AR, Casal JI. Proteomics. 2007;7:2522–2529. doi: 10.1002/pmic.200700026. [DOI] [PubMed] [Google Scholar]
- 30.Xia Y, Chrisman PA, Pitteri SJ, Erickson DE, McLuckey SA. J. Am. Chem. Soc. 2006;128:11792–11798. doi: 10.1021/ja063248i. [DOI] [PubMed] [Google Scholar]
- 31.Hakansson K, Cooper HJ, Hudgins RR, Nilsson CL. Curr. Org. Chem. 2003;7:1503–1525. [Google Scholar]
- 32.O'Connor PB, Lin C, Cournoyer JJ, Pittman JL, Belyayev M, Budnik BA. J. Am. Soc. Mass Spectrom. 2006;17:576–585. doi: 10.1016/j.jasms.2005.12.015. [DOI] [PubMed] [Google Scholar]