

Abstract
Multiple hippocampal processes and cognitive functions change with aging or Alzheimer's disease, but the potential triggers of these aging cascades are not well understood. Here, we quantified hippocampal expression profiles and behavior across the adult lifespan to identify early aging changes and changes that coincide with subsequent onset of cognitive impairment. Well powered microarray analyses (N = 49 arrays), immunohistochemistry, and Morris spatial maze learning were used to study male F344 rats at five age points. Genes that changed with aging (by ANOVA) were assigned to one of four onset age ranges based on template pattern matching; functional pathways represented by these genes were identified statistically (Gene Ontology). In the earliest onset age range (3–6 months old), upregulation began for genes in lipid/protein catabolic and lysosomal pathways, indicating a shift in metabolic substrates, whereas downregulation began for lipid synthesis, GTP/ATP-dependent signaling, and neural development genes. By 6–9 months of age, upregulation of immune/inflammatory cytokines was pronounced. Cognitive impairment first appeared in the midlife range (9–12 months) and coincided and correlated primarily with midlife upregulation of genes associated with cholesterol trafficking (apolipoprotein E), myelinogenic, and proteolytic/major histocompatibility complex antigen-presenting pathways. Immunolabeling revealed that cholesterol trafficking proteins were substantially increased in astrocytes and that myelination increased with aging. Together, our data suggest a novel sequential model in which an early-adult metabolic shift, favoring lipid/ketone body oxidation, triggers inflammatory degradation of myelin and resultant excess cholesterol that, by midlife, activates cholesterol transport from astrocytes to remyelinating oligodendrocytes. These processes may damage structure and compete with neuronal pathways for bioenergetic resources, thereby impairing cognitive function.
Keywords: spatial learning, brain aging, Alzheimer's, cholesterol, inflammation, myelination
Introduction
Alterations in multiple molecular processes develop in the brain during aging or Alzheimer's disease (AD), some of which correlate with cognitive impairment. Well established brain aging changes include increased glial reactivity, inflammation and oxidative stress, and decreased mitochondrial function (Landfield et al., 1981; Green and Simpkins, 2000; Lee et al., 2000; Peters et al., 2000; Wyss-Coray and Mucke, 2002; Lu et al., 2004; McGeer and McGeer, 2004; Mrak and Griffin, 2005; Norris et al., 2005; Butterfield et al., 2006; Frank et al., 2006; Finch and Morgan, 2007; Gemma and Bickford, 2007; Brinton, 2008; Gibson et al., 2008). Altered lysosomal activity (Bi et al., 2000; Nixon and Cataldo, 2006) and synaptic remodeling (Dickey et al., 2003; Lynch et al., 2006; Mufson et al., 2006; Burger et al., 2007) also are seen consistently.
Additionally, in recent microarray studies of hippocampal gene expression, we identified several other aging-dependent upregulated processes, including lipid catabolism, proteolysis, cholesterol transport, and myelinogenesis in normally aging rats (Blalock et al., 2003, 2005; Rowe et al., 2007) and tumor suppressor/oligodendrocyte growth factor pathways in incipient AD subjects (Blalock et al., 2004). Increased or continuing brain myelination with aging also has been found in monkeys (Peters et al., 2000) and humans (Bartzokis et al., 2007).
Many of these aging-sensitive genes/processes were correlated with cognitive dysfunction, but upregulated cholesterol trafficking pathways in normal brain aging (Rowe et al., 2007) may have particularly important implications for cognitive impairment. That is, high plasma cholesterol is a significant risk factor for cognitive impairment, whereas statin use may have a protective effect (Petanceska et al., 2002; Wolozin, 2004; Yaffe et al., 2004; Sparks et al., 2005; Craft, 2006). Furthermore, the ApoE4 allele of the gene for the cholesterol trafficking lipoprotein, apolipoprotein E (ApoE), is a major risk factor for idiopathic AD (Roses, 2006), and cholesterol pathways may modulate amyloid clearance (see Discussion).
However, data on the key early events in cognitive dysfunction are lacking. It is evident (Bi et al., 2000; Markowska and Savonenko, 2002; Blalock et al., 2003; Gant et al., 2006) that many brain and cognitive aging changes begin by midlife (∼12 months of age in F344 rats), but few if any studies have attempted to identify the earliest detectable aging changes in expression or the changes that develop concurrently with the onset of cognitive impairment. Although early or concurrent changes cannot establish cause and effect, or separate detrimental from compensatory effects, they can generate unique insights and testable hypotheses on processes that may drive brain and cognitive aging.
Here, we used well powered microarray analyses (N = 49), semiquantitative immunohistochemistry (IHC), and Morris water maze (MWM) testing to analyze hippocampal gene/protein expression and cognitive function in rats across the adult lifespan. This task assesses spatial memory (Morris, 1984; Markowska and Savonenko, 2002; Gallagher et al., 2003) and is highly dependent on dorsal hippocampal function and plasticity (Moser and Moser, 1998; Moser et al., 1998). Thus, our results provide new insight into the temporal relationships between cognitive aging and molecular changes in a critical brain region.
Materials and Methods
Subjects.
Fifty-one male F344 rats (3, 6, 9, 12, and 23 months old; N = 9 per age group except N = 15 in the 23-month-old group) were obtained from the National Institute of Aging aging rodent colony maintained by Harlan. Animals were maintained on ad libitum food and water and housed individually. Animals were allowed to acclimate to new surroundings for 1 month before starting behavioral studies. All protocols and procedures were in accordance with Institutional Animal Care and Use Committee-approved procedures.
Morris water maze training.
Animals were trained on the MWM task, with methods similar to those described previously (Rowe et al., 2007). Briefly, the maze consisted of a black tub 1.9 m in diameter filled with 26 ± 2°C water, placed in the middle of a darkened room, and equipped with three prominent black and white cues (70 × 70 cm). Walls, ceiling, and floor were painted black. A Plexiglas escape platform (15 cm diameter) was located in the center of one of the quadrants of the pool. The animals were trained over nine trials (three trials per day for the 3 d training phase) to find the submerged platform. An intertrial interval of 1.25 min was used during which 30 s was spent on the platform and 45 s was spent in a heated holding chamber. Start quadrant was changed daily, and animals were never placed in the quadrant containing the escape platform. The platform stayed in the same quadrant for the duration of the study until the fourth day, when the platform was removed (retention or probe day). On the sixth day, animals were tested on a single cued trial (visible platform plus cue on platform) to assess visual acuity and noncognitive effects on performance. Animal motion was visually tracked with the Videomex software (Columbus Instruments) and a digital camera (Columbus Instruments). Latency to find the hidden platform, averaged across the eighth and ninth training trials on day 3, and latency to enter an area centered above the platform and twice its area (annulus) on the probe trial were used to assess retention. Training latencies appear to stabilize by day 3 under our conditions and reliably identify cognitive impairment (Rowe et al., 2007). Path length data for both training and probe trials were primarily consistent with latency measures (Table 1). Two 23-month-old animals were removed from the study because of poor health (one died at the end of training and one was too weak to complete training). Seven additional animals (four 23-month-old, two 12-month-old, and one 6-month-old) failed to meet criterion on both the cued and retention tasks (i.e., did not find the platform in 60 s) and were excluded from behavioral analysis because of potential sensorimotor deficits unrelated to cognition.
Table 1.
MWM results for animal age groups
| Age (months) | Training |
Probe trial |
||||
|---|---|---|---|---|---|---|
| Latency (s) | Path (cm) | Velocity (cm/s) | Latency (s) | Path (cm) | Velocity (cm/s) | |
| 3 | 15.7 ± 3.5 | 554 ± 143 | 35.4 ± 3.3 | 16.5 ± 4.5 | 525 ± 155 | 30.9 ± 1.2 |
| 6 | 19 ± 5.7 | 580 ± 171 | 30.1 ± 0.9 | 19.4 ± 7 | 625 ± 233 | 32.2 ± 1.3 |
| 9 | 20 ± 5.8 | 647 ± 168 | 34.4 ± 0.9 | 23.2 ± 6.2 | 823 ± 205 | 37.3 ± 1.1 |
| 12 | 40.3 ± 6.8* | 1152 ± 187* | 29.1 ± 1.1* | 27.3 ± 8.3 | 827 ± 237 | 35.4 ± 2.3 |
| 23 | 49.9 ± 3.8* | 1110 ± 069* | 21.9 ± 1.2* | 44.1 ± 6.6* | 1120 ± 165 | 25.6 ± 1.0* |
| p value | 0.000065 | 0.016091 | 0.000032 | 0.043672 | 0.287534 | 0.000017 |
Retention data from last two training trials (left) and probe trial (right). Latency, Time from entry to goal; Path, swim distance to goal; Velocity, swim speed;
p value from one-way ANOVA test (n = 42); *also significant post hoc pairwise Tukey's contrast versus 3 month group.
To minimize stress and hypothermia, two factors that might influence gene expression and/or MWM performance, we gradually (2 weeks) acclimated the animals to the facility and handlers. In addition, a minimum of 30 min acclimation time in the MWM room began each daily session. Hypothermia was minimized by warming the water and placing heat lamps near animal cages between and after each trial.
Tissue preparation.
On day 7, the day after the last (cued) trial, subjects were anesthetized deeply (intraperitoneal pentobarbital, 80 mg/kg) and killed by intracardial perfusion with cold saline. Brains were rapidly removed and hemisected. The right hemisphere went to immunohistochemistry. The hippocampus of the left hemisphere was dissected away, and the CA1 region was subdissected out under a stereoscope, as described previously (Blalock et al., 2003). The CA1 region of each animal was placed in a separate, labeled 1.5 ml centrifuge tube, immediately placed in dry ice, and stored at −80°C until used for microarray studies (see below).
Microarrays.
RNA was extracted from each CA1 tissue block, as described previously (Blalock et al., 2003), using TRIzol reagent (Invitrogen), precipitated with ethanol, reconstituted in RNase-free water, and quantified and checked for RNA integrity with an Agilent 2100 bioanalyzer. For each CA1 sample, 5 μg of total RNA was processed to generate biotin-labeled cRNA following the standard protocol in the Affymetrix expression analysis technical manual, of which 20 μg of labeled-cRNA was then applied to a rat RAE230A GeneChip (Affymetrix) for hybridization (one chip per animal). Scanned microarray images were analyzed using the Microarray Suite 5.0 (MAS5; Affymetrix) algorithm. A total of 49 GeneChips were used in this study. The study is uploaded to the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) under accession number GSE9990, and quality control results are reported (supplemental Table D, available at www.jneurosci.org as supplemental material). Probe set annotations were downloaded from Affymetrix and integrated with signal intensity and presence call data for additional analysis (see below, Statistics).
Immunohistochemistry.
Microarray-identified candidate proteins for which well characterized specific antibodies are available were selected for IHC, and semiquantitative analysis was performed on the hippocampus in three coronal sections from each animal in the young (3 month), midlife (12 month), and aged (23 month) groups as described previously (Kadish and Van Groen, 2002). Briefly, after perfusion with cold saline, half of the brain was fixed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, overnight. Brains were transferred to 30% sucrose solution the following day for cryoprotection and cut on microtome into 30-μm-thick coronal sections. Sections planned for cathepsin D staining were pretreated in boiling Na citrate, pH 6.0, for 30 min before incubation with primary antibody. For single immunoperoxidase staining, tissues were transferred to primary antibody solution for 18 h [an anti-S100A4 rabbit (1:500; NeoMarkers), anti-ApoE goat (1:400; Santa Cruz Biotechnology), anti-SOAT-1 rabbit (1:500; Cayman Chemicals), anti-cathepsin D goat (1:1000; Santa Cruz Biotechnology), and mouse anti-oligodendrocytic receptor interacting protein (RIP) (1:100,000; Millipore Bioscience Research Reagents)] in Tris-buffered saline with 0.5% Triton X-100 (TBS-T) added. After incubation, sections were rinsed three times in TBS-T and transferred to the appropriate secondary antibody [goat anti-mouse biotin (1:500; Sigma); goat anti-rabbit biotin (1:500; Abcam); rabbit anti-goat (1:500; Abcam)] for 2 h and transferred to a solution containing mouse ExtrAvidin (Sigma). Sections were rinsed (three times in TBS-T) and incubated for 3 min with nickel-enhanced diaminobenzidine (Kadish and Van Groen, 2002). For double-immunofluorescence staining, sections were incubated with a mixture of anti-GFAP mouse (1:1000; Sigma) and one of the following: anti-S100A4 (NeoMarkers), ApoE (Santa Cruz Biotechnology), cathepsin D (Santa Cruz Biotechnology) or SOAT-1 (Cayman Chemicals), and then with goat anti-mouse FITC (1:500; Jackson ImmunoResearch) and donkey anti-rabbit cyanine 3 (Cy3) (1:500; Jackson ImmunoResearch) or donkey anti-goat Cy3 (1:500; Jackson ImmunoResearch). Grayscale images of the appropriate areas were acquired with an Olympus Optical DP70 digital camera. For each antibody, all sections were processed in parallel. Optical density of the region of interest was measured using ImagePro Plus (Media Cybernetics) on a standardized area at three different septotemporal levels at similar lateromedial positions.
Statistics.
Behavioral data were analyzed by one-way ANOVA with post hoc protected all-pairwise Tukey's comparisons with control for multiple testing error. Semiquantitative immunohistochemical density data were averaged across sections to create a single measure per animal, and differences between young and aged animals were assessed by ANOVA or unpaired heteroschedastic t test (as noted in Results). Microarray data were prestatistically filtered as described previously (Blalock et al., 2003). Unique, annotated gene probe sets rated present on at least six chips in the study were retained for statistical analysis. The presence call cutoff of six was chosen based on factorial analysis, indicating that there was a <5% probability that six presence calls would be found in a set of 49 chips by chance (E. Blalock, unpublished observations). Outlier signal intensity (expression) data (>2 SD from the group mean for each probe set in each age group) were treated as missing values. Data were analyzed by one-way ANOVA for significant differences, and the extent to which the error of multiple testing might have contributed was estimated by the false discovery rate (FDR) (Hochberg and Benjamini, 1990). Each gene found significant at p ≤ 0.05 by ANOVA [aging-related genes (ARGs)] was then correlated with 10 idealized templates (for upregulated patterns, see Fig. 1), to determine the aging-related “shape” of change for each gene. Each template was an artificial construct made up of five numbers, one for each age (3, 6, 9, 12, and 23 months). For example, the Δ3–6 template, designed to detect ARGs that changed from 3 to 6 months of age and then showed stable expression from 6 to 23 months (Fig. 1), was constructed with a 3 month value of 0 and then 6, 9, 12, and 23 month values of 1 (0,1,1,1,1). In contrast, the Δ6–23 template was constructed to reflect a later onset of change and a progressive increase across age (0,0,1,2,3). For each identified ARG, mean signal intensity values for each of the five age groups were correlated to each of the 10 idealized templates. A positive correlation indicated an upregulated gene and a negative correlation a downregulated gene. A gene was assigned to the template with which it had the highest Pearson's r value, if that r value exceeded |0.87|. Thus, some genes rated significant by ANOVA failed to meet criterion and were not assigned to any template. Genes that met criteria were allocated to an age-of-onset based on the template to which they were assigned (Fig. 1) (see Results). Functional processes represented by the assigned ARGs were determined using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) overrepresentation clustering analysis tool (Dennis et al., 2003) on the Gene Ontology (GO) databases of Biological Process, Cellular Component, and Molecular Function (Ashburner et al., 2000). To avoid redundancy, only a single illustrative process for each cluster of similar significantly overrepresented processes [p < 0.05, Expression Analysis Systematic Explorer (EASE) score], populated by between 3 and 50 genes, is reported in Results. Furthermore, for the mid-aged (12 months, N = 7) and aged (23 months, N = 9) animals that reached criteria on the Morris water maze (see above), ANOVA-significant genes were also tested by post hoc Pearson's test for correlations with behavioral variables. These correlations were performed within the 12- and 23-month-old groups separately, as well as within the 12- plus 23-month-old groups combined. Importantly, the same filtered microarray dataset used for ANOVA analysis was used here. Thus, if outliers were removed in the ANOVA, those outlier values also were not included in the correlation analysis, which is highly sensitive to the effects of outliers.
Figure 1.
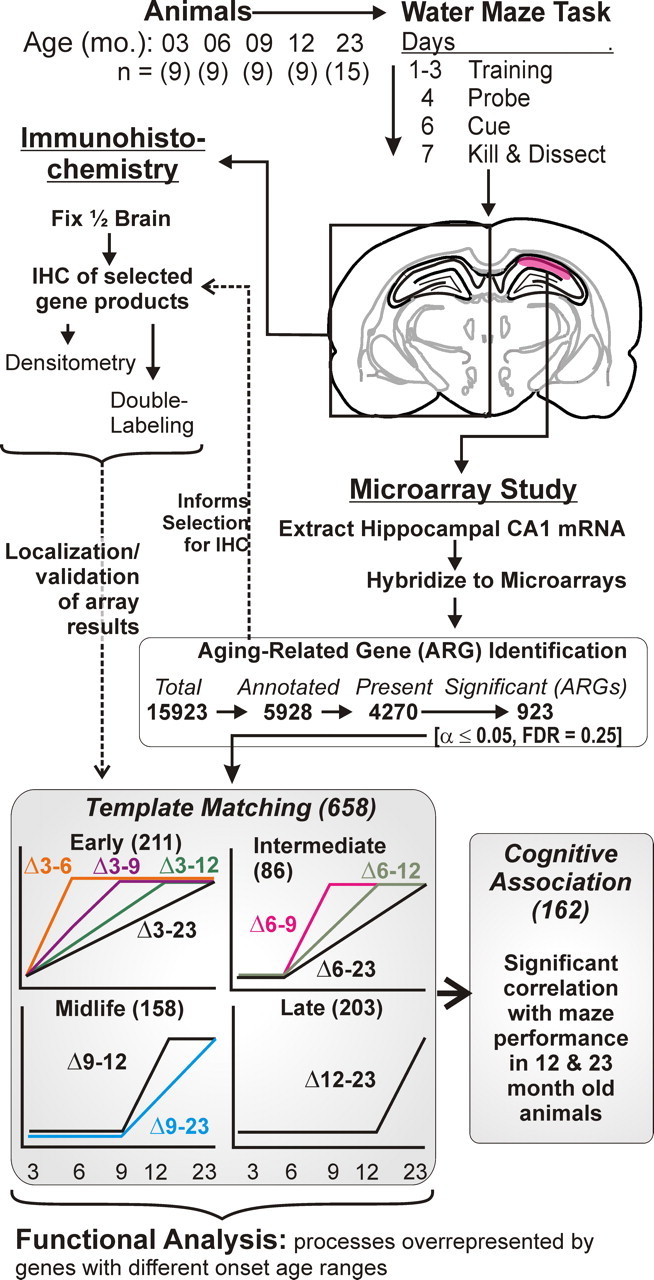
Overview of experimental protocol and microarray analysis procedure. Top, Animals (N = 9–15) from five age groups (3–23 months) were tested on the Morris water maze. Top middle, Brains were removed and one hemisphere was used for immunohistochemistry, and the hippocampal CA1 region (red) of the other hemisphere was subdissected and used for microarray studies. Bottom middle, For microarray analyses, genes were filtered to exclude ESTs and absence calls. The remaining genes/probe sets were tested across age groups by one-way ANOVA. Genes significant by this procedure were designated ARGs. Dashed arrow from ARG identification to selected IHC analyses: ARG identification was used to inform selection for immunohistochemical analysis. Bottom, Based on highest correlation with 1 of 10 idealized templates, ARGs were assigned to one of four basic ages of onset (see Materials and Methods). Note that only upregulated templates were used; downregulated ARGs within each template were identified based on negative, rather than positive, correlations (see Materials and Methods). Templates were then grouped according to onset age ranges (early, intermediate, midlife, and late) determined from the age point at which the initial deflection in expression from the 3 month group was detected. For all ARGs assigned an onset range, correlation of gene expression and MWM performance in the 12- and 23-month-old animals was tested (Pearson's test). Functional process overrepresentation analysis was performed for ARGs in each onset age range using EASE/DAVID analysis procedures interrogating the Gene Ontology (see Materials and Methods).
Results
Figure 1 provides a schematic overview of the protocols used in the present studies. F344 male rats in five age groups were trained and tested for cognitive performance, and brains were divided, with half being used for IHC and the other half being used to identify aging changes in hippocampal gene expression, as described below.
Morris water maze testing
MWM performance data for 42 animals that met behavioral criteria (of an original 49 rats; see Materials and Methods) in five age groups are shown in Table 1. Two primary measures of retention (cognitive function) were obtained: latency to find the hidden platform, averaged across the eighth and ninth training trials on day 3, and latency to enter the platform annulus on the probe trial. Both of these indexes assessed retention performance near the end of training. The average training latency on trials 8 and 9 (day 3) was selected as the primary index of retention for the gene expression–behavioral correlation analyses described below, because training latency stabilizes by day 3 under our conditions and reliably identifies cognitive impairment (Rowe et al., 2007). Path length data for both training and probe trials were primarily consistent with the latency measures (Table 1). Performance on the last two training trials showed a clear effect of age, with both the 12 month and 23 month groups differing significantly from the 3 month group. Performance on the probe retention trial appeared less age sensitive, with only the 23 month group differing from the 3 month group.
Statistical identification of aging-related genes
The CA1 region from one hippocampus of each animal was processed on an individual Affymetrix RAE230A microarray. After filtering out absence calls and expressed sequence tags (ESTs) (Blalock et al., 2003), 4270 gene probe sets were retained for testing (Fig. 1). Each was tested by one-way ANOVA across all five groups to determine which genes changed with aging (p ≤ 0.05). A total of 923 genes were found by ANOVA to differ with age, for a false discovery rate of ∼0.25 (FDR: expected false positives from multiple testing/total observed positives). This FDR is reasonable for a microarray study, in which added confidence is provided by coregulation (overrepresentation) of related genes in the same functional category (Mirnics and Pevsner, 2004; Blalock et al., 2005; Rowe et al., 2007). Moreover, in the present study, microarray results were confirmed by semiquantitative immunohistochemistry of corresponding protein expression for five identified genes (see below).
Estimation of age of onset for changed expression
To identify the age of onset, defined as the age range in which changed expression was first detectable for an ARG, we used 10 idealized templates of frequent profiles to extrapolate the aging change back to the age range of earliest apparent divergence from its 3-month-old baseline (Fig. 1 shows the 10 templates used for upregulated genes; negative correlations to the 10 templates were used for downregulated genes). Based on the idealized template (Fig. 1) with which it correlated most highly, each gene was assigned to one of four onset-of-change age ranges: early-adult onset (change beginning between 3 and 6 months old); intermediate onset (change beginning between 6 and 9 months); midlife onset (change beginning between 9 and 12 months); and late onset (change beginning between 12 and 23 months).
The numbers of ARGs assigned to each onset age range are shown in Figure 1 and Table 2. The complete list of gene symbols/names of upregulated and downregulated genes in each onset range, along with mean expression values at each age, is organized by template pattern in supplemental Table A and organized alphabetically in supplemental Table B (both available at www.jneurosci.org as supplemental material). As in our previous studies, more ARGs were found to be upregulated than downregulated (Table 2). More genes were assigned to the early-onset range than to any other, although late-onset genes ranked a close second. Figure 2 provides a heat map of highly significant genes assigned to each age range and shows the standardized average pattern for each age range. It can be seen that these standardized averages closely resemble the idealized templates used for original correlation and assignment (Fig. 1).
Table 2.
Assignments of genes to onset-age ranges
| Age range | Upregulated | Downregulated |
|---|---|---|
| Early | 115 (26; 22.6%) | 92 (17; 18.5%) |
| Intermediate | 56 (15; 26.8%) | 37 (10; 27.0%) |
| Midlife | 95 (33; 34.7%*) | 60 (16; 26.7%) |
| Late | 105 (27; 25.7%) | 98 (18; 18.4%) |
Numbers of ARGs assigned to each onset range (Fig. 1) are shown. Parentheses, Numbers and percentages of ARGs within each age range that were also significantly correlated with MWM performance. Note: Percentage of midlife upregulated genes correlated with behavior (34.7%) was significantly higher (*p < 0.05, binomial test) than the percentages in every other upregulated age range.
Figure 2.
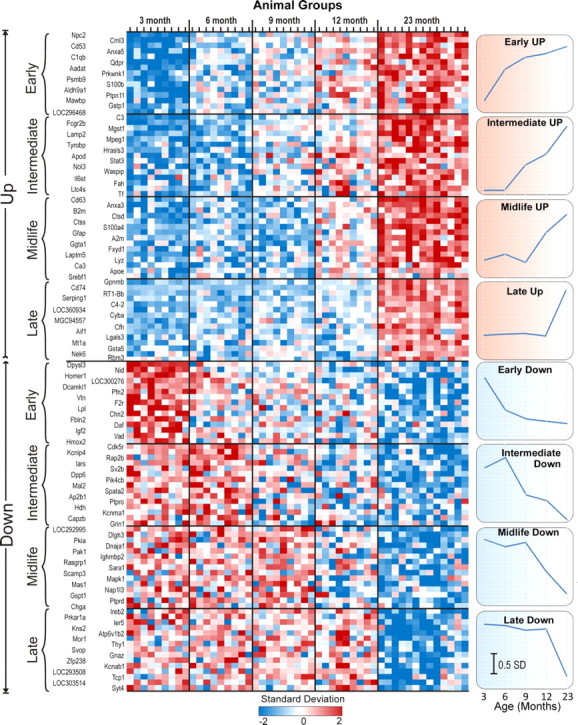
Heat map of aging-related genes with assigned onset age. Individual subjects are listed in columns and grouped by age, and genes are listed in rows and grouped by change direction and age of onset. For each of the four age-of-onset ranges, the 15 most significant upregulated (top) and downregulated (bottom) gene symbols are displayed. The expression level of each ARG is standardized across all subjects. This results in a mean of 0, and each individual subject's ARG expression level is expressed in SDs from that mean level. This does not affect statistical results but allows genes with different signal intensity ranges and similar patterns of expression to be displayed on the same heat map. Results for each ARG shown are color coded from low (blue, 2 SDs below mean), through average (white, 0 SDs from mean) to high (red, 2 SDs above mean; see color bar) expression. Right, Standardized averages for all ARGs in each upregulated and downregulated age range of onset (note that these averages use all of the genes assigned to the range rather than just the 15 displayed in the figure). Points represent mean ± SEM (error bars plotted but too small to see). Scale bar, 0.5 SDs.
Cognitive performance correlations
To control for age as a potentially confounding factor in correlation, we focused primarily on within-group same-age correlations. Only the 12 month and 23 month groups showed evidence of cognitive impairment, and, therefore, cognitive performance was tested for correlation with ARGs only within each of those two age groups. In addition, because they did not differ on day 3 retention latency (Table 1), these two groups were combined for an overall correlation test between performance and expression of each gene. The numbers of genes that correlated with cognitive performance (p ≤ 0.05) on at least one of the three tests (within 12 month group, within 23 month group, and within combined 12 and 23 month group) are shown in parentheses in Table 2 and are listed individually by onset age range in supplemental Table C (available at www.jneurosci.org as supplemental material). Upregulated ARGs with midlife-onset range were more likely to be correlated with MWM performance (which also showed midlife onset of change) than ARGs in any of the other onset ranges (Table 2) (p < 0.05 for binomial test of midlife range against every other upregulated age range). An example of one such correlation (12 plus 23 combined) is illustrated in Figure 3 for Apoe, which showed the highest average r value across all three within-group correlation tests of all upregulated ARGs.
Figure 3.

Apoe mRNA expression positively correlated with MWM impairment. Individual microarray-derived Apoe gene expression measures (y-axis) are plotted as a function of retention latency during water maze performance (x-axis) for 12- and 23-month-old animals. There was a significant (p = 0.003; Pearson's test) correlation across 12 plus 23 month subjects (solid line indicates regression, with dashed lines representing 95% confidence intervals). Correlations within the 12- and 23-month-old groups separately were also significant (12 months, p = 0.04; 23 months, p = 0.05). r values for correlations within 12 months, 23 months, and 12 plus 23 months combined data are shown in the graph.
Determination of functional processes represented by aging-related genes
To determine which pathways/processes began to be activated or suppressed at different stages of adult life, we identified the functional categories in the GO database (Ashburner et al., 2000) that were represented by a statistically higher proportion of aging-related genes than would be predicted by chance (overrepresented categories), using DAVID analysis (Dennis et al., 2003) of gene functional assignments. Such overrepresentation often reflects a high degree of coregulation within a pathway (Mirnics and Pevsner, 2004; Blalock et al., 2005). Genes for each onset range (early, intermediate, midlife, and late) and each direction of change (up or down) were uploaded and analyzed separately. The functional annotation clustering tool of DAVID identifies overrepresented functional processes and also groups such processes based on overlap among populating genes. To reduce redundancy, we are reporting (Table 3) only the process that appears to be the most informative and characteristic for each cluster.
Table 3.
Functional processes identified for each onset age range

Functional processes identified for each onset age range. Significantly overrepresented upregulated (left) and downregulated (right) biological processes (α < 0.05 by DAVID overrepresentation analysis; see Materials and Methods) and associated symbols of genes populating the processes, for each onset age range (A, early; B, intermediate; C, midlife; D, late). Underlined gene symbols are ARGs that were also significantly correlated with MWM performance. Italicized genes were added to categories based on literature review.
Table 3 shows these overrepresented functional processes for upregulated and downregulated genes for each age of onset, along with the lists of gene symbols populating all such processes (each gene symbol shown is also listed alphabetically with gene name/protein name in supplemental Table B, available at www.jneurosci.org as supplemental material). GO annotation/assignment is still a work in progress, and many genes have not yet been fully assigned. Accordingly, we have added some ARGs omitted by DAVID analysis to processes that appeared to be appropriate based on literature review. These added gene symbols are shown in italics. Also added (and italicized) under the early-onset range are two sets of lipid-related genes reflecting upregulated and downregulated lipid metabolic processes. These processes were not identified in DAVID but are suggested by the literature to represent coregulated functional groupings.
Table 3 also highlights (with underlining) all ARGs in the overrepresented processes that showed at least one significant within-group correlation with cognitive impairment (full list in supplemental Table C, available at www.jneurosci.org as supplemental material). Note that, as indicated in Table 2, these are particularly numerous among upregulated midlife-onset categories.
Altered functional processes identified in different age ranges of adult lifespan (Table 3)
Early-adult onset (3–6 months old)
Upregulated aging-related genes that began to change soon after reproductive maturity were statistically associated with ribosomal assembly, growth, lysosomal, stress/inflammatory, and protein/lipid degradative pathways, whereas downregulated ARGs were associated with neuronal development and energy-dependent signaling/transporter pathways. This pattern appears to reflect a shift in energy sources, as indicated by altered regulation of genes for lipid and amino acid catabolism. ARGs important in lipid/cholesterol synthesis (e.g., Cyp51, Elovl6) or uptake into cells (Lpl) were primarily downregulated, whereas those important in lipid degradation (Acadl, Decr1) or extrusion/transport (Abca2, Npc2) were generally upregulated. Increased Pdk2 expression was included in the lipid degradation category (Table 3), because this increase favors a shift from glucose to free fatty acid (FFA) oxidation in generating substrates for mitochondrial oxidative phosphorylation. This shift appears similar to a “Randle-cycle shift” (Randle, 1998; Holness and Sugden, 2003) (see Discussion), evidence of which has also been seen previously in aging brain (Rowe et al., 2007; Brinton, 2008).
Although changes in early-onset genes were detected beyond an age range generally defined as adulthood in rodents (3 months), it nevertheless seems possible that some changes from 3 to 6 months could reflect the end of late developmental processes rather than the onset of aging changes. However, such developmental expression patterns seemingly should be expected to plateau by 6 months of age. Although some early-onset genes fulfilled this developmental pattern, the large majority fit linear or progressive templates more consistent with aging-related patterns of expression (Fig. 1, Table 2) (supplemental Table A, available at www.jneurosci.org as supplemental material). Therefore, our data indicate that the onset of some genomic aging processes occurs very early in adulthood, during or soon after reproductive maturation (i.e., between 3 and 6 months old).
Intermediate onset (6–9 months old)
Although this onset age range contained the fewest ARGs and functional processes Tables 2, 3), upregulated ARGs in this phase prominently included key cytokines and regulators of the innate immune/inflammatory and complement pathways, as well as regulators of proteolytic processes. Downregulated ARGs represented categories of ion channels/transport (e.g., Grin1 encoding NMDAR1) and vesicle transport. Interestingly, several of the upregulated inflammatory ARGs (IL6st, Stat3) are critical signaling molecules in the IL6/leukemia inhibitory factor pathways that have been implicated in both astrocytic differentiation (Mi et al., 2001) and oligodendrocytic myelination (Ishibashi et al., 2006). Thus, these changes could foreshadow the evidence of astrocyte reactivity and myelinogenesis seen in subsequent phases (see below).
Midlife onset (9–12 months old)
The first evidence of cognitive impairment appeared in the midlife phase (Table 1), as did the highest number and proportion of (upregulated) ARGs correlated with cognitive performance (Table 2). Upregulated ARGs were overrepresented for processes of astrocyte reactivity/cytoskeletal structure (Gfap, S100a4, Cd81, Arpb1c), cation/iron homeostasis (myelinogenesis), glycosyl transferase activity, and, notably, lipid metabolism, the latter enriched with genes involved in cholesterol esterification and its transport by lipoproteins or intracellular storage (e.g., Apoe, Clu/Apoj, Lcat, Pcyt1a, Gm2a, Srebf1). As noted (Fig. 3), Apoe was the gene most consistently correlated with cognitive impairment among all upregulated ARGs (supplemental Table C, available at www.jneurosci.org as supplemental material). New sets of inflammation/immunity and protein processing ARGs also began to upregulate during the midlife-onset phase, in particular, ARGs with antigen-presenting roles (B2m, Ctss, Hla-dmb, Psme2, Psmb8, RT1-Aw2, and RT1-M3) that perhaps reflect a later phase of inflammatory damage and elevated proteolysis. Concurrently, there was downregulation of additional ARGs for energy-dependent signaling, vesicle transport, GTPase activity, and neurite outgrowth, a category including behaviorally correlated App, the gene for amyloid precursor protein.
Late onset (12–23 months old)
The late-life period was characterized by onset of numerous additional genes in categories already exhibiting change, namely in upregulated immune/inflammatory and ribosomal/biosynthetic processes and downregulated energy-dependent signaling, transporter, and cytoskeletal processes. This pattern appeared to reflect expansion of alterations in functional processes already set in motion.
Immunohistochemical cell-type localization of key midlife-upregulated processes
To elucidate the cell-type origins of key changes in expression, semiquantitative IHC was performed on six proteins that were of particular interest because they were representative of midlife-upregulated processes that coincided with onset of cognitive decline and/or had some relationship to cholesterol trafficking. The proteins selected for quantitative IHC (with names, corresponding gene symbols, onset ages, and primary protein function) were as follows: ApoE (apolipoprotein E, Apoe, midlife-up; cholesterol transport); CathD (cathepsin-D, Ctsd, midlife-up; lysosomal protease); S100A4 (S100a4, midlife-up; white matter astrocyte marker, response to injury); GFAP (glial fibrillary acidic protein, Gfap, midlife-up; structural marker of astrocyte reactivity); and ApoD (apolipoprotein D, Apod, intermediate-up; sterol transporter in oligodendrocytes, response to stress). In addition, we quantified SOAT1/ACAT1 (sterol O-acyltransferase/acyl-coenzyme A: cholesterol acyltransferase, Soat1; catalyzes cholesterol esterification for intracellular storage and transport). Although the gene Soat1 was not rated present in this study (possibly because it is expressed primarily in astrocytes in or near white matter (Fig. 4), which was trimmed and reduced in these dissected CA1 microarray samples, as described previously), it was found previously to be an ARG in non-trimmed hippocampal samples (Rowe et al., 2007), and the SOAT1 IHC here clearly showed a major effect of aging by midlife (Fig. 4). We also used the RIP antibody against myelin to quantify the degree of hippocampal myelination with aging. RIP immunostaining provided sensitive visualization of aging changes in myelinated fibers (Fig. 5), which was confirmed by several other myelin antibodies we compared.
Figure 4.
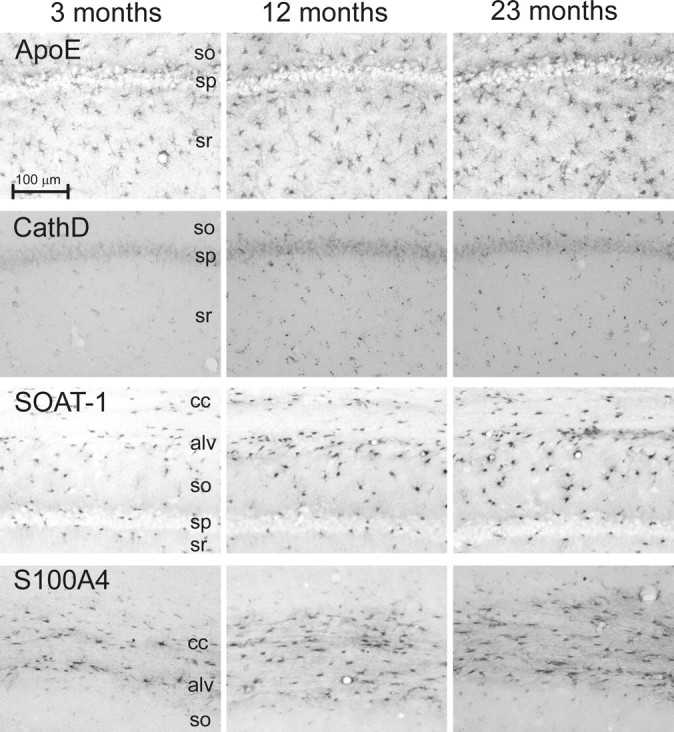
IHC patterns of selected proteins. Micrographs from brain specimens contralateral to those used for microarray for four proteins, ApoE, CathD, SOAT1, and S100A4 across three ages (3, 12, and 23 months) are shown. These proteins were selected for validation of aging differences and identification of cell-type localization based on relevance to astrocyte reactivity and cholesterol trafficking (Table 3). All were significantly upregulated by semiquantitative IHC (p < 0.05; see Results). Note preferential upregulation of SOAT1 and S100A4 in astrocytes near or in white matter tracts (alv and cc). sp, Stratum pyramidale; so, stratum oriens; sr, stratum radiatum; cc, corpus callosum; alv, alveus. Scale bar, 100 μm.
Figure 5.
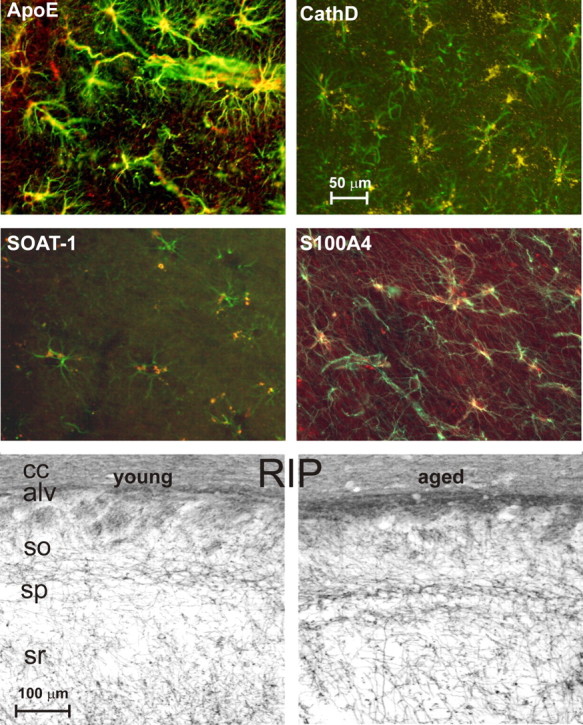
Double-labeled colocalization with astrocytic GFAP. Top four panels, Double-labeled images from brain specimens contralateral to the ones used for microarray analysis showing overlapping expression (in yellow) of GFAP (an astrocyte marker, in green) with ApoE, CathD, SOAT-1, or S100A4 staining (red). ApoE, CathD, and SOAT-1 specimens are from dorsal hippocampus, and S100A4 staining is from corpus callosum. Note the extensive distribution of APOE in astrocyte somata and blood vessel end feet, the more punctuate distribution of CathD and SOAT1, likely in lysosomes and storage granules, and S100A4 widely distributed in cell anatomical processes. Bottom two panels, Photomicrographs of dorsal hippocampal specimens contralateral to those used for microarray stained with RIP, a myelin/oligodendrocyte marker. Note the widely increased staining density with age. Semiquantitative analysis also showed that RIP staining was significantly increased across age (p < 0.05) (see Results). sp, Stratum pyramidale; so, stratum oriens; sr, stratum radiatum; cc, corpus callosum; alv, alveus.
All four of the proteins studied across 3-, 12-, and 23-month-old animals by IHC exhibited clear increases with aging (ApoE, p = 0.018; CathD, p = 2.44−7; S100A4, p = 9.23−13; GFAP, p = 1.44−8; one-way ANOVA). In addition, SOAT1 was highly significant (p = 1.45−6, t test) (Rowe et al., 2007), and Apod was clearly elevated with age (determined by visual rating alone because there was essentially no ApoD immunostaining in young animals, whereas aged animals showed numerous stained somata in white matter tracts). For additional validation, results for all four proteins that were quantified in three age groups were tested for rank-order correlation with the microarray-based measures of corresponding gene expression, and all were significantly correlated (Spearman's test: N = 23–26; ApoE, p = 6.55−3; CathD, p = 2.57−7; S100A4, p = 6.37−11; GFAP, p = 2.09−7).
The IHC results showed clearly that many of the key midlife changes took place in astrocytes. Upregulation of cholesterol trafficking (ApoE, SOAT1), structural reactivity (S100a4), and lysosomal activation (punctate CathD) occurred primarily in astrocytes (although CathD was also upregulated in a non-punctate manner in neurons) (Figs. 4, 5). The quantitative effects were readily apparent in photomicrographs (Fig. 4). Double-labeled images (Fig. 5) clearly revealed predominant colocalization with astrocytic GFAP of ApoE, CathD, SOAT1, and S100A4 (the latter two primarily in astrocytes of the white matter and adjacent stratum oriens) (Fig. 4). Moreover, the ApoD results indicated upregulated transport in oligodendrocytes with aging, and the RIP data revealed an increase in myelin staining with age (p = 0.042, t test). Increased RIP immunostaining with aging can be seen in myelinated axons traversing the CA1 strata radiatum and oriens and in white matter tracts (Fig. 5), consistent with our microarray data showing widespread aging-related upregulation of myelin protein genes (Blalock et al., 2003; Rowe et al., 2007), as well as with EM data (Peters et al., 2000).
Discussion
This is the first study to elucidate aging changes in hippocampal expression comprehensively across the adult lifespan. The resulting temporal perspective allows identification of early aging processes that may be candidates for trigger mechanisms and identifies midlife-onset changes that may be candidates for a role in the onset of cognitive decline. The correlative data presented here cannot, of course, provide direct evidence of a causal relationship nor can they directly distinguish detrimental aging changes from compensatory/beneficial or incidental changes. However, close correlation is one of the key predictions of a causal hypothesis, and failure to find correlation can eliminate many potential candidates from additional testing, an important consideration in global studies.
Sequence of aging changes
Early-adult range downregulated genes appeared to reflect a decrease in lipid/cholesterol synthesis and neurite outgrowth, whereas early-adult upregulated genes reflected activation of stress, growth, and ribosomal assembly (see Results) (Table 3). Of particular note was the upregulation of genes for lysosomal processes and catabolic generation of ketogenic amino acids (AAs) (Qdpr, Aadat, Maob) and FFAs (Acadl, Decr1, Pdk2), also found previously in hippocampus of aged rats (Rowe et al., 2007). This pattern strongly suggests a metabolic shift favoring oxidation of FFAs and ketone bodies (KBs) while conserving glucose by gluconeogenesis, ketogenesis, and glycogen synthesis, capabilities that astrocytes appear to share uniquely with hepatocytes (Rowe et al., 2007). This age-related metabolic shift (Rowe et al., 2007; Brinton, 2008) can be likened to a shift in the Randle cycle, which regulates the competitive balance between FFAs and glucose as substrates for the tricarboxylic acid cycle. Pyruvate dehydrogenase (PDH) activation favors glucose oxidation, whereas its inactivation by PDH kinases (e.g., Pdk2) favors FFA oxidation and glucose conservation (Randle, 1998; Holness and Sugden, 2003). Because lipoproteins or FFAs do not readily cross the blood–brain barrier, increased energy substrates would likely have to be obtained from extracellular structures, debris, and myelin (see below), consistent with upregulation of lysosomal pathways (Table 3). However, additional studies will be required to determine whether this shift represents a genetically “programmed” change, an adaptation to another process (e.g., membrane degradation from oxidative stress) or an incidental change.
In the following intermediate age range (6–9 months old), elevated expression was pronounced for inflammatory cytokines and complement molecules (Table 3). Many inflammatory/oxidative processes apparently originate in microglia, and their activation might mediate degradation of the myelin sheath and other membranes. Because myelin is highly enriched in phospholipids, cholesterol, and proteins, this degradation, in turn, would generate increased FFAs and AAs as potential fuel substrates.
The midlife age range (9–12 months old) was the period in which evidence of cognitive impairment first appeared (Table 1), consistent with previous studies in rodents and other species that detected onset of cognitive decline around or before the midpoint of the lifespan (∼12 months old in F344 rats) (Markowska and Savonenko, 2002; Disterhoft and Oh, 2006). Interestingly, midlife is also the period in which we previously first detected emergence of Ca2+-related electrophysiological markers of hippocampal aging (Gant et al., 2006) and in which initial changes in long-term potentiation have been found (Lynch et al., 2006). Furthermore, midlife upregulated ARGs exhibited the highest proportion of genes correlated with cognitive impairment (Tables 2, 3). Consequently, it appears possible that the midlife range is a critical convergent phase in which expression changes begin to interact with electrophysiological processes and negatively impact cognitive functions.
Immune response genes activated in the midlife phase differed from those in earlier periods by the predominance in midlife of upregulated ARGs important in antigen presentation [e.g., immunoproteasome subunits, major histocompatibility complex I (MHC I) and MHC II molecules] (Table 3). Increased expression of MHC II molecules has been seen previously in aged brain (Blalock et al., 2003; McGeer and McGeer, 2004; Frank et al., 2006) and primarily reflects activation of microglia, the resident antigen-presenting cells of the CNS. This midlife upregulation of antigen-presenting ARGs may reflect a new level or phase of intensified tissue damage and proteolysis, but additional studies will be needed to assess its functional implications.
Upregulation also began in midlife for a significant number of myelinogenic/oligodendrocytic genes, including Mobp and Mog, and several iron-regulating genes (Fth1, Ftl1, Mt3) correlated with cognitive function. Oligodendrocytes are the cells with the highest concentrations of iron in the brain, and iron is critical for myelinogenesis (Connor and Menzies, 1996; Bartzokis et al., 2007). Therefore, the increased expression of myelinogenic/iron-related genes may well be related to the aging-related increase in immunostaining of myelin in corpus callosum and hippocampus (Fig. 5). Previous work in monkeys, humans, and rodents also found increased or continuing activation of myelination programs in aging brains (Peters et al., 2000; Blalock et al., 2003; Bartzokis et al., 2007; Rowe et al., 2007). In addition, human and monkey studies suggest that myelin degeneration and rarefaction may develop with advanced age, perhaps indicating a late stage of vulnerability in the more extensively myelinated brains of primates.
A substantial number of cholesterol and phospholipid trafficking genes/proteins also showed initial upregulation in the midlife range (e.g., Apoe, Clu/ApoJ, Lcat, Srebf1, Pcyt1a) (Table 3). These findings importantly extend previous studies (Blalock et al., 2003; Rowe et al., 2007) by showing that much of this cholesterol pathway upregulation occurs in midlife and generally coincides with onset of cognitive impairment. In addition, some cholesterol-related ARGs were positively correlated with cognitive dysfunction, particularly Apoe, which was the upregulated gene most consistently correlated with cognitive impairment in this study (Fig. 3) (supplemental Table C, available at www.jneurosci.org as supplemental material).
During maturation, much of the excess free cholesterol in brain appears to be disposed of via metabolism to oxysterols and transport to neurons or extrusion into blood vessels (Dietschy and Turley, 2001; Björkhem, 2006). However, it is unclear why cholesterol trafficking pathways might exhibit a new phase of upregulation at midlife. In the liver, many cholesterol trafficking genes are known targets of the nuclear liver X receptor (LXR), and an LXR–ApoE pathway also appears to be present in brain, induced primarily in astrocytes by high cholesterol or its metabolites (Petanceska et al., 2003; Liang et al., 2004; Abildayeva et al., 2006). Therefore, one possible explanation for a midlife phase of activated cholesterol trafficking is that a new source of excess free cholesterol develops, possibly as a byproduct of lysosomal degradation of cholesterol-rich myelin fragments, in turn activating upregulation of ApoE and other cholesterol trafficking genes/proteins in astrocytes (Figs. 4, 5). In addition, the data also suggest a novel pathway for the disposal of this putative midlife cholesterol excess. That is, upregulation of myelinogenic programs, cholesterol transport molecules in oligodendrocytes (Apod, Abca2), and apparent myelination (Fig. 5) suggest that transport of cholesteryl esters in ApoE-containing lipoproteins from astrocytes to remyelinating oligodendrocytes may mediate incorporation and long-term storage of excess cholesterol in new myelin.
Functional implications of upregulated cholesterol trafficking
Studies in AD mouse models have shown that pharmacologic or genetic enhancement of brain LXR–ApoE cholesterol transport/storage pathways increases clearance and/or reduces deposition of β-amyloid in brain (Holtzman et al., 2000; Rebeck et al., 2002; Puglielli et al., 2003; Cao et al., 2007; Koldamova and Lefterov, 2007; Landreth et al., 2008). Conversely, however, elevated ApoE expression may be associated with brain degeneration under some conditions (Gallardo et al., 2008). Moreover, high dietary cholesterol (associated with elevated AD risk) stimulates, whereas statins reduce, ApoE expression in astrocytes (Petanceska et al., 2003). Furthermore, the findings here provide new evidence that elevated ApoE expression in hippocampus is correlated positively with cognitive impairment in normal brain aging (Fig. 3). Accordingly, whether upregulation of ApoE and cholesterol transport pathways is beneficial or harmful may depend on tradeoffs between the damage caused by accumulation of a toxic substance (e.g., cholesterol, β-amyloid) versus the metabolic costs of upregulating the energy-expensive cholesterol trafficking pathway to clear the substance. Thus, the level of activity in the cholesterol trafficking pathway may be an important target for regulating the intersection between normal brain aging and AD.
Sequential model of hippocampal/cognitive aging
Together, the findings suggest a novel sequential model of hippocampal aging, in which early shifts in bioenergetic processes lead to inflammatory/oxidative degradation of myelin and other membranes, generating energy substrates. However, this degradation also releases excess cholesterol that by midlife activates cholesterol transport from astrocytes to oligodendrocytes for cholesterol storage/disposal via myelinogenesis. These energy-expensive glial responses damage neurons and compete for energy with neuronal signaling pathways, inducing cognitive impairment in the most susceptible subjects (Fig. 6). In this view, astrocytes function in brain analogously to hepatocytes in conserving glucose and regulating cholesterol trafficking, myelin functions in the brain analogously to peripheral adipose tissue in the periphery, serving as a source for FFAs, and elevated inflammatory processes function somewhat analogously to peripheral stress pathways that generate FFA/AA substrates. Alternative models could, of course, account for these results, including those based on the converse sequence, in which increased metabolic utilization of FFAs is an adaptive response to initial inflammatory/oxidative damage and lipid degradation. Regardless of whether the present view is correct, however, these data on lifespan aging changes should provide an important new basis for developing testable models of brain aging cascades.
Figure 6.
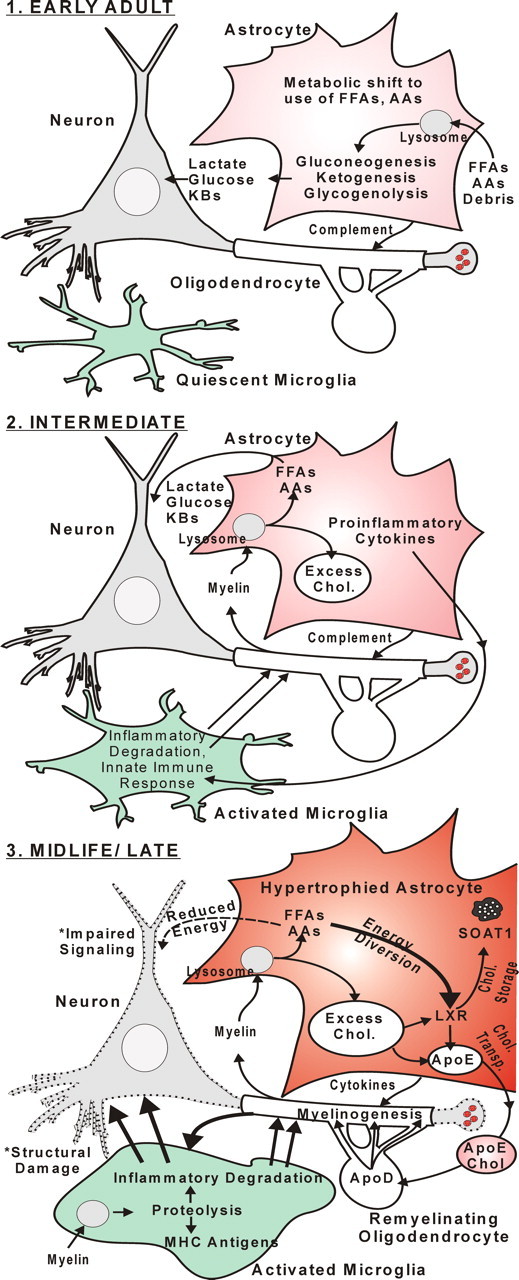
Schematic model of sequential hippocampal aging changes. EARLY ADULT, Early age-related alterations in hippocampal gene expression suggest an aging-related metabolic shift from glucose to FFAs/AAs/KBs as predominant fuel source. This shift appears orchestrated primarily in astrocytes with upregulation of genes in pathways for lipid and protein catabolism and upregulation of the lysosomal pathway. INTERMEDIATE, Astrocytes generate increased FFAs and AAs by releasing cytokines and complement components that trigger microglial inflammatory degradation of the lipid- and protein-rich myelin sheath, generating excess cholesterol as a byproduct. MIDLIFE/LATE, Increased excess cholesterol triggers upregulation of astrocytic ApoE, cholesterol trafficking, and myelinogenic programs that enable incorporation of excess cholesterol from ApoE-containing lipoproteins into remyelinating oligodendrocytes for storage/disposal. Elevated proteolysis activates antigen-presenting pathways in microglia. Together, the increased inflammation and energy-expensive glial activation induce damage in neurons (dashed outline) and compete for energy with neuronal signaling pathways, resulting in cognitive impairment.
Footnotes
This work was supported in part by National Institute on Aging Grants PO1 AG10836 and R37 AG04542. We thank Michael Bridges for important assistance in the behavioral studies.
References
- Abildayeva K, Jansen PJ, Hirsch-Reinshagen V, Bloks VW, Bakker AH, Ramaekers FC, de Vente J, Groen AK, Wellington CL, Kuipers F, Mulder M. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem. 2006;281:12799–12808. doi: 10.1074/jbc.M601019200. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Mintz J. Human brain myelination and amyloid beta deposition in Alzheimer's disease. Alzheimers Dement. 2007;3:122–125. doi: 10.1016/j.jalz.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Yong AP, Zhou J, Gall CM, Lynch G. Regionally selective changes in brain lysosomes occur in the transition from young adulthood to middle age in rats. Neuroscience. 2000;97:395–404. doi: 10.1016/s0306-4522(00)00021-x. [DOI] [PubMed] [Google Scholar]
- Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260:493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer's disease: statistical reliability and functional correlation. Ageing Res Rev. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer's disease. Adv Drug Deliv Rev. 2008;60:1504–1511. doi: 10.1016/j.addr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger C, López MC, Feller JA, Baker HV, Muzyczka N, Mandel RJ. Changes in transcription within the CA1 field of the hippocampus are associated with age-related spatial learning impairments. Neurobiol Learn Mem. 2007;87:21–41. doi: 10.1016/j.nlm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Cao G, Bales KR, DeMattos RB, Paul SM. Liver X receptor-mediated gene regulation and cholesterol homeostasis in brain: relevance to Alzheimer's disease therapeutics. Curr Alzheimer Res. 2007;4:179–184. doi: 10.2174/156720507780362173. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL. Relationship of iron to oligodendrocytes and myelination. Glia. 1996;17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Craft S. Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord. 2006;20:298–301. doi: 10.1097/01.wad.0000213866.86934.7e. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003;23:5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Finch CE, Morgan TE. Systemic inflammation, infection, ApoE alleles, and Alzheimer disease: a position paper. Curr Alzheimer Res. 2007;4:185–189. doi: 10.2174/156720507780362254. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Bizon JL, Hoyt EC, Helm KA, Lund PK. Effects of aging on the hippocampal formation in a naturally occurring animal model of mild cognitive impairment. Exp Gerontol. 2003;38:71–77. doi: 10.1016/s0531-5565(02)00159-6. [DOI] [PubMed] [Google Scholar]
- Gallardo G, Schlüter OM, Südhof TC. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–308. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–3490. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma C, Bickford PC. Interleukin-1beta and caspase-1: players in the regulation of age-related cognitive dysfunction. Rev Neurosci. 2007;18:137–148. doi: 10.1515/revneuro.2007.18.2.137. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Karuppagounder SS, Shi Q. Oxidant-induced changes in mitochondria and calcium dynamics in the pathophysiology of Alzheimer's disease. Ann N Y Acad Sci. 2008;1147:221–232. doi: 10.1196/annals.1427.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I, Van Groen T. Low levels of estrogen significantly diminish axonal sprouting after entorhinal cortex lesions in the mouse. J Neurosci. 2002;22:4095–4102. doi: 10.1523/JNEUROSCI.22-10-04095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Lefterov I. Role of LXR and ABCA1 in the pathogenesis of Alzheimer's disease: implications for a new therapeutic approach. Curr Alzheimer Res. 2007;4:171–178. doi: 10.2174/156720507780362227. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science. 1981;214:581–584. doi: 10.1126/science.6270791. [DOI] [PubMed] [Google Scholar]
- Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics. 2008;5:481–489. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, Bales KR, DeMattos RB, May PC, Li SD, Jiang XC, Eacho PI, Cao G, Paul SM. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J Neurochem. 2004;88:623–634. doi: 10.1111/j.1471-4159.2004.02183.x. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. Synaptic plasticity in early aging. Ageing Res Rev. 2006;5:255–280. doi: 10.1016/j.arr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Markowska AL, Savonenko AV. Protective effect of practice on cognition during aging: implications for predictive characteristics of performance and efficacy of practice. Neurobiol Learn Mem. 2002;78:294–320. doi: 10.1006/nlme.2002.4064. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- Mi H, Haeberle H, Barres BA. Induction of astrocyte differentiation by endothelial cells. J Neurosci. 2001;21:1538–1547. doi: 10.1523/JNEUROSCI.21-05-01538.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Pevsner J. Progress in the use of microarray technology to study the neurobiology of disease. Nat Neurosci. 2004;7:434–439. doi: 10.1038/nn1230. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Moser EI, Krobert KA, Moser MB, Morris RG. Impaired spatial learning after saturation of long-term potentiation. Science. 1998;281:2038–2042. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- Moser MB, Moser EI. Functional differentiation in the hippocampus. Hippocampus. 1998;8:608–619. doi: 10.1002/(SICI)1098-1063(1998)8:6<608::AID-HIPO3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Potential inflammatory biomarkers in Alzheimer's disease. J Alzheimers Dis. 2005;8:369–375. doi: 10.3233/jad-2005-8406. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Counts SE, Che S, Ginsberg SD. Neuronal gene expression profiling: uncovering the molecular biology of neurodegenerative disease. Prog Brain Res. 2006;158:197–222. doi: 10.1016/S0079-6123(06)58010-0. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis. 2006;9:277–289. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petanceska SS, DeRosa S, Olm V, Diaz N, Sharma A, Thomas-Bryant T, Duff K, Pappolla M, Refolo LM. Statin therapy for Alzheimer's disease: will it work? J Mol Neurosci. 2002;19:155–161. doi: 10.1007/s12031-002-0026-2. [DOI] [PubMed] [Google Scholar]
- Petanceska SS, DeRosa S, Sharma A, Diaz N, Duff K, Tint SG, Refolo LM, Pappolla M. Changes in apolipoprotein E expression in response to dietary and pharmacological modulation of cholesterol. J Mol Neurosci. 2003;20:395–406. doi: 10.1385/JMN:20:3:395. [DOI] [PubMed] [Google Scholar]
- Peters A, Moss MB, Sethares C. Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J Comp Neurol. 2000;419:364–376. doi: 10.1002/(sici)1096-9861(20000410)419:3<364::aid-cne8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998;14:263–283. doi: 10.1002/(sici)1099-0895(199812)14:4<263::aid-dmr233>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Rebeck GW, Kindy M, LaDu MJ. Apolipoprotein E and Alzheimer's disease: the protective effects of ApoE2 and E3. J Alzheimers Dis. 2002;4:145–154. doi: 10.3233/jad-2002-4304. [DOI] [PubMed] [Google Scholar]
- Roses AD. On the discovery of the genetic association of Apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9:361–366. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]
- Rowe WB, Blalock EM, Chen KC, Kadish I, Wang D, Barrett JE, Thibault O, Porter NM, Rose GM, Landfield PW. Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J Neurosci. 2007;27:3098–3110. doi: 10.1523/JNEUROSCI.4163-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DL, Petanceska S, Sabbagh M, Connor D, Soares H, Adler C, Lopez J, Ziolkowski C, Lochhead J, Browne P. Cholesterol, copper and Abeta in controls, MCI, AD and the AD cholesterol-lowering treatment trial (ADCLT) Curr Alzheimer Res. 2005;2:527–539. doi: 10.2174/156720505774932296. [DOI] [PubMed] [Google Scholar]
- Wolozin B. Cholesterol, statins and dementia. Curr Opin Lipidol. 2004;15:667–672. doi: 10.1097/00041433-200412000-00007. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease: a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, Tylavsky FA, Newman AB. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA. 2004;292:2237–2242. doi: 10.1001/jama.292.18.2237. [DOI] [PubMed] [Google Scholar]