

Abstract
In vitro studies have suggested that the TatBC complex serves as the receptor for signal peptides targeted for export via the twin-arginine translocation (Tat) pathway. Substitution of the hallmark twin-arginine dipeptide with two lysines abrogates export of physiological substrates in all organisms. We report the isolation and characterization of suppressor mutations that allow export of an ssTor(KK)-GFP-SsrA tripartite fusion. We identified two amino acid suppressor mutations in the first cytoplasmic loop of TatC. In addition, two other amino acids in the first cytoplasmic loop exhibit epistatic suppression. Surprisingly, we also identified a suppressor mutation predicted to lie within the second periplasmic loop of TatC, a region that is not expected to interact directly with the signal peptide. The suppressor mutations allowed export of the native Esherichia coli Tat substrate trimethylamine N-oxide reductase with a twin-lysine substitution in its signal sequence. The cytoplasmic suppressor mutations conferred SDS sensitivity and partial filamentation, indicating that Tat export of authentic substrates was impaired.
Keywords: twin-arginine translocase, TatC, signal peptide recognition, flow cytometry, suppressors
The bacterial twin-arginine (RR) translocation (Tat) pathway is responsible for the transport of cofactor-containing proteins and other folded polypeptides across the cytoplasmic membrane. Proteins exported via Tat contain long signal peptides featuring a conserved RR motif with a consensus sequence of S/T-R-R-x-F-L-K.1 In Escherichia coli, the functional Tat translocon is composed of the membrane proteins TatA, TatB and TatC. Among Gram-positive bacteria, a few organisms can be found containing only two identifiable tat genes, encoding TatA and TatC homologues and lacking a tatB gene.2-5 In Escherichia coli, TatA is purified as a series of high molecular weight homo-oligomers that form pore-like structures of varying diameters. Based on these observations, Gohlke et al. proposed that TatA forms the protein-conducting pore that assembles on demand, the size of which is determined by the dimensions of the translocated protein substrate.6 Biochemical analyses have revealed that TatB copurifies in a complex with TatC and that this complex plays a key role in the targeting of Tat substrates to the translocon.7-9 Based on cross-linking of in vitro translocated proteins into inverted membrane vesicles, Alami et al. discovered that TatC in particular exhibits extensive contact with the signal peptide and recognizes the RR motif.7
We sought to obtain genetic evidence that supports the role of TatC in the recognition of the RR motif and to further define the TatC epitopes that are important for this process. Substitution of the nearly invariant RR dipeptide by two lysines completely abolishes the export of all physiological Tat proteins tested so far.10,11 However, there is evidence for very low level export of heterologous proteins such as colicin V2 and the maltose binding protein (MBP) when fused to the trimethylamine N-oxide reductase (TorA) signal peptide in which the RR motif had been mutated to a KK dipeptide (TorA(KK)).12
Here we report the isolation and characterization of TatC suppressor mutations that allow export of precursor proteins fused to a defective TorA(KK) signal peptide. These results provide additional genetic evidence that TatC serves as the receptor for Tat signal peptides. In addition, the suppressor mutations we have obtained help define regions in TatC that play a role in the recognition of the RR motif. As this manuscript was about to be submitted for publication, Kreutzenbeck et al. reported the isolation of suppressor mutations in a plasmid encoding the TatABCE protein conferring export of an ssTorA-MBP fusion in which the RR dipeptide had been substituted by a lysine-glutamine sequence.12 Kreutzenbeck et al. presented evidence showing that the suppressor mutations permitted near wild-type (w.t.) level of export of ssTorA-MBP containing a w.t. RR dipeptide. In contrast, mutants we report here appear to represent a different class of suppressors that allow export of TorA(KK) signal peptides but impair translocation of native Tat proteins.
Isolation of suppressor TatC variants by fluorescence-activated cell sorting
ssTorA fused to the fluorescent reporter GFP-SsrA allows the facile and quantitative detection of Tat export by flow cytometry.11 The ssTorA-GFP-SsrA (TGS) fusion is encoded by the plasmid pTGS11 under the control of the arabinose promoter. The presence of the SsrA tag on green fluorescence protein (GFP) ensures that any fusion protein that fails to be exported via the Tat pathway is degraded by ClpXP.13 Thus, cell fluorescence arises only from TGS that has been sequestered away from the proteolytic machinery following Tat-dependent export into the periplasmic space (Fig. 1).11,14 E. coli B1lk0 (ΔtatC)15 cells carry a complete deletion of the tatC gene and therefore exhibit background fluorescence with GFP-SsrA; in addition, they are sensitive to anionic detergents such as SDS and form filaments. B1lk0 cells were transformed with the plasmid pBR322-tatC, which encodes the tatC gene with its own ribosome-binding site downstream of the constitutive tat promoter. B1lk0 pBR322-tatC cells also containing pTGS exhibited a sevenfold increase in GFP-SsrA fluorescence and normal cell morphology and were resistant to SDS (Fig. 1 and data not shown). Mutation of the RR dipeptide within TGS to a KK sequence reduced the cell fluorescence to the background level, but as expected did not have any effect on cell morphology or SDS resistance (Fig. 1).11
Fig. 1.
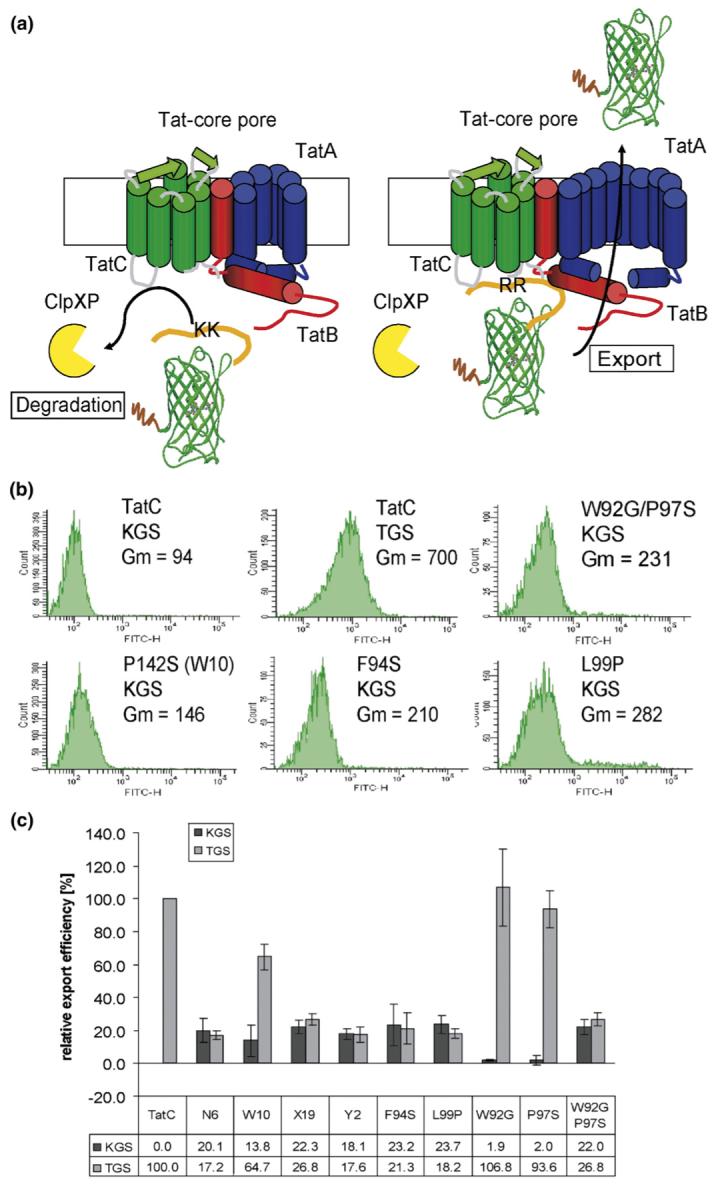
FACS analysis of cells expressing reporter fusions. (a) Schematic analysis of the screen for the isolation of suppressors. (b) Fluorescence histograms of E. coli Blk0 cells expressing either TGS (pTGS) or KGS (pKGS). tatC with its own promoter sequence was amplified from pFAT41717 using primers EMS21 (GCGGCGGACGTCTCTAGAGGAAGTGCAGCCGCAACTGG) and EMS22 (GCGGCGCCTGCAGGGTCGACTTATTCTTCAGTTTTTTCGCTTTCTGC) and then was inserted into pBR322 between the AatII and SalI sites. A 0.6% nucleotide substitution library of tatC was generated by error-prone PCR16 using primers TatB217 and EMS22; the mutagenized gene product was used to replace tatC gene in the pBR322-tatC construct. For immunostaining, an octahistidine tag was added by PCR amplification with the primers TatB2 and EMS23 (CGCCGCGTCGACTTAATGGTGATGGTGATGGTGTTCTTCAGTTTTTTCGCTTTCTGC). Cells were cultivated in LB after subculturing 1/20 from saturated overnight cultures. Cultures were induced in mid-logarithmic phase with 0.02% arabinose, and after a 5-h induction period, cells were washed once with PBS, diluted 100 times and analyzed by flow cytometry on a BD FACSort™ Cell Sorter or FACSAria. The fluorescence histograms of single amino acid suppressors and a reconstructed double amino acid substitution exporting KGS are indicated; data shown were acquired by FACSAria (Gm, geometric mean fluorescence). (c) Average relative export efficiencies of isolated variants and single point mutations exporting KGS and the w.t. TorA signal peptide fused to GFP-SsrA. The geometric mean fluorescence of the negative control (B1lk0 pBR322-tatC with pKGS) was subtracted and the results were normalized by comparing the values obtained to the fluorescence in B1lk0 pBR322-tatC expressing pTGS. Relative export efficiency was the average of three independent measurements from three separate cultures for each clone; error bars represent the standard deviation of the relative export efficiency.
The tatC gene was subjected to random mutagenesis by error-prone PCR.16 The resulting DNA was ligated into pBR322-tatC in place of the w.t. tatC gene. The ligation product was transformed into XL-1 blue cells (Stratagene) resulting in a library of 2×106 independent transformants. The plasmid preparation of mutagenized pBR322-tatC was then introduced into E. coli B1lk0 cells transformed with a plasmid encoding ssTorA(KK)-GFP-SsrA (pKGS). Sequencing of six clones selected at random revealed a mutation rate of 0.6% nucleotide substitutions per gene.
B1lk0 cells expressing the tatC alleles and KGS were then screened by flow cytometry and clones with increased fluorescence were isolated. No clones with fluorescence comparable to the wild type (around sevenfold higher than background) were obtained even after repeated attempts. However, four clones (B1lk0 with pTatC-N6, pTatC-W10, pTatC-X19 and pTatC-Y2) that reproducibly exhibited about 2- to 2.5-fold higher fluorescence over background were obtained and studied further (see Fig. 1 and Figure ST2, Supplemental Data, for sequence alignment). The elevated fluorescence phenotype was dependent on the expression of both the TatC variant and the KGS reporter protein, suggesting that the mutations act as suppressors of the export defect caused by the KK substitution in the ssTorA signal peptide.
With the exception of pTatC-W10, which contained the single amino acid substitution P142S, all isolated tatC genes contained multiple amino acid changes. Several of the respective single amino acid mutations (W92G, F94S, P97S, L99P, A160S, V167D, L172P, C179R and K192N) as well as the double amino acid substitution W92G/P97S were generated by site-specific mutagenesis and their ability to export KGS was analyzed by fluorescence-activated cell sorting (FACS). TatC carrying the amino acid substitutions F94S and L99P conferred cell fluorescence comparable to clones pTatC-X19 and pTatC-Y2, respectively. B1lk0 carrying pTatC-N6 contained the amino acid substitutions W92G, P97S and P172L. While neither the amino acid change in pTatC-W92G nor pTatC-P97S displayed export of KGS above background, the double substitution variant W92G/P97S exhibited a fluorescence profile comparable to that of pTatC-N6 (Fig. 1C). None of the other tested single amino acid substitutions conferred fluorescence above background in cells co-transformed with pKGS. To investigate the effect of mutations on the substrate specificity for the w.t. TorA signal peptide, we analyzed the fluorescence of the mutants expressing TGS (Fig. 1C). We calculated the relative export efficiency after subtracting the background fluorescence of B1lk0 cells transformed with pBR322-tatC and pKGS. The export efficiency of the w.t. signal peptide fusion for all suppressing variants except for the clone carrying pTatC-W10 (containing the single mutation P142S) was around the same amount as for KGS, indicating a partial impairment of export. The TatC P142S variant conferred around 65% transport efficiency of TGS compared to the w.t. TatC plasmid.
The level of suppression conferred by the various single amino acid TatC variants may also be related to the expression level of the respective proteins. In pBR322-tatC, tatC expression occurs from the weak tat promoter and a pBR322 origin. Attempts to detect TatC produced from pBR322-tatC using Western blot analysis with existing polyclonal sera17-19 were unsuccessful, evidently because of the low level of tatC expression. To circumvent this problem, TatC was fused to an octahistidine tag (His8). Consistent with previous observations, we confirmed that a C-terminal His8 fusion does not interfere with the function of w.t. TatC or with that of the suppressor variants (data not shown).17,18 Western blot analysis of TatC variants in total cell lysate revealed that the level of expression of pTatC-F94S-His8 and pTatC-W10-His8 (P142S) was identical with that of w.t. pTatC-His8, whereas pTatC-L99P-His8 accumulated at somewhat lower levels (Figure S2). Thus, at least for two-thirds of the single amino acid TatC variants, the degree of suppression does not stem from differences in expression level.
Further, we examined whether the putative suppressors identified in the screen above could export a native E. coli protein having a KK substitution in the RR dipeptide. The enzyme TorA reduces trimethylamine N-oxide (TMAO) to trimethylamine. TorA contains a bismolybdopterin guanine dinucleotide cofactor, which is incorporated into the apo protein within the cytoplasm, a process that requires the chaperone TorD.20,21 Following cofactor incorporation, the TorA holoenzyme is exported into the periplasm by the Tat machinery.22 Reduction of TMAO has to occur on the periplasmic side of the inner membrane in order to assure electron transfer from TorC to TorA onto TMAO.23
In E. coli GB22KK-C (torA R11K,R12K; ΔdmsABC∷ KanR, ΔtatC∷SpecR) the RR dipeptide in the signal peptide of the chromosomal torA gene has been replaced with a KK sequence10 and there is a complete tatC deletion marked with a spectinomycin resistance cassette.17 Strain GBKK22-C also lacks dsmABC gene that encodes the only other enzyme that is known to reduce TMAO.10 The TorA protein in GBKK22-C cannot be exported, but it is competent for cofactor assembly and, as a result, the active enzyme accumulates in the cytoplasm.10 GBKK22-C cells expressing the TatC variants encoded by pTatC-N6, pTatC-W10 (P142S), pTatC-X19, pTatC-Y2, the TatC single amino acid substitution construct pTatC-L99P or the double amino acid substitution pTatC-W92G/P97S were grown anaerobically for 22-24 h in Cohen and Rickenberg medium,24 harvested, and fractionated by the osmotic shock procedure.25 The efficiency of the fractionations was evaluated by monitoring the level of GroEL and DsbA as cytoplasmic and periplasmic markers, respectively. No GroEL could be detected by Western blot analysis in the periplasmic fractions, whereas DsbA was localized in the periplasmic fraction as expected (data not shown). Periplasmic and cytoplasmic proteins were then resolved by native electrophoresis and the gel was stained with methyl viologen in the presence of the reducing agent dithionite. Reduced methyl viologen served as an artificial electron donor for TMAO reductase, resulting in a readily detectable color change from blue to transparent upon oxidation of TMAO by the mature TorA enzyme26 (Fig. 2). In E. coli MC4100 cells, about 50% of the active TMAO reductase was localized in the periplasmic fraction. No periplasmic TMAO reductase band was detected in E. coli GBKK22-C expressing w.t. TatC from pBR322-tatC (Fig. 2). Enzymatically active TMAO reductase was clearly detected in periplasmic fractions of cells co-expressing the TatC suppressor plasmids except for cells with pTatC-W10 (Fig. 2). However, we noticed that cells expressing the plasmid pTatC-W10 carrying the tatC P142S allele grew anaerobically with glycerol (0.6%) and TMAO (0.4%) as the final electron acceptor. Since periplasmic TMAO reductase activity is required for growth on TMAO, TatC P142S variants must be capable of exporting TorA having a KK mutation in its signal sequence. Consequently, it is possible that the failure to detect soluble TorA in the periplasmic fraction of these cells is due to the retention of the enzyme on the surface of the spheroplasts. Alternatively, it is possible that a very low level of exported TMAO that cannot be detected by the assay we used is still sufficient to allow growth under these conditions.
Fig. 2.

Distribution of active TMAO reductase in cytoplasmic and periplasmic fractions of cells expressing tatC suppressors. Cell cultures (15 ml) were grown for 22-24 h anaerobically in Cohen and Rickenberg medium24 as previously described36 with 0.4% glucose and 0.4% TMAO (Sigma-Aldrich). Cells were normalized based on OD600 to a total of OD600 5 and then fractionated by the cold osmotic shock procedure.25 Briefly, cells were resuspended in fractionation buffer (30 mM Tris-HCl, pH 8.0, 20% (w/v) sucrose, 1 mM EDTA, pH 8.0), incubated at room temperature for 10 min, and then after centrifugation were resuspended in ice-cold MgSO4 (5 mM). Spheroplasts were sonicated. Of the obtained fractions, 12.5% were loaded, and proteins were separated under native condition on a 7.5% Tris gel at pH 6.5. Gels were incubated with 20 ml of 100 mM sodium phosphate buffer (pH 7); 1 mM methyl viologen was added and reduced by an excess of a 1% dithionite solution in 0.1 mM NaOH. Gels were incubated under mild shaking until stained blue; 10 ml of a 2 M TMAO in the same phosphate buffer was added to visualize TorA activity as can be seen in the transparent lanes. MC4100 and B1lk0 (ΔtatC) containing pBR322-tatC were used as positive controls. GB22KK-C cells contain the gene encoding TorA(R11K,R12K) in the chromosome and the deletion of dmsABC∷kan.17
Effects of tatC suppressor alleles on SDS resistance and cell separation
In E. coli, the N-acetylmuramoyl-l-alanine amidases AmiA and AmiC are exported by Tat.2 These enzymes cleave the peptide moiety of the N-acetylmuramic acid in peptidoglycan. Mutations that block the Tat pathway and prevent the export of AmiA and AmiC result in sensitivity to SDS2 and partially inhibit cell division.27 In amiC-deficient strains, the daughter cells separate poorly and, as a result, up to 30% of the population forms chains.28 amiA mutants exhibit a milder defect in septation with only 5-10% of the cell population forming short chains. amiA and amiC double mutants show a much more pronounced cell-chain phenotype than amiC mutants.27-29 Light microscopy of stationary-phase cells expressing the tatC suppressor genes in B1lk0 (ΔtatC) revealed different degrees of cell-chain formation (Fig. 3). The TatC P142S variant gave the least defect in cell separation with several short chains (3-4 cells) observed in addition to a few single cells. B1lk0 cells containing pTatC-N6 showed a higher percentage of longer chains (>4 cells per chain). The TatC W92G/P97S substitutions that recapitulate the cell fluorescence and TMAO activity phenotypes of cells carrying pTatC-N6, which contains these two amino acid changes next to the substitution P172L, showed a lower percentage of long chains. The TatC variants, encoded by pTatC-X19, pTatC-Y2, pTatC-F94S and pTatC-L99P, showed severe impairment in cell division. Collectively, these data indicate that none of the suppressor mutations completely restored a normal cell morphology, suggesting that normal Tat export of proteins involved in cell division, presumably AmiA and AmiC, is at least partially impaired.
Fig. 3.
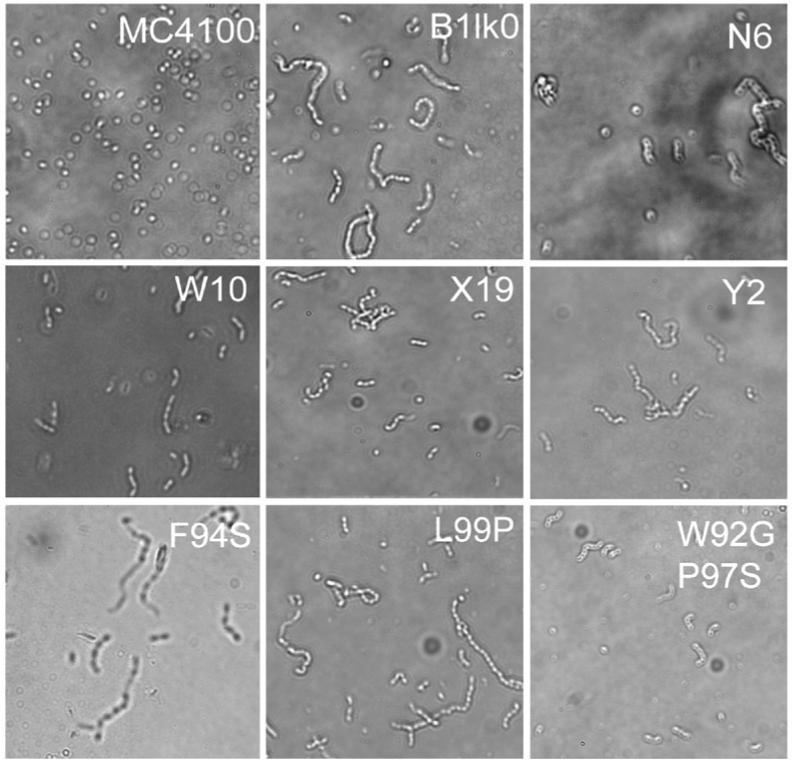
Morphology of TatC suppressors. Light microscopy photographs of stationary phase of B1lk0 transformed with plasmids expressing the indicated tatC suppressor alleles. Cells were grown in LB medium at 37 °C.
A functional Tat export system is required for normal cell envelope formation as determined by the ability of the cells to grow in the presence of the anionic detergent SDS. W.t. E. coli cells are almost unaffected by the presence of 2% SDS. The tatC strain B1lk0 fails to form colonies on agar plates with 2% SDS, but upon transformation with pBR322-tatC completely restored SDS resistance. However, cells expressing the tatC suppressor alleles failed to form colonies on LB plates with 2% SDS. The only exception was the TatC P142S substitution, which showed normal colony formation on 2% SDS (data not shown). These observations provide further evidence that the TatC suppressor mutations do not support the export of the native Tat proteins that are responsible for detergent resistance.
Discussion
Substitution of the prototypical RR sequence found in Tat signal peptides by a lysine dipeptide prevents translocation via the Tat pathway in all organisms examined so far, presumably because this mutation inhibits the interaction of the signal peptide with tatC.7 A search for mutations in the tatC gene that allow export of KGS, conferring increased cell fluorescence, ultimately led to the isolation of two different types of suppressor tatC alleles having a single amino acid substitution each. Two single amino acid suppressor mutations located on the cytoplasmic site of TatC conferred 20-25% of the cell fluorescence relative to the level observed with the w.t. tatC regardless of the signal peptide (RR or KK) used. We also identified two mutations, W92G and P97S, that acted in an epistatic manner to suppress the export defect resulting from the substitution of the RR dipetide by KK. Neither of these two amino acid substitutions alone conferred export of signal peptides with a KK dipeptide. In addition, the two single amino acid suppressors and the W92G/P97S allele were shown to confer export of TMAO reductase (TorA) fused to a signal peptide containing the KK substitution. Finally, the cells displayed defects in septation and in SDS sensitivity. On the other hand, the amino acid substitution P142S constitutes a different kind of suppressor in that it confers around 15% relative export efficiency for KGS but a significantly higher relative export efficiency for TGS (∼65%). This finding and the cell’s higher resistance to SDS suggest that export of Tat substrates containing w.t. signal peptides is not greatly impaired by the TatC P142S mutation. While we could not detect TMAO activity in the periplasmic space of GB22KK-C with the TatC P142S variant encoded by pTatC-W10, the finding that these cells can grow anaerobically with TMAO as a final electron acceptor, a process that depends on the presence of TMAO reductase in the periplasm, revealed that export of the enzyme was taking place.
The topology of TatC within the membrane has been determined experimentally using numerous reporter fusions (Fig. 4).30-32 According to this model, the first types of suppressor mutations, F94S, L99P and W92G/P97S, are located at the end of the second transmembrane domain and at the beginning of the first cytoplasmic loop of TatC, respectively. The first cytoplasmic loop, also referred to as the second cytoplasmic domain of TatC, corresponds to a highly conserved region in the bacterial and chloroplast TatC family and plays a critical role in Tat export.17,33 In earlier studies, it was reported that the mutation L99A severely reduces the export of authentic Tat substrates to less than 5%. However, the effect of this mutation with respect to KK export had not been tested.17 Similarly it has been reported that an F94A substitution completely abolishes export.
Fig. 4.

Predicted location of the TatC suppressor mutations. The topological model of TatC was obtained from Buchanan et al.,17 Behrendt et al.32 and Ki et al.31
The mutation P142S is predicted to be located in the second periplasmic loop, raising the question of how it is able to affect the recognition of the signal peptide, since this event is expected to take place or at least be initiated in the cytoplasm. However, in a search for tatC mutants that allow export of a defective ssTorA(KQ)-MBP fusion, Kreutzenbeck et al. reported the isolation of the amino acid substitution E8K located at the N terminus of TatB, a domain that is predicted to be localized in the periplasm.12 The finding that mutations in periplasmic residues in TatC or in TatB can impact the recognition of the signal sequence highlights the complexity of the organization of the Tat translocon. As has been noted by Kreutzenbeck et al., mutations in residues exposed to the periplasmic side could result in a reorganization of the tertiary structure of TatC or TatB. For the thylakoid Tat pathway, signal peptide binding was reported to occur in a binding pocket deep inside the membrane domains of the TatB/TatC (Hfc106/cpTatC) receptor complex.8 Thus, it is conceivable that mutations in a periplasmic domain may affect recognition events that occur either deep within the membrane or perhaps in proximity of the periplasmic site. Alternatively, we cannot rule out the possibility that during the translocation cycle TatB and also some domains of TatC may undergo dramatic conformational changes, as has been proposed to be the case for TatA.34
The tatC mutations we identified in the present study exhibit different phenotypes relative to those reported recently by Kreutzenbeck et al.12 The latter were isolated based on their ability to confer growth of cells expressing ssTorA(KQ)-MBP on maltose plates but they were also shown to allow efficient recognition or even higher membrane translocation rates of w.t. ssTorA-MBP. We utilized a more stringent screen in which export could be quantitatively determined based on cell fluorescence and isolated mutants that suppressed the export defect of KGS. We found two different types of suppressors: one type of suppressor variants exhibited an ∼80% reduction in the export of TGS and reduced ability to export authentic Tat substrates as manifested by septation defects and SDS sensitivity. These suppressor mutations, namely, substitutions F94S, L99P and W92G/P97S, are located in the first cytoplasmic loop of TatC, unlike the majority of Kreutzenbeck’s mutations, which localized within the first few N-terminal residues of the protein. In addition, we isolated the mutation P142S, which is located within a putative periplasmic loop and has a more moderate effect on the export of authentic substrates, thus leading to SDS resistance but partial defects in septation.
The suppressors we characterized in the present study, F94S, L99P, W92G/P97S and P142S, lie in the center of the protein sequence at opposite sides of the third transmembrane domain. All suppressor mutations resulted in nonconservative substitutions that are likely to have a significant effect on protein conformation. Interestingly, the TatC single point mutations identified by Kreutzenbeck et al. and the ones reported in the present study were all located within the N-terminal half of the protein.12 This suggests that the N-terminal of TatC is likely to be particularly important for the recognition and interactions with the signal peptide. In agreement with these observations, recent in vitro cross-linking studies also highlighted the significance of the N terminus of TatC for signal peptide recognition.35
As was the case with the mechanistic elucidation of the Sec export pathway, the synergism between genetic and biochemical analysis is expected to lead to deep insights that would be unlikely to arise solely by either approach.
Supplementary Material
Acknowledgements
We would like to thank Dr. Philip A. Lee for helpful discussions and Dr. Danielle Tullman-Ercek for comments on the manuscript. We also would like to express our gratitude to Dr. Frank Sargent for the gift of the strain GB22KK-C, Dr. Jon Beckwith for the gift of the DsbA antibody and Dr. Ben Berks and Dr. Tracy Palmer for the gift of TatC antibodies and helpful discussions. The research was funded by National Institutes of Health 1 R01 GM069872 and by the Foundation for Research.
Abbreviations used
- Tat
twin-arginine translocation
- MBP
maltose-binding protein
- GFP
green fluorescence protein
- FACS
fluorescence-activated cell sorting
- KGS
ssTorA(KK)-GFP-SsrA
- TGS
ssTorA-GFP-SsrA
- pKGS
plasmid encoding ssTorA(KK)-GFP-SsrA
- pTGS
plasmid encoding ssTorA-GFP-SsrA
- KK
twin-lysine
- RR
twin-arginine
- TorA
trimethylamine N-oxide reductase
- TMAO
trimethylamine N-oxide
- w.t.
wild type
References
- 1.Berks BC. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- 2.Ize B, Gerard F, Zhang M, Chanal A, Voulhoux R, Palmer T, et al. In vivo dissection of the Tat translocation pathway in Escherichia coli. J. Mol. Biol. 2002;317:327–335. doi: 10.1006/jmbi.2002.5431. [DOI] [PubMed] [Google Scholar]
- 3.Jongbloed JD, Antelmann H, Hecker M, Nijland R, Bron S, Airaksinen U, et al. Selective contribution of the twin-arginine translocation pathway to protein secretion in Bacillus subtilis. J. Biol. Chem. 2002;277:44068–44078. doi: 10.1074/jbc.M203191200. [DOI] [PubMed] [Google Scholar]
- 4.Pop O, Martin U, Abel C, Muller JP. The twin-arginine signal peptide of PhoD and the TatAd/Cd proteins of Bacillus subtilis form an autonomous Tat translocation system. J. Biol. Chem. 2002;277:3268–3273. doi: 10.1074/jbc.M110829200. [DOI] [PubMed] [Google Scholar]
- 5.Yen MR, Tseng YH, Nguyen EH, Wu LF, Saier MH., Jr Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Arch. Microbiol. 2002;177:441–450. doi: 10.1007/s00203-002-0408-4. [DOI] [PubMed] [Google Scholar]
- 6.Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, et al. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc. Natl. Acad. Sci. USA. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Muller M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell. 2003;12:937–946. doi: 10.1016/s1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- 8.Gerard F, Cline K. The thylakoid proton gradient promotes an advanced stage of signal peptide binding deep within the Tat pathway receptor complex. J. Biol. Chem. 2007;282:5263–5272. doi: 10.1074/jbc.M610337200. [DOI] [PubMed] [Google Scholar]
- 9.Dabney-Smith C, Mori H, Cline K. Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J. Biol. Chem. 2006;281:5476–5483. doi: 10.1074/jbc.M512453200. [DOI] [PubMed] [Google Scholar]
- 10.Buchanan G, Sargent F, Berks BC, Palmer T. A genetic screen for suppressors of Escherichia coli Tat signal peptide mutations establishes a critical role for the second arginine within the twin-arginine motif. Arch. Microbiol. 2001;177:107–112. doi: 10.1007/s00203-001-0366-2. [DOI] [PubMed] [Google Scholar]
- 11.DeLisa MP, Samuelson P, Palmer T, Georgiou G. Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J. Biol. Chem. 2002;277:29825–29831. doi: 10.1074/jbc.M201956200. [DOI] [PubMed] [Google Scholar]
- 12.Kreutzenbeck P, Kroger C, Lausberg F, Blaudeck N, Sprenger GA, Freudl R. Escherichia coli twin arginine (Tat) mutant translocases possessing relaxed signal peptide recognition specificities. J. Biol. Chem. 2007;282:7903–7911. doi: 10.1074/jbc.M610126200. [DOI] [PubMed] [Google Scholar]
- 13.Levchenko I, Seidel M, Sauer RT, Baker TA. A specificity-enhancing factor for the ClpXP degradation machine. Science. 2000;289:2354–2356. doi: 10.1126/science.289.5488.2354. [DOI] [PubMed] [Google Scholar]
- 14.Tullman-Ercek D, DeLisa MP, Kawarasaki Y, Iranpour P, Ribnicky B, Palmer T, Georgiou G. Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J. Biol. Chem. 2007;282:8309–8316. doi: 10.1074/jbc.M610507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogsch EG, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J. Biol. Chem. 1998;273:18003–18006. doi: 10.1074/jbc.273.29.18003. [DOI] [PubMed] [Google Scholar]
- 16.Fromant M, Blanquet S, Plateau P. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal. Biochem. 1995;224:347–353. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- 17.Buchanan G, Leeuw E, Stanley NR, Wexler M, Berks BC, Sargent F, Palmer T. Functional complexity of the twin-arginine translocase TatC component revealed by site-directed mutagenesis. Mol. Microbiol. 2002;43:1457–1470. doi: 10.1046/j.1365-2958.2002.02853.x. [DOI] [PubMed] [Google Scholar]
- 18.de Leeuw E, Granjon T, Porcelli I, Alami M, Carr SB, Muller M, et al. Oligomeric properties and signal peptide binding by Escherichia coli Tat protein transport complexes. J. Mol. Biol. 2002;322:1135–1146. doi: 10.1016/s0022-2836(02)00820-3. [DOI] [PubMed] [Google Scholar]
- 19.Punginelli C, Maldonado B, Grahl S, Jack R, Alami M, Schroder J, et al. Cysteine scanning mutagenesis and topological mapping of the Escherichia coli twin-arginine translocase TatC component. J. Bacteriol. 2007;189:5482–5494. doi: 10.1128/JB.00647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilbert M, Mejean V, Giudici-Orticoni MT, Samama JP, Iobbi-Nivol C. Involvement of a mate chaperone (TorD) in the maturation pathway of molybdoenzyme TorA. J. Biol. Chem. 2003;278:28787–28792. doi: 10.1074/jbc.M302730200. [DOI] [PubMed] [Google Scholar]
- 21.Pommier J, Mejean V, Giordano G, Iobbi-Nivol C. TorD, a cytoplasmic chaperone that interacts with the unfolded trimethylamine N-oxide reductase enzyme (TorA) in Escherichia coli. J. Biol. Chem. 1998;273:16615–16620. doi: 10.1074/jbc.273.26.16615. [DOI] [PubMed] [Google Scholar]
- 22.Santini CL, Ize B, Chanal A, Muller M, Giordano G, Wu LF. A novel sec-independent periplasmic protein translocation pathway in Escherichia coli. EMBO J. 1998;17:101–112. doi: 10.1093/emboj/17.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gon S, Giudici-Orticoni MT, Mejean V, Iobbi-Nivol C. Electron transfer and binding of the c-type cytochrome TorC to the trimethylamine N-oxide reductase in Escherichia coli. J. Biol. Chem. 2001;276:11545–11551. doi: 10.1074/jbc.M008875200. [DOI] [PubMed] [Google Scholar]
- 24.Cohen GN, Rickenberg HV. Not available. Ann. Inst. Pasteur. (Paris) 1956;91:693–720. [PubMed] [Google Scholar]
- 25.Stanley NR, Palmer T, Berks BC. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 2000;275:11591–11596. doi: 10.1074/jbc.275.16.11591. [DOI] [PubMed] [Google Scholar]
- 26.Silvestro A, Pommier J, Giordano G. The inducible trimethylamine-N-oxide reductase of Escherichia coli K12: biochemical and immunological studies. Biochim. Biophys. Acta. 1988;954:1–13. doi: 10.1016/0167-4838(88)90049-0. [DOI] [PubMed] [Google Scholar]
- 27.Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, et al. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol. Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- 28.Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol. Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Priyadarshini R, Popham DL, Young KD. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J. Bacteriol. 2006;188:5345–5355. doi: 10.1128/JB.00476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drew D, Sjostrand D, Nilsson J, Urbig T, Chin CN, de Gier JW, von Heijne G. Rapid topology mapping of Escherichia coli inner-membrane proteins by prediction and PhoA/GFP fusion analysis. Proc. Natl. Acad. Sci. USA. 2002;99:2690–2695. doi: 10.1073/pnas.052018199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ki JJ, Kawarasaki Y, Gam J, Harvey BR, Iverson BL, Georgiou G. A periplasmic fluorescent reporter protein and its application in high-throughput membrane protein topology analysis. J. Mol. Biol. 2004;341:901–909. doi: 10.1016/j.jmb.2004.05.078. [DOI] [PubMed] [Google Scholar]
- 32.Behrendt J, Standar K, Lindenstrauss U, Bruser T. Topological studies on the twin-arginine translocase component TatC. FEMS Microbiol. Lett. 2004;234:303–308. doi: 10.1016/j.femsle.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 33.Allen SC, Barrett CM, Ray N, Robinson C. Essential cytoplasmic domains in the Escherichia coli TatC protein. J. Biol. Chem. 2002;277:10362–10366. doi: 10.1074/jbc.M109135200. [DOI] [PubMed] [Google Scholar]
- 34.Gouffi K, Gerard F, Santini CL, Wu LF. Dual topology of the Escherichia coli TatA protein. J. Biol. Chem. 2004;279:11608–11615. doi: 10.1074/jbc.M313187200. [DOI] [PubMed] [Google Scholar]
- 35.Holzapfel E, Eisner G, Alami M, Barrett CM, Buchanan G, Luke I, et al. The entire N-terminal half of TatC is involved in twin-arginine precursor binding. Biochemistry. 2007;46:2892–2898. doi: 10.1021/bi062205b. [DOI] [PubMed] [Google Scholar]
- 36.Sawers RG, Ballantine SP, Boxer DH. Differential expression of hydrogenase isoenzymes in Escherichia coli K-12: evidence for a third isoenzyme. J. Bacteriol. 1985;164:1324–1331. doi: 10.1128/jb.164.3.1324-1331.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.