

Abstract
The receptor for advanced glycation end products (RAGE) is a pattern recognition receptor (PRR) that interacts with diverse endogenous ligands. Ligation of RAGE triggers a series of cellular signaling events, including the activation of transcription factor NF-κB, leading to the production of pro-inflammatory cytokines, and causing inflammation. While acute inflammation serves to resolve pathogen infection and stresses, which promote tissue repair, persistent inflammation results in maladaptive tissue remodeling and damage. RAGE signaling has been implicated in multiple detrimental human illnesses including diabetes, atherosclerosis, arthritis, and Alzheimer’s disease. In addition, prolonged inflammation often serves as the precursor for arterial remodeling that underlies the exponential increase of age-associated arterial diseases. Despite the significant progress and exciting discoveries in RAGE research, little is known on the biochemistry of RAGE and the signaling mechanism of RAGE remains poorly defined. The biological impact of RAGE signaling in clinical situations and aging-associated diseases also remains to be fully realized. This review attempts to provide a comprehensive summary on both recent findings and missing pieces of the RAGE puzzle.
Keywords: receptor for advanced glycation end products, RAGE, RAGE Ligands, Toll-Like Receptors, Innate Immunity, Signaling, Inflammation, Aging, Vascular Diseases, Review
2. INTRODUCTION
Human innate immune responses are initiated through a group of PRRs termed Toll-like receptors (TLR). Ligation of TLRs with corresponding patterned ligands triggers a series of cellular signaling events, resulting in the production of pro-inflammatory cytokines and local acute inflammation. Cytokine-mediated inflammation contains the invading pathogens and activates the adaptive immune mechanisms, leading to pathogen clearance (1, 2). Until recently, this evolutionally ancient mechanism was thought to serve as the first line of defense that enables organisms to fight against pathogen infections. However, inflammation has dual effects that can be both beneficial and adverse. The latter case occurs when physiological homeostasis is perturbed. Under such situation, the acute inflammatory response sustains, and chronic inflammation often results in significant tissue damage, leading to chronic inflammatory disease states.
It is still unclear how the body environment regulates immune clearance versus chronic inflammation. Although pathogen infections often contribute to or exacerbate inflammatory disease states, a clear etiology and the mechanistic link between pathogen infections and the subsequent development of chronic inflammatory diseases have not been established. Increasing evidence, however, suggests that endogenous “danger signals” generated by cell necrosis, tissue injury, oxidative and environmental stresses, as well as aging, may provide the basis for, or contribute to, the development of chronic inflammation-related diseases, e. g. diabetes, atherosclerosis, arthritis, and Alzheimer’s disease (3-5). Two recent findings support this view. First, TLRs also interact with endogenous ligands, e.g. high mobility group box 1 protein (HMGB1, also termed amphoterin) (6, 7). HMGB1 remains intracellular under normal physiological conditions, but is released into extracellular milieu as a “danger signal” either during necrosis or under stressed conditions (8-10). Activation of leukocytes also results in active secretion of “danger signals” including HMGB1 from these cells (10). Another group of calcium binding cellular factors S100 (also termed calgranulin) seems to play a similar role (11). The list of endogenous ligands that interact with the classical Toll-like receptors is still growing (3, 12, 13). Second, a novel inflammatory signaling receptor, RAGE, which interacts with a wide range of endogenous ligands, has been identified (14, 15).
RAGE, like TLRs, is expressed in tissues and cell types that are critical for immune surveillance including lung, liver, vascular endothelium, monocytes, dendritic cells, and neurons. RAGE and TLRs have overlapping ligands: HMGB1 is also a major ligand that transduces signals via RAGE to cause inflammation. In addition to signaling, ligand-activated RAGE also serves as an adhesion receptor that interacts with integrins and facilitates the recruitment of pro-inflammatory leukocytes to the sites of inflammation, further enhancing the inflammatory state (16, 17). Genetic studies also support the role that RAGE plays in innate immunity: Ager ( the gene encoding for RAGE) knockout mice exhibit resistance to septic shock, suggesting that RAGE, like TLRs, is also important for innate immune responses (18). The genomic location of the Ager gene also suggests its possible role in innate immunity: like the majority of components in the innate immune system, Ager is encoded within the major histocompatibility (MHC) class III locus on human chromosome 6 and mouse chromosome 17 (19, 20). Most recently, RAGE has been found to complex with TLR9 and this complex detects pathogen DNA. The formation of RAGE-TLR9 complex is mediated by HMGB1 (21). This finding implies that RAGE can “team up” with other PRRs to facilitate the detection of either pathogens, or perhaps endogenous “danger signals”. Similar to that of TLRs, RAGE signaling also impacts the adaptive immunity, either directly or indirectly (22, 23). Together, these findings suggest that RAGE functions as a “noncanonical Toll” that plays a role that is broadly involved in innate immunity and disease development (24).
While ligands of TLRs known thus far are mainly derived from pathogens, most, if not all, identified RAGE ligands do not originate from pathogens. In addition to HMGB1 and S100s, advanced glycation end products (AGEs) are the major in vivo ligands that activate RAGE to elicit inflammation (25). AGEs are proteins, lipids and polynucleotides modified by nonenzymatic glycation and oxidation (26). These crosslinked and highly stable complexes are resistant to enzymatic degradation (27). Examples of well-characterized and widely studied AGEs inclu ε N-carboxymethyl-lysine (CML), pentosidine, and methylglyoxal derivatives (28). Advanced glycation and oxidation increases during aging and these processes are accelerated under physiopathological conditions such as hyperglycemia (29-31). Thus, AGEs accumulated during aging may function as persistent “endogenous danger signals” that set the stage for the manifestation of various complications including those associated with cardiovascular diseases and diabetes in older persons (32-34). In addition to their internal origin, AGEs are also derived from external sources such as heat-treated food (35) and environmental pollutions (36). Given such wide range of ligands and its sites of expression, RAGE is likely to serve as the focal point that integrates endogenous and exogenous danger signals that contribute to innate immunity-elicited and aging-associated diseases.
RAGE ligand AGEs are formed by the Maillard reaction– a complicated, non-enzymatic, and irreversible process that links reducing sugar groups to proteins, lipids and nucleic acids (37). Such adducts are not only generated during pathological conditions such as hyperglycemia and diabetes, but also during normal aging process, as various forms of AGE have been detected in the aging tissues (38, 39). RAGE signaling has also been implicated in the Alzheimer’s disease (40). Although conceivable, the role of RAGE signaling in aging and aging-related diseases has not yet fully realized and well studied.
Arterial aging is characterized by intima-media thickening accompanied by central arterial lumen increase (41). The accompanying structural and cellular changes in arterial wall are characterized by endothelial dysfunction, vascular inflammation, smooth muscle cell hyperplasia, and increased collagen and elastin degradation. These changes are often associated with persistent inflammation that results in an increase of the stiffness of the arterial wall in aging humans or animal models. Arterial aging is a dominant risk factor for the development of cardiovascular diseases. Both AGEs and RAGE have been detected in the intima, media and adventitia of the aorta, and aged tissues exhibit a clear increase of AGEs, suggesting their age-dependent accumulation (42). A significant increase of AGEs accumulation has been found in vascular tissues from diabetic patients (42) and, in hypertensive subjects (43), an increased level of AGEs is associated with increased arterial stiffness, independent of the blood pressure level.
3. BASIC STRUCTURE OF RAGE
Unlike TLRs, which share leucine-rich repeats in their extracellular portions, RAGE is a member of the immunoglobulin (Ig) superfamily (15). Although the three-dimensional structure of RAGE has not been solved, based on structural similarities to Ig, a basic picture can be drawn: RAGE is a Type I membrane protein that has three Ig domains, a single transmembrane spinning helix, and a short, highly acidic C-terminal cytosolic domain that is required for the signal transduction (Figure 1) (11). The portion most distal to the transmembrane anchor is the V-type domain that functions as the ligand-binding module (44). Following the V-domain are two C-type domains. The functions of these two C-type domains are unclear, although it has been proposed that the C1 domain adjacent to the V domain may participate in ligand binding as well (45). It has been proposed that RAGE can form cis homodimers (45, 46), or heterodimers with Mac-1 (also termed CD11B/CD18, or αMβ2), a component of β2-integrin, either in cis, or trans configurations (16, 17). However, since the former conclusion was based on studies of soluble RAGE (sRAGE, i.e. the RAGE lacking membrane anchor and the cytosolic domain) expressed and purified from E.coli, the molecular status of RAGE in its native state is still unknown. The domain (s)/region (s) required for either cis- or trans- oligomerization have also not been clearly defined. It is also unclear whether ligand binding will induce oligomerization of RAGE, as observed in several other Ig superfamily members (47, 48), or modifications of its signal domain.
Figure 1.

The basic structure of RAGE.
4. RAGE SIGNALING AND RAGE-MEDIATED INFLAMMATION
RAGE is not the only receptor that interacts with AGEs. Other PRRs, such as macrophage scavenger receptors have been reported to interact with AGEs (49, 50). However, while macrophage scavenger receptors show effective endocytosis upon ligation – a process that disposes and clears the bound AGEs, RAGE ligation, in parallel to TLR ligation, mainly promotes signaling and cellular perturbation, leading to inflammation (51). This property, along with a family of diverse endogenous ligands, has made RAGE not only a possible alternative of the TLR, but also a distinct player in the development and progression of inflammatory diseases and aging.
4.1. Signaling through RAGE
Similar to TLRs, activation of RAGE relays the cell surface signals to various intracellular pathways including the NF-κB pathway, which is responsible for a myriad of transcriptional programs leading to the production of pro-inflammatory cytokines. In addition to NF-κB, among the known pathways that can be activated by RAGE are phosphoinositide 3-kinases (PI3K)/Akt (also termed as protein kinase B), and mitogen-activated protein kinase members (MAPKs) (52, 53). The latter group includes Jun-N-terminal kinase (JNK), p38, and extracellular signal-regulated kinases (ERK) (54, 55) (Figure 2). These signaling pathways often network to regulate the overall cellular responses to various stimuli, stresses and environmental cues (56).
Figure 2.

Signaling via RAGE. RAGE is activated by its ligand, and signals are relayed to three potential pathways. Among the identified signaling routes, NF-κB pathway is best studied and is likely to play major role in RAGE signaling-mediated inflammation. Depending upon ligand type, these pathways are either selectively activated, or, crosstalk, to regulate cellular activities.
Because the expression of RAGE itself is also controlled by NF-κB transcription factors (57), the activation of the RAGE-NF-κB signaling route results in an increased cell surface expression of RAGE. Such a “feed forward” loop amplifies the initial signal and further enhances inflammation (33, 58). The key step that activates NF-κB is the stimulus-dependent, residue-specific phosporylation of NF-κB inhibitors, IκBs. This phosphorylation is a prerequisite for the subsequent ubiquitination and proteasomal degradation of the inhibitor (59). This unique regulatory mechanism does not require de novo cellular protein synthesis. The kinase complex that is responsible for the specific phosphorylation of IκB is the IKK signalsome (60). Although signals generated by different stimulus-receptor pairs are relayed via different upstream cellular effectors, they eventually converge at the IKK signalsome, which serves as the linchpin for the final few common acts to dismantle ΙκB inhibitors that release, and activate NF-κB (61).
While various cellular effectors, especially those within the “first lap of the relay” that transmit signals from TLRs, T-cell and TNFα receptors to NF-κB have been successfully dissected and well studied, none of the cellular effectors in the RAGE signaling pathway has been identified. Thus, despite studies clearly demonstrate that RAGE ligation activates NF-κB (11, 17), the molecular mechanisms of such activation remain unknown. Since RAGE possesses a unique cytosolic signal domain that does not share homologies with any of the aforementioned receptors, one can hardly intuit that RAGE employs the same identified initial effectors of these receptors as the first lap of the signal relay. It is possible though, that RAGE may “cut into the line” by directly interacting with a common downstream factor to pass the signal to the IKK signalsome (24).
4.2. Consequences of RAGE signaling
Experimental evidence and observations strongly suggest that RAGE signaling results in profound inflammation (24, 62, 63). If RAGE is a “noncanonical Toll”, does RAGE signaling simply manifest similar inflammatory consequences as TLRs, and thus reflect the functional redundancy in the innate immune system? Available experimental evidence apparently suggests otherwise. Ager-/- mice, although being resistant to septic shock, exhibit osteosclerotic-like phenotypes with increased bone mass and bone mineral density, and decreased bone resorptive activity (18, 64). Such deficiencies are due to the functional disruption of the development of osteoclast precursor cells. Osteoclasts, like monocytes, are derived from hematopoietic stem cells in the bone marrow, and signaling via RAGE in osteoclasts has been shown to affect actin cytoskeletal reorganization and cell adhesion. These steps are critical for the development of the osteoclast precursor cells towards maturity (64). In addition, RAGE also plays a homeostatic role in lung development (65). These observations argue strongly that RAGE signaling, in addition to its “non-canonical Toll” tasks, plays a distinct role in normal physiology.
Within the signaling realm, the role of RAGE seems to be unique in several aspects. First, although RAGE shares certain ligand group such as HMGB1 with TLRs, it has not been reported that other RAGE ligands such as AGEs, S100 and amyloid β-peptides also interact with TLRs. Different ligands may elicit distinct signaling patterns from the same receptor, and the selectivity of a ligand-receptor interaction in vivo is often subject to the exquisite temporal and spatial regulation. For example, the outcome of RAGE stimulation by S100B or by S100A6 differs: while S100B activates PI3/AKT kinase and NF-κB, S100A6 selectively activates JNK pathway, despite the fact both ligands are structurally identical and belong to the same calcium-binding polypeptide superfamily (52). Second, it is possible that RAGE and TLRs have different affinity to the same ligand. Endogenous ligands may have a lower dissociation constant (Kd) when interacting with RAGE than with TLRs, whereas ligands originating from pathogens bind TLRs tighter than they bind RAGE. Such ‘fine-tuning” by the specific ligand-receptor affinity, and the formation of diverse signaling complexes, may modulate the strength of the signal and determine which cellular signaling pathway is activated. Hence, the overall cellular response is tightly regulated. A parallel argument has been made that cytokines interleukin-4 and -13 influence the cell signaling specificity by forming diverse signaling complexes with different ligand-receptor affinity (66). Third, factors in a signaling pathway often exhibit multi-faceted of interactions that lead to either “crosstalk” with other pathways or with components of the regulatory circuitry. RAGE possesses a short, but highly charged, cytosolic signaling domain that bears no homologies with TLR signal domains. This unique feature may render RAGE able to attract distinct cellular factors, and hence open novel signaling and regulatory avenues.
How RAGE signaling elicits acute versus chronic inflammation is yet a remaining puzzle. Two hypotheses are summarized in Figure 3. One hypothesis proposes that the oligomerization status of RAGE ligands may play a role in eliciting acute versus chronic inflammatory responses (51). Specifically, it has been suggested that monomeric RAGE ligands generate the acute response, whereas oligomerization of RAGE ligands triggers persistent signaling, resulting in chronic inflammation. In line with this hypothesis, it has been shown that the S100B tetramer is much more effective in promoting cell survival than its dimeric counterpart, and that tetramaric S100B binds sRAGE in vitro with higher affinity than the dimeric form (46). To boot, it has been demonstrated that only multimeric forms of S100 family members can trigger RAGE-mediated signaling and cellular activities (67, 68). Nonetheless, although these observations argue that oligomeric forms of RAGE ligands are required for eliciting signaling and cellular activities, they do not distinguish between acute and chronic inflammation, and experimental evidence for this hypothesis is yet to come. Alternatively, it can also be hypothesized, on the basis of the aforementioned receptor-ligand affinity, that RAGE, may “team up” with TLRs to generate acute inflammation, leading to rapid clearance of the invading pathogen (21). In contrast, in responding to persistent endogenous danger signals with a low Kd, the receptor amounts chronic inflammation. As noted, in vivo temporal-spatial regulation may be a critical determinant of whether acute or chronic inflammation ensues. Thus, while both hypotheses are ligand-selectivity based, the former stresses the configuration of ligands regardless their origin, suggesting additional cellular regulations on the formation of the ligand’s molecular patterns; the latter, in contrast, emphasizes the specific origin of ligands and implies that the molecular pattern of a ligand is an inherent property. Systematic kinetic studies of RAGE-ligand interactions and the duration of the signaling events may help to resolve the two models, and provide some clues to understand this critical regulatory mechanism.
Figure 3.
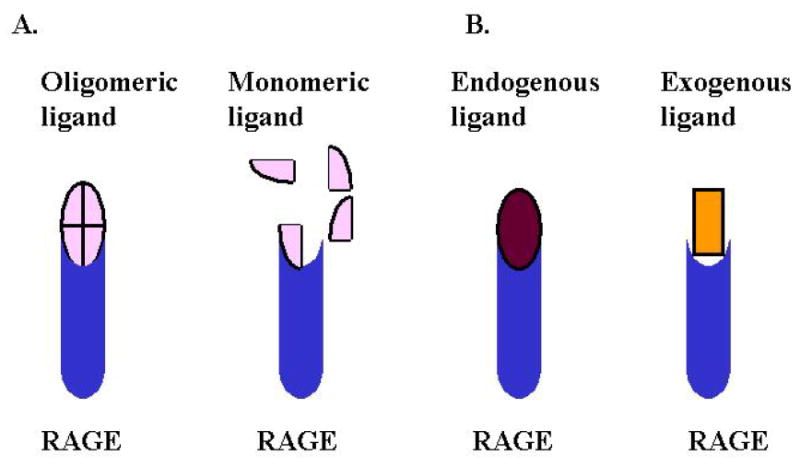
Two possible strategies that differentiate RAGE-mediated chronic versus acute inflammatory responses. A. The configuration of RAGE ligand regulates chronic versus acute inflammation through RAGE signaling. Oligomeric ligands have higher affinity to RAGE and elicit persistent signaling. In contrast, monomeric ligands bind RAGE poorly and triggers acute response only. B. Ligand origin determines chronic versus acute inflammation. While endogenous ligands possess patterns that have higher affinity to RAGE and trigger chronic inflammation, the exogenous ligands have patterns with lower affinity to RAGE, leading to acute responses and clearance.
5. RAGE-MEDIATED LEUKOCYTE RECRUITMENT AND EXTRAVASATION
In addition to signaling, ligand-activated RAGE also functions as an adhesion receptor in leukocytes to facilitate their extravasation across the endothelial barrier (16, 17). Leukocyte recruitment and infiltration through the vascular endothelium are primary processes of inflammation, and are also the early steps of atherogenesis (69). Leukocyte recruitment to the endothelium is significantly impaired in Ager-/- mice when they were artificially induced by acute peritonitis effected by thioglycollate (16). Recruitment of leukocytes can be restored by vascular endothelium-specific expression of RAGE in Ager-/- mice. Further, RAGE-mediated leukocyte adhesion to endothelial cells in vitro is mediated by a direct interaction between RAGE and the β2-integrin subunit Mac-1. Such interactions are augmented by the addition of S100B ligand. Hence, this RAGE-Mac-1-dependent leukocyte recruitment may be involved in the ICAM-1 (intercellular adhesion molecule 1)-independent leukocyte transmigration as observed in the ICAM-1 deficient mice (70). The role of RAGE ligands in leukocyte transmigration is further elucidated in another in vivo animal model system in which administration of HMGB1 induces rapid neutrophil recruitment to the endothelium in an ICAM-1 or fibrinogen-dependent manner (17). Curiously, although S100B and HMGB1 both promote interactions between RAGE and Mac-1, S100B-induced recruitment requires the expression of RAGE on endothelial cells (trans dimerization of RAGE and Mac-1 between leukocytes and endothelial cells), whereas HMGB1-induced neutrophil recruitment requires the expression of both RAGE and Mac-1 on neutrophils but not on endothelial cells (cis dimerization of RAGE and Mac-1 on neutrophils) (16, 17). RAGE-HMGB-1 interactions also enhance integrin-dependent homing of endothelial progenitor cells to ischemic locations during vascular tissue repair (71).
The molecular mechanisms underlining such diverse cell recruitment strategies induced by different RAGE ligands are obscure. HMGB1 seems to participate in the adhesion process directly, by bridging RAGE and Mac-1, rather than simply promoting RAGE expression via signaling, because ε N-carboxymethyl-lysine (CML), an AGE species that activates RAGE, does not enhance RAGE-Mac-1 trans-interactions (16). It is still possible, though, that HMGB1 promotes inflammation indirectly by eliciting cell signaling via other receptors such as TLRs 2 and 4 (6, 7), leading to the expression of adhesion molecules that are required for such recruitment on either leukocytes or endothelia. Although it has been shown that HMBG1 resembles chemotactic factors and induces cellular lamellipodium formation (17, 72, 73), whether RAGE mediates diapedisis, per se, also remains unclear. Given the pathological significance of leukocyte transmigration in atherogenesis and arterial aging, further studies on the molecular mechanism of RAGE-mediated cell migration are certainly of importance.
6. RAGE AND ARTERIAL AGING AND DISEASES
Age-associated alterations in large arteries have been well documented in both human and animal models (74-77). Clinical and experimental evidence suggest that these changes are a dominant risk factor for the development of cardiovascular diseases in older persons (4, 75). It has also been reported that the aged human arterial wall exhibits a proinflammatory profile, with elevation of several cellular matrix proteins including matrix metalloproteinases and monocyte chemoattractant protein-1 (78). Upregulation of these proteins contributes to the development of cardiovascular diseases. Since the RAGE ligand, HMGB1, also functions as a chemoattractant that facilitates the RAGE-dependent transmigration of leukocytes across vascular endothelium (71, 73, 79, 80), it is likely that RAGE, in addition to triggering inflammation, also participates in the subsequent cellular remodeling events that may lead to the abnormal proliferation of vascular cells, or to the formation of the atherosclerotic plaques. The former is a main cause for the age-associated thickening of the arterial wall, which may be a fertile soil in which arterial diseases can develop. In addition, AGEs may exert biological effects other than signaling through RAGE by adhering to extracellular matrix, thereby adversely affecting the structural integrity of the vessel wall and the underlying basement membrane (62). Notwithstanding these circumstantial clues linking RAGE signaling and RAGE-mediated leukocyte transmigration to arterial aging, direct pathological studies on these aspects are yet to be undertaken.
6.1. RAGE signaling and arterial aging and diseases
The innate immune system has been implicated in cardiovascular diseases (81). How does RAGE signaling, which also mediates innate immune response, contribute to arterial aging and diseases? Activation of RAGE by its ligands results in a “feed forward” signaling mode that upregulates RAGE expression and amplifies the cell signals. In diabetic apolipoprotein E (apo E) deficient mice, RAGE signaling mediates prolonged vascular inflammation, and enhances the expression of vascular cell adhesion molecule (VCAM)-1 and tissue factor, leading to an exacerbation of the inflammatory state (82). In addition, blockade of RAGE stabilizes established atherosclerotic plaques in diabetic apoE null mice (83). By functioning as a co- or counter-receptor of the adhesion molecules, RAGE facilitates the recruitment of leukocytes to the injured vascular tissues. This recruitment process establishes a basis for the subsequent remodeling that may alter the integrity of vascular structure and function, thus promoting atherogenesis.
RAGE signaling may also be connected to other signaling events that contribute to vascular inflammation. For example, tumor necrosis factor (TNF)-α and 17β-estradiol (E2) can enhance RAGE expression in human vascular endothelial cells, probably through the activation of NF-κB (84). AGEs also induce the expression of vascular endothelial growth factor (VEGF) in cultured microvascular endothelial cells, which confers additional cell signaling events and potentially stimulates angiogenesis in vivo (85). Alagebrium (also known as ALT7-11) is an AGE cross linkage breaker that has been demonstrated to improve ventricular function and arterial compliance in both animal studies and in human trials (86, 87). Alagebrium not only reduces RAGE expression but also collagen accumulation in the arterial tissue. A recent study has further demonstrated that alagebrium improves endothelial function in patients with isolated systolic hypertension (88), and that this is correlated with a reduction in inflammatory markers, suggesting that an anti-inflammatory effect may be a mechanism for the beneficial effect. Similarly, reducing arterial stiffness and breaking the AGE-RAGE nexus by alagebrium may help the overall reduction of inflammation and arterial aging.
6.2. Soluble RAGE
Besides production of RAGE, the AGER gene can also be alternatively spliced to produce several RAGE isoforms. The mRNAs encoding both N- and C-terminally truncated forms of RAGE have been detected in human vascular endothelial cells, pericytes, and lung (89, 90). The N-terminally truncated RAGE lacks the V-domain in the Ig-like extracellular portion, and hence does not bind AGEs. This feature may contribute to the retardation of migration of endothelial cell overexpressing N-terminally truncated RAGE (90). The observation suggests that N-terminally truncated RAGE may regulate endothelial cell migration, although the actual role of this isoform in vivo is subjected to further investigation. The C-terminally truncated RAGE is often referred as endogenous secretory RAGE (esRAGE). This variant receptor does not contain the transmembrane, and thus is secreted into the extracellular milieu as a soluble protein. In addition to esRAGE, the native, cell surface RAGE can be proteolytically cleaved to generate soluble RAGE (sRAGE) (91). Therefore, in plasma, the population of soluble RAGE includes both esRAGE and sRAGE generated by proteolytic “shedding” of native RAGEportion and the cytosolic signaling domain. The two species only differ in a small portion of their C-termini. The total circulating esRAGE and sRAGE, the cleaved RAGE products, are often collectively referred to as sRAGE, regardless of their origin.
How the two processes, which both lead to a higher level of soluble RAGE in plasma, are regulated in vivo is currently unclear. Alternative splicing can be regulated either by genomic structure of the AGER gene, and can be influenced by other trans cellular factors. Inhibition of angiotensin converting enzyme (ACE) in rats increases the sRAGE level in kidney, and this is associated with a decrease in RAGE expression (92). As noted, the molecular mechanisms underlying this phenomenon are unclear, and the regulation of sRAGE/RAGE ratio by angiotensin is likely to be indirect. Vigorous investigation on the mechanism of sRAGE production is required to provide important insights that may foster the development of therapeutic strategies to treat and prevent RAGE-linked diseases, and to advance our understanding of arterial aging.
6.3. Role of sRAGE in vascular diseases and aging
Because all sRAGE species contain an intact N-terminal portion that encompasses the entire Ig-like domains, they maintain the full capacity of ligand binding, yet are devoid of signaling functions. These properties confer upon sRAGEs the status of a natural decoy for the plasma membrane-anchored RAGE (93, 94), and a potential therapeutic tool for the treatment of inflammatory diseases including diabetes and cardiovascular diseases. In mouse diabetes models, administration of recombinant sRAGE suppressed formation of atherosclerotic lesions at aortic sinus (94). Vascular inflammatory phenotypes such as accelerated expression of VCAM-1, tissue factor, or matrix metalloproteinases in mice are also prevented by sRAGE treatment (95). Administration of sRAGE also helps to stabilize established atherosclerotic lesions in either diabetic or non-diabetic apoE-/- mice (83). Since AGEs may also form adducts with extracellular matrix, they may affect the structural integrity of the vessel in addition to signaling via RAGE (62). Thus, sRAGE may also prevent such “non-specific” adverse effects of AGEs, especially during aging.
Various clinical studies have also been carried out to define whether the plasma level of sRAGE (or esRAGE) is associated with cardiovascular and metabolic diseases (96). In general, plasma sRAGE/esRAGE level appears to be lower in patients with hypertension, atherosclerosis, cardiovascular diseases, and end-stage renal disease, suggesting that sRAGE and/or esRAGE level can serve as biomarkers for these diseases (96). These observations, as well as studies in animal models, suggest that genetically engineered recombinant sRAGE can be used as a potential therapeutic blocker for the treatment and prevention of inflammatory and metabolic diseases, or for modulating the inflammatory complications.
The plasma level of sRAGE also appears to affect human longevity (97). A high level of plasma sRAGE is associated with extreme human longevity, suggesting a role of sRAGE as an anti-aging reagent (97). But since this study measured the total soluble RAGE in the plasma of the subjects, it is unclear whether such elevation is due to the contribution of splicing (esRAGE) or to proteolytic cleavage of the wildtype RAGE (sRAGE).
7. CONCLUSIONS AND PERSPECTIVES
Since its discovery, RAGE has been implicated in several widespread chronic diseases that share a common etiology of chronic inflammation. Increasing evidence suggests that RAGE, as a PRR, functions as a “noncanonical Toll” that may not only act as a member of the innate immune system for the defense against infections, but also respond to “endogenous danger signals” generated by injuries and stresses, leading to persistent inflammation. RAGE signaling-mediated chronic inflammation may also be an important contributing factor for arterial aging. Prior research has established the role of RAGE in various dire inflammatory diseases, identified diverse range of RAGE ligands, and discovered possible corresponding signaling pathways. More recent work has revealed the role of RAGE in inflammatory cell recruitment and extravasation across endothelial barrier, and has further implicated the receptor in the pathogenic process that results in vascular complications. The pivotal role of RAGE in pathophysiological processes suggests that the receptor, as well as its signaling network, is an attractive target for therapeutic interventions. In spite of these exciting discoveries, a large void of RAGE signaling mechanisms prevails, as little is known about these signaling mechanisms and none of the players within the signaling pathways has been discovered. This, together with the lack of structure-function information of RAGE, continue to stymie our understanding of RAGE-mediated acute versus chronic inflammation. The AGER gene transcription regulation, e.g. the mechanism of alternative splicing, or post-translational regulation, e.g. the proteolytic cleavage of RAGE at cell surface, is also obscure. Future efforts on the biology of RAGE must include systematic biochemical and molecular approaches that can reveal the mechanistic aspects of RAGE-signaling and RAGE-ligand interactions. Insights drawn from such studies should help to derive more effective strategies that may harness RAGE signaling towards beneficial, rather than adverse, consequences. These efforts may also help to develop novel strategies to modulate disease development and to shed light on our understanding of human arterial aging.
Acknowledgments
We thank Dr. Rui-Ping Xiao for a reading of the manuscript, and her encouragement for the RAGE project. We are grateful for Dr. Kenneth Boheler for his critical review and specific suggestions. Special thanks go to John Pang for editing the manuscript. Our research is supported by the NIH grant RO1CA94102 (L. L.), and funding from the National Institute on Aging Intramural Research Program (E. G. L).
References
- 1.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R, Janeway C., Jr Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173:89–97. doi: 10.1034/j.1600-065x.2000.917309.x. [DOI] [PubMed] [Google Scholar]
- 3.Harris HE, Raucci A. Alarmin (g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–8. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–46. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 5.Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8:11–3. doi: 10.1038/ni0107-11. [DOI] [PubMed] [Google Scholar]
- 6.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 7.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 8.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 9.Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, Andrassy M, Marso SP, Duda S, Arnold B, Liliensiek B, Nawroth PP, Stern DM, Schmidt AM, Naka Y. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111:959–72. doi: 10.1172/JCI17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scaffidi PT, Misteli ME. Bianchi: Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 12.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 13.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–9. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 14.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–712. [PMC free article] [PubMed] [Google Scholar]
- 15.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–5004. [PubMed] [Google Scholar]
- 16.Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198:1507–15. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–39. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hammerling GG, Nawroth PP, Arnold B. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–50. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauptmann G, Bahram S. Genetics of the central MHC. Curr Opin Immunol. 2004;16:668–72. doi: 10.1016/j.coi.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, Inoko H, Ikemura T. Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics. 1994;23:408–19. doi: 10.1006/geno.1994.1517. [DOI] [PubMed] [Google Scholar]
- 21.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 22.Moser B, Szabolcs MJ, Ankersmit HJ, Lu Y, Qu W, Weinberg A, Herold KC, Schmidt AM. Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am J Transplant. 2007;7:293–302. doi: 10.1111/j.1600-6143.2006.01617.x. [DOI] [PubMed] [Google Scholar]
- 23.Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, Schmidt AM, Brown C, Stern A, LaFaille J, Chess L, Stern DM, Jiang H. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med. 2003;9:287–93. doi: 10.1038/nm831. [DOI] [PubMed] [Google Scholar]
- 24.Lin L. RAGE on the Toll Road? Cell Mol Immunol. 2006;3:351–8. [PubMed] [Google Scholar]
- 25.Schmidt AM, Yan SD, Brett J, Mora R, Nowygrod R, Stern D. Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J Clin Invest. 1993;91:2155–68. doi: 10.1172/JCI116442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thornalley PJ. Dietary AGEs and ALEs and risk to human health by their interaction with the receptor for advanced glycation endproducts (RAGE)--an introduction. Mol Nutr Food Res. 2007;51:1107–10. doi: 10.1002/mnfr.200700017. [DOI] [PubMed] [Google Scholar]
- 27.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344(Pt 1):109–16. [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas MC, Forbes JM, MacIsaac R, Jerums G, Cooper ME. Low-molecular weight advanced glycation end products: markers of tissue AGE accumulation and more? Ann N Y Acad Sci. 2005;1043:644–54. doi: 10.1196/annals.1333.073. [DOI] [PubMed] [Google Scholar]
- 29.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–21. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 30.Cerami A, Vlassara H, Brownlee M. Protein glycosylation and the pathogenesis of atherosclerosis. Metabolism. 1985;34:37–42. doi: 10.1016/s0026-0495(85)80008-1. [DOI] [PubMed] [Google Scholar]
- 31.Cerami A, Vlassara H, Brownlee M. Role of nonenzymatic glycosylation in atherogenesis. J Cell Biochem. 1986;30:111–20. doi: 10.1002/jcb.240300203. [DOI] [PubMed] [Google Scholar]
- 32.Alves M, Cunha DA, Calegari VC, Saad MJ, Boschero AC, Velloso LA, Rocha EM. Nuclear factor-κB and advanced glycation end-products expression in lacrimal glands of aging rats. J Endocrinol. 2005;187:159–66. doi: 10.1677/joe.1.06209. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt AM, Yan SD, Wautier JL, Stern D. Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999;84:489–97. doi: 10.1161/01.res.84.5.489. [DOI] [PubMed] [Google Scholar]
- 34.Simm A, Casselmann C, Schubert A, Hofmann S, Reimann A, Silber RE. Age associated changes of AGE-receptor expression: RAGE upregulation is associated with human heart dysfunction. Exp Gerontol. 2004;39:407–13. doi: 10.1016/j.exger.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 35.Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, Vlassara H. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. 2007;62:427–33. doi: 10.1093/gerona/62.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gursinsky T, Ruhs S, Friess U, Diabate S, Krug HF, Silber RE, Simm A. Air pollution-associated fly ash particles induce fibrotic mechanisms in primary fibroblasts. Biol Chem. 2006;387:1411–20. doi: 10.1515/BC.2006.177. [DOI] [PubMed] [Google Scholar]
- 37.Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol. 1992;23:1–34. doi: 10.1016/s1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed N, Ahmed U, Thornalley PJ, Hager K, Fleischer G, Munch G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J Neurochem. 2005;92:255–63. doi: 10.1111/j.1471-4159.2004.02864.x. [DOI] [PubMed] [Google Scholar]
- 39.Glenn JV, Beattie JR, Barrett L, Frizzell N, Thorpe SR, Boulton ME, McGarvey JJ, Stitt AW. Confocal Raman microscopy can quantify advanced glycation end product (AGE) modifications in Bruch’s membrane leading to accurate, nondestructive prediction of ocular aging. FASEB J. 2007;21:3542–52. doi: 10.1096/fj.06-7896com. [DOI] [PubMed] [Google Scholar]
- 40.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 41.Virmani R, Avolio AP, Mergner WJ, Robinowitz M, Herderick EE, Cornhill JF, Guo SY, Liu TH, Ou DY, O’Rourke M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am J Pathol. 1991;139:1119–29. [PMC free article] [PubMed] [Google Scholar]
- 42.Soulis T, Thallas V, Youssef S, Gilbert RE, McWilliam BG, Murray-McIntosh RP, Cooper ME. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia. 1997;40:619–28. doi: 10.1007/s001250050725. [DOI] [PubMed] [Google Scholar]
- 43.McNulty M, Mahmud A, Feely J. Advanced glycation end-products and arterial stiffness in hypertension. Am J Hypertens. 2007;20:242–7. doi: 10.1016/j.amjhyper.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 44.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM. N (epsilon)- (carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–9. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 45.Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry. 2007;46:6957–70. doi: 10.1021/bi7003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ostendorp T, Leclerc E, Galichet A, Koch M, Demling N, Weigle B, Heizmann CW, Kroneck PM, Fritz G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007;26:3868–78. doi: 10.1038/sj.emboj.7601805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soroka V, Kolkova K, Kastrup JS, Diederichs K, Breed J, Kiselyov VV, Poulsen FM, Larsen IK, Welte W, Berezin V, Bock E, Kasper C. Structure and interactions of NCAM Ig1-2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure. 2003;11:1291–301. doi: 10.1016/j.str.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Tarbouriech N, Ruggiero F, de Turenne-Tessier M, Ooka T, Burmeister WP. Structure of the Epstein-Barr virus oncogene BARF1. J Mol Biol. 2006;359:667–78. doi: 10.1016/j.jmb.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 49.Peiser L, Mukhopadhyay S, Gordon S. Scavenger receptors in innate immunity. Curr Opin Immunol. 2002;14:123–8. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- 50.Vlassara H, Brownlee M, Cerami A. Novel macrophage receptor for glucose-modified proteins is distinct from previously described scavenger receptors. J Exp Med. 1986;164:1301–9. doi: 10.1084/jem.164.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herold K, Moser B, Chen Y, Zeng S, Yan SF, Ramasamy R, Emond J, Clynes R, Schmidt AM. Receptor for advanced glycation end products (RAGE) in a dash to the rescue: inflammatory signals gone awry in the primal response to stress. J Leukoc Biol. 2007;82:204–12. doi: 10.1189/jlb.1206751. [DOI] [PubMed] [Google Scholar]
- 52.Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem. 2007;282:31317–31. doi: 10.1074/jbc.M703951200. [DOI] [PubMed] [Google Scholar]
- 53.Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, Johnson RJ, Lan HY. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18:176–8. doi: 10.1096/fj.02-1117fje. [DOI] [PubMed] [Google Scholar]
- 54.Stern D, Yan SD, Yan SF, Schmidt AM. Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev. 2002;54:1615–25. doi: 10.1016/s0169-409x(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 55.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–60. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Lin A. Wiring the cell signaling circuitry by the NF-κ B and JNK1 crosstalk and its applications in human diseases. Oncogene. 2007;26:3267–78. doi: 10.1038/sj.onc.1210417. [DOI] [PubMed] [Google Scholar]
- 57.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–97. [PubMed] [Google Scholar]
- 58.Foell D, Wittkowski H, Roth J. Mechanisms of disease: a ’DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol. 2007;3:382–90. doi: 10.1038/ncprheum0531. [DOI] [PubMed] [Google Scholar]
- 59.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. Rapid proteolysis of I κ B-alpha is necessary for activation of transcription factor NF-κB. Nature. 1993;365:182–5. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 60.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 61.Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 62.Chavakis T, Bierhaus A, Nawroth PP. RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect. 2004;6:1219–25. doi: 10.1016/j.micinf.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–55. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou Z, Immel D, Xi CX, Bierhaus A, Feng X, Mei L, Nawroth P, Stern DM, Xiong WC. Regulation of osteoclast function and bone mass by RAGE. J Exp Med. 2006;203:1067–80. doi: 10.1084/jem.20051947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lizotte PP, Hanford LE, Enghild JJ, Nozik-Grayck E, Giles BL, Oury TD. Developmental expression of the receptor for advanced glycation end-products (RAGE) and its response to hyperoxia in the neonatal rat lung. BMC Dev Biol. 2007;7:15. doi: 10.1186/1471-213X-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zdanov A, Wlodawer A. A New Look at Cytokine Signaling. Cell. 2008;132:179–181. doi: 10.1016/j.cell.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 67.Kiryushko D, Novitskaya V, Soroka V, Klingelhofer J, Lukanidin E, Berezin V, Bock E. Molecular mechanisms of Ca (2+) signaling in neurons induced by the S100A4 protein. Mol Cell Biol. 2006;26:3625–38. doi: 10.1128/MCB.26.9.3625-3638.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem. 2007;282:4218–31. doi: 10.1074/jbc.M608888200. [DOI] [PubMed] [Google Scholar]
- 69.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 70.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–101. [PubMed] [Google Scholar]
- 71.Chavakis E, Hain A, Vinci M, Carmona G, Bianchi ME, Vajkoczy P, Zeiher AM, Chavakis T, Dimmeler S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. 2007;100:204–12. doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 72.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–82. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 74.Cerami A, Vlassara H, Brownlee M. Glucose and aging. Sci Am. 1987;256:90–6. doi: 10.1038/scientificamerican0587-90. [DOI] [PubMed] [Google Scholar]
- 75.Pepe S, Lakatta EG. Aging hearts and vessels: masters of adaptation and survival. Cardiovasc Res. 2005;66:190–3. doi: 10.1016/j.cardiores.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 76.Spinetti G, Wang M, Monticone R, Zhang J, Zhao D, Lakatta EG. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler Thromb Vasc Biol. 2004;24:1397–402. doi: 10.1161/01.ATV.0000134529.65173.08. [DOI] [PubMed] [Google Scholar]
- 77.Wang M, Takagi G, Asai K, Resuello RG, Natividad FF, Vatner DE, Vatner SF, Lakatta EG. Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension. 2003;41:1308–16. doi: 10.1161/01.HYP.0000073843.56046.45. [DOI] [PubMed] [Google Scholar]
- 78.Wang M, Zhang J, Jiang LQ, Spinetti G, Pintus G, Monticone R, Kolodgie FD, Virmani R, Lakatta EG. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50:219–27. doi: 10.1161/HYPERTENSIONAHA.107.089409. [DOI] [PubMed] [Google Scholar]
- 79.Palumbo R, Bianchi ME. High mobility group box 1 protein, a cue for stem cell recruitment. Biochem Pharmacol. 2004;68:1165–70. doi: 10.1016/j.bcp.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 80.Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A, Cossu G, Bianchi ME. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–9. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–54. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 82.Kislinger T, Tanji N, Wendt T, Qu W, Lu Y, Ferran LJ, Jr, Taguchi A, Olson K, Bucciarelli L, Goova M, Hofmann MA, Cataldegirmen G, D’Agati V, Pischetsrieder M, Stern DM, Schmidt AM. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2001;21:905–10. doi: 10.1161/01.atv.21.6.905. [DOI] [PubMed] [Google Scholar]
- 83.Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–35. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 84.Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-κB, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem. 2000;275:25781–90. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- 85.Yamagishi S, Yonekura H, Yamamoto Y, Katsuno K, Sato F, Mita I, Ooka H, Satozawa N, Kawakami T, Nomura M, Yamamoto H. Advanced glycation end products-driven angiogenesis in vitro. Induction of the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. J Biol Chem. 1997;272:8723–30. doi: 10.1074/jbc.272.13.8723. [DOI] [PubMed] [Google Scholar]
- 86.Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, deGroof RC, Lakatta EG. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104:1464–70. doi: 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- 87.Vaitkevicius PV, Lane M, Spurgeon H, Ingram DK, Roth GS, Egan JJ, Vasan S, Wagle DR, Ulrich P, Brines M, Wuerth JP, Cerami A, Lakatta EG. A cross-link breaker has sustained effects on arterial and ventricular properties in older rhesus monkeys. Proc Natl Acad Sci U S A. 2001;98:1171–5. doi: 10.1073/pnas.98.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zieman SJ, Melenovsky V, Clattenburg L, Corretti MC, Capriotti A, Gerstenblith G, Kass DA. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J Hypertens. 2007;25:577–83. doi: 10.1097/HJH.0b013e328013e7dd. [DOI] [PubMed] [Google Scholar]
- 89.Malherbe P, Richards JG, Gaillard H, Thompson A, Diener C, Schuler A, Huber G. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Brain Res Mol Brain Res. 1999;71:159–70. doi: 10.1016/s0169-328x(99)00174-6. [DOI] [PubMed] [Google Scholar]
- 90.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, Reinhart TA, Oury TD. Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE) J Biol Chem. 2004;279:50019–24. doi: 10.1074/jbc.M409782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Forbes JM, Thorpe SR, Thallas-Bonke V, Pete J, Thomas MC, Deemer ER, Bassal S, El-Osta A, Long DM, Panagiotopoulos S, Jerums G, Osicka TM, Cooper ME. Modulation of soluble receptor for advanced glycation end products by angiotensin-converting enzyme-1 inhibition in diabetic nephropathy. J Am Soc Nephrol. 2005;16:2363–72. doi: 10.1681/ASN.2005010062. [DOI] [PubMed] [Google Scholar]
- 93.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105:1117–24. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–31. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 95.Wendt T, Harja E, Bucciarelli L, Qu W, Lu Y, Rong LL, Jenkins DG, Stein G, Schmidt AM, Yan SF. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis. 2006;185:70–7. doi: 10.1016/j.atherosclerosis.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 96.Choi KM, Yoo HJ, Kim HY, Lee KW, Seo JA, Kim SG, Kim NH, Choi DS, Baik SH. Association between endogenous secretory RAGE, inflammatory markers and arterial stiffness. International Journal of Cardiology. doi: 10.1016/j.ijcard.2007.10.047. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 97.Geroldi D, Falcone C, Minoretti P, Emanuele E, Arra M, D’Angelo A. High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J Am Geriatr Soc. 2006;54:1149–50. doi: 10.1111/j.1532-5415.2006.00776.x. [DOI] [PubMed] [Google Scholar]