

Abstract
Introduction
Our group has previously reported genetic studies associating polymorphisms in the low density lipoprotein receptor related protein 8 (LRP8) gene with myocardial infarction. The aim of this study was to define the role of platelet surface LRP8 in thrombosis.
Materials and Methods
Flow cytometry, aggregometry, intravital microscopy and tail bleeding assays were used to examine platelet function and hemostasis in LRP8-deficient mice and littermate controls.
Results
We demonstrated that activation of platelets from both LRP8+/- and LRP8-/- mice was reduced in vitro in response to either ADP or thrombin. In vivo, LRP8-hemizygous and LRP8-/- mice demonstrated 200% and 68% increased time for carotid occlusion in response to FeCl3 injury, respectively. Moreover, lipidated apoE3, a ligand for LRP8, inhibited platelet activation in a dose-dependent fashion. This inhibition was markedly attenuated in LRP8-/- but not LRP8+/- mice and did not result from membrane cholesterol efflux or a nitric oxide dependent pathway. Tail bleeding times were unaffected in both genotypes.
Conclusions
Our results suggest that LRP8 is capable of altering thrombosis without affecting normal hemostasis through mechanisms both dependent on and independent of apoE. This suggests a means whereby clot formation could be affected in humans with LRP8 gene variants.
Keywords: ApoER2, hemostasis, LRP8, platelet, thrombosis
Introduction
Multiple risk factors have been elucidated for myocardial infarction (MI), but the most significant and least understood of the epidemiologic factors associated with MI is family history [1]. Coronary artery disease (CAD) and MI are recognized to have polygenic modes of inheritance and complex interaction with environmental factors. Our group has previously reported the results of a genetic linkage study for MI susceptibility genes in which we detected a novel MI susceptibility locus on chromosome 1p34-36 [2]. Sixteen candidate genes in this region were subsequently evaluated; however, the only single nucleotide polymorphisms (SNPs) significantly associated with MI were in or near the low density lipoprotein receptor related protein 8 (LRP8, also termed apoE receptor 2 or apoER2) gene [3]. Moreover, the positive associations between SNPs or rare haplotypes of LRP8 and MI has been replicated in three other large case-control populations [3].
LRP8 is a member of the low density lipoprotein receptor (LDLR) family [4]; however, LRP8 contains an additional exon that encodes a novel 59 amino acid segment in the cytoplasmic tail, including three potential copies of the minimal consensus sequence for the Src homology 3 (SH3)-binding motif [5]. Interactions between adapter molecules and the cytoplasmic tail of LRP8 have been shown to have an important role in signal transduction in the brain [6]; and LRP8 has been shown to localize to caveolae in CHO cell monolayers [7], further supporting a role in cellular signaling. LRP8-deficient mice have defects in sperm maturation [8], and mice deficient in both LRP8 and the VLDL receptor have defects in cortical and cerebellar neuronal layering, similar to that observed in mice deficient in Reelin or mouse Disabled 1 [6, 9]. Known apolipoprotein/lipoprotein ligands for LRP8 include apoE and LDL [5, 10]; however, LRP8 is not thought to play a role in the endocytosis and degradation of lipoproteins [11, 12].
Although three splice variants of LRP8 have been identified on human platelets [5, 13], the function of this protein on platelets remains incompletely characterized. Lipidated apoE has been shown to inhibit human platelet reactivity by stimulating intracellular nitric oxide (NO) synthase, eventually resulting in elevated levels of anti-aggregatory cGMP [14]. It has been suggested that this effect is mediated through LRP8 [5]. The purpose of this study was to characterize the function of LRP8 in platelets and provide data that could explain the association between polymorphisms in LRP8 and myocardial infarction.
Materials and Methods
Mice
B6;129S6-Lrp8tm1Her/J mice were purchased from The Jackson Laboratory (Bar Harbor, Maine), and heterozygotes were mated to generate LRP8+/+, LRP8+/- and LRP8-/- littermates. Mice were maintained on a mixed C57BL/6 and 129S6 background, and male animals were used for experimentation. All experiments were performed in accordance with protocols approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
Isolation and purification of platelets
While anesthetized with ketamine (170 mg/kg)/xylazine (5 mg/kg), 800 μL of blood was drawn from the inferior vena cavae into a syringe containing 100 μL of acid citrate dextrose (85 mM trisodium citrate dehydrate, 65 mM citric acid monohydrate and 111 mM glucose, pH 4.6) and 100 μg prostaglandin E1 (Sigma-Aldrich, St. Louis, MO). Blood was then diluted 1:1 in Tyrode's Buffer (137 mM sodium chloride, 12 mM sodium bicarbonate and 2.5 mM potassium chloride, pH 7.2). Platelet rich plasma (PRP) was collected by two sequential centrifugations at 138× g, and platelets were sedimented at 863× g and resuspended in 250 μL of Tyrode's Buffer containing 0.1% glucose and 0.35% BSA before gel-filtration through a sepharose 2B (Sigma-Aldrich) column. All centrifugations were performed at room temperature (24°C). Final concentrations of 1 mM MgCl2 and 2 mM CaCl2 were added to the platelet rich fraction.
Preparation of apoE3/DMPC complexes
1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) large unilamellar vesicles (LUVs) were prepared by extrusion, as previously described [15]. Human recombinant apoE3 (Invitrogen, Carlsbad, CA) was then added at a lipid to protein ratio of 3.75:1 (w/w) and incubated at room temperature overnight to form apoE3/DMPC complexes [16]. DMPC only controls were prepared similarly, using PBS instead of apoE, and therefore had equivalent DMPC content.
Flow cytometry
Flow cytometry was performed on 1×106 fresh, gel-filtered platelets using 5 μL phycoerythrin (PE)-conjugated JON/A or P-selectin antibody (Emfret, Eibelstadt, Germany) in a 20 μL final volume. The JON/A antibody selectively binds to activated mouse integrin αIIbβ3. Platelets were pre-incubated for 5 minutes with PBS, 10-30 μg/mL of apoE3/DMPC complexes or DMPCs alone. Activation was then initiated through the addition of either 1 μM ADP (Chrono-log, Havertown, PA) or 6.9 mU/mL of human thrombin (Sigma-Aldrich). 300 nM murine fibrinogen (Sigma-Aldrich) was added to all ADP reactions. Incubation occurred in the dark at room temperature for 30 minutes following the addition of ADP and for 15 minutes following the addition of thrombin. Agonist concentrations and time courses were selected from a standard curve such that approximately 50-60% of maximal activation was achieved. Reactions were quenched by addition of 400 μL of PBS and quantitated immediately using a Guava EasyCyte flow cytometer (Guava Technologies, Hayward, CA).
For studies of the role of nitric oxide, platelets were pretreated with 1.2 mM N-nitro-L-arginine methyl ester (L-NAME) (Sigma-Aldrich) for 30 minutes prior to treatment with 20 μg/mL of apoE3/DMPC complexes and 6.9 mU/mL of thrombin, as described, and activation was quantitated using the JON/A antibody.
Aggregometry
Aggregometry was performed using approximately 1×108 gel-filtered platelets/ml in a final reaction volume of 250 μL. Aggregation was stimulated using 0.5 μM ADP or 50 mU/mL of thrombin. 300 nM fibrinogen was added to platelet suspensions before stimulation with ADP. Tyrode's buffer containing 0.1% glucose and 0.35% BSA was used to establish baseline turbidimetric readings. All assays were performed using a Chrono-log whole blood aggregometer and data was recorded using AGROLINK software.
Intravital microscopy
Intravital microscopy was performed on similarly-sized 8 week old mice. Mice were anesthetized, and the right jugular vein and left carotid artery were exposed through a midline cervical incision. The carotid artery was suspended on a piece of black plastic in order to provide contrast for the microscope, and the vessel was stripped of adventitia. 100 μL Rhodamine 6G (0.5 mg/mL, Sigma-Aldrich) was injected directly into the right jugular vein in order to fluorescently label platelets. A 1 × 2 mm piece of filter paper was saturated with a 12.5% FeCl3 solution and was then applied to the left carotid artery for 1 min. The filter paper was removed, and the vessel was rinsed with saline. Clot formation was observed in real-time under a Leica DMLFS fluorescent microscope (Leica, Issaquah, WA) using a Leica water immersion objective at 20× magnification and an attached Gibraltar Platform (EXFO, Quebec, Canada). The time to complete occlusion of flow was determined through visual inspection using real time video image capture with a QImaging Retigo Exi 12-bit mono digital camera (Surrey, Canada) and Streampix version 3.17.2 software (Norpix, Montreal, Canada). The assay end points were defined as 1) cessation of blood flow for more than 30 seconds or 2) 45 minutes without carotid occlusion (in which case the time was recorded as 45 minutes).
Cholesterol efflux assays
Gel-filtered platelets resuspended in 250 μL of Tyrode's buffer containing 0.1% glucose and 0.35% BSA were incubated with 1 μCi of 3H-cholesterol (Amersham Biosciences, Piscataway, NJ) for 1 hr at room temperature. The platelets were then pelleted, resuspended in fresh buffer, and washed again by filtration through a sepharose 2B column. Incubations with 30 μg/mL of apoE3/DMPC complexes or equivalent DMPC alone were performed in a final volume of 40 μL for a duration of 25 minutes at room temperature. 10 mM methyl-β-cyclodextrin was used as a positive control. Subsequently, sepharose 2B beads suspended in PBS were added to the reaction as carriers and platelets were sedimented at 863 g. Supernatants were collected, pellets were lysed in 100 μL of lysis solution (1% SDS (w/w), 10 mM sodium phosphate and 5 mM DTT), and radioactivity was measured in a scintillation counter. Percent efflux was calculated by dividing the DPM in the supernatant over the total DPM in the reaction.
Tail bleeding assays
Tails of anesthetized, 7 week old mice were equilibrated for 5 min in 37 °C PBS. 5 mm of the distal tail was then amputated and the tail was returned to the PBS. The time to cessation of bleeding was recorded as the bleeding time. The assay endpoint for bleeding times was 10 minutes (in which case the time was recorded as 10 minutes).
Statistical Analysis
All data is shown as mean ± standard deviation, unless otherwise stated. Kolmogorov-Smirnov tests were used to determine if data was normally distributed. Comparisons of three groups were performed by ANOVA, unless the data was not normally distributed, in which case the non-parametric Kruskal-Wallis test was used. All statistics were performed using Prism 4.0 software (GraphPad Software, Inc., San Diego, CA).
Results
Activation of LRP8-deficient platelets is reduced in vitro in response to ADP and thrombin
We examined activation of gel-filtered platelets in the absence of apoE using both flow cytometry and turbidimetric aggregometry. Activation of αIIbβ3 integrin, measured by flow cytometry, was significantly reduced in both LRP8+/- and LRP8-/- mice in response to both ADP (Figure 1A) and thrombin (Figure 1B). 1 μM ADP activated 21.2 ± 3.3% of platelets isolated from LRP8+/+ mice (n=6) but only activated 12.1 ± 4.2% and 15.9 ± 2.0% of platelets from LRP8+/- (n=6) and LRP8-/- mice (n=6), respectively (p<0.01 and p<0.05 for LRP8+/- and LRP8-/- platelets compared to wild-type controls). Likewise, 6.9 mU/mL thrombin activated a mean of 66.0 ± 5.9% of platelets isolated from LRP8+/+ mice (n=8), whereas it only activated 55.3 ± 5.3% and 53.1 ± 11.4% of platelets from LRP8+/- (n=7) and LRP8-/- mice (n=6), respectively (p<0.05 for both LRP8+/- and LRP8-/- platelets when compared to LRP8+/+ platelets). In contrast, thrombin mediated P-selectin expression in the LRP8 mutant mice was not statistically different across genotypes (data not shown).
Figure 1. Reduced expression of activated αIIbβ3 integrin on the surface of LRP8+/- and LRP8-/- platelets in response to ADP and thrombin by flow cytometry.
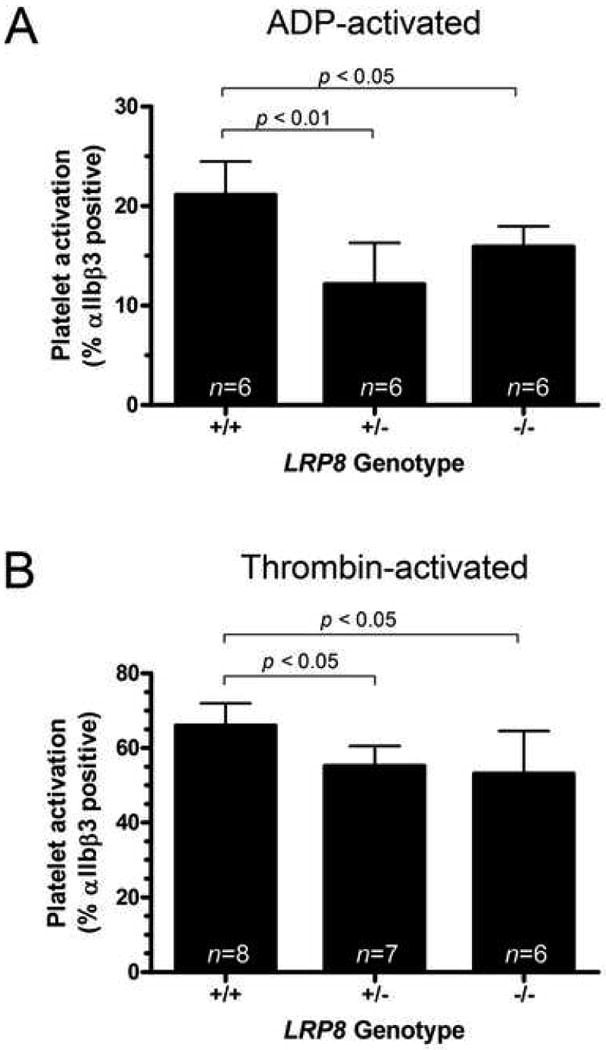
Gel filtered platelets from mice of each of the LRP8 genotypes were activated with either 1 μM ADP (A) or 6.9 mU/mL thrombin (B) at room temperature.
Results generated with turbidimetric aggregometry were consistent with the activated αIIbβ3 expression differences seen using flow cytometry. Both the rate and extent of platelet aggregation, as evidenced by the initial slope of the curve and the maximum percent aggregation achieved were significantly different in response to both ADP and thrombin (Table 1). The initial slope and maximum aggregation were decreased in LRP8+/- and LRP8-/- mice. Representative aggregation curves are shown for both ADP (Figure 2A) and thrombin (Figure 2B).
Table 1. Platelet aggregometry analytic metrics.
| 1 LRP8+/+ | 1 LRP8+/- | 1 LRP8-/- | 2 Overall p-value | 3 Dunnett's p-value | |
|---|---|---|---|---|---|
| ADP (n=5) | |||||
| 4 Slope | 1.45 (0.25) | 0.85 (0.23) | 0.85 (0.21) | 0.016 | <0.05 |
| 5% Aggregation | 47.40 (8.70) | 33.20 (10.07) | 31.40 (7.41) | 0.022 | <0.05 |
| Thrombin (n=4) | |||||
| 4 Slope | 1.18 (0.48) | 0.33 (0.15) | 0.24 (0.14) | 0.033 | <0.05 |
| 5% Aggregation | 42.50 (17.70) | 25.25 (16.33) | 20.75 (13.95) | 0.013 | <0.05 |
Values expressed as mean (SEM)
P-values generated using a repeated measures ANOVA
P-values for LRP8+/- and LRP8-/- vs. LRP8+/+ by Dunnett's post-test.
Absolute value of the slope of the results between minutes 1-2 of aggregation calculated by linear regression
Maximum aggregation in response to agonist
Figure 2. Effects of LRP8-deficiency on platelet aggregation.
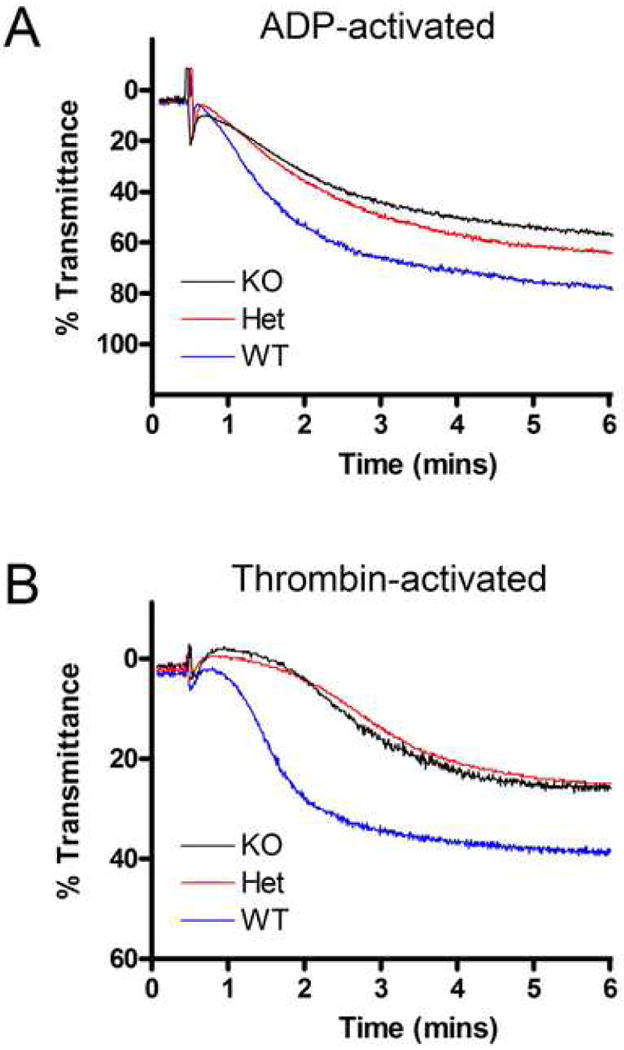
Representative in vitro platelet aggregation stimulated by either 0.5 μM ADP (A) or 50 mU/mL thrombin (B). Aggregation of gel filtered platelets was measured on a Chrono-log whole blood aggregometer, and data were recorded as percent transmittance (% aggregation).
Clot formation and vessel occlusion are altered in vivo
Both intravital microscopy following injury with ferric chloride and tail bleeding assays were performed to evaluate thrombosis in vivo in LRP8-deficient mice. Carotid occlusion times in response to ferric chloride injury, as measured by intravital microscopy, were significantly different between the LRP8+/- and LRP8+/+ mice (Figure 3A). The median times to occlusion were 9.5 minutes, 29.0 minutes, and 16.0 minutes in LRP8+/+, LRP8+/-, and LRP8-/- mice, respectively. Thus, the occlusion times for the hemizygous LRP8+/- and the LRP8-/- mice were approximately 200% and 68% greater, respectively, than those for the wild-type mice. A non-parametric ANOVA comparing occlusion times between genotypes yielded an overall p-value of 0.044, and a Dunn's post-test demonstrated a significant difference between LRP8+/+ and LRP8+/- mice with a p-value <0.05. Representative still images of early clot formation for each genotype are shown (Figure 3B). Qualitatively, the LRP8+/- mice formed smaller thrombi throughout the time course than either LRP8+/+ or LRP8-/- mice. Compared to the LRP8+/+ mice, the LRP8-/- mice had smaller thrombi at 2 minutes post injury, but typically the thrombus size was only slightly reduced at 4 minutes and later time points.
Figure 3. Thrombus formation assessed by intravital microscopy is altered in vivo.
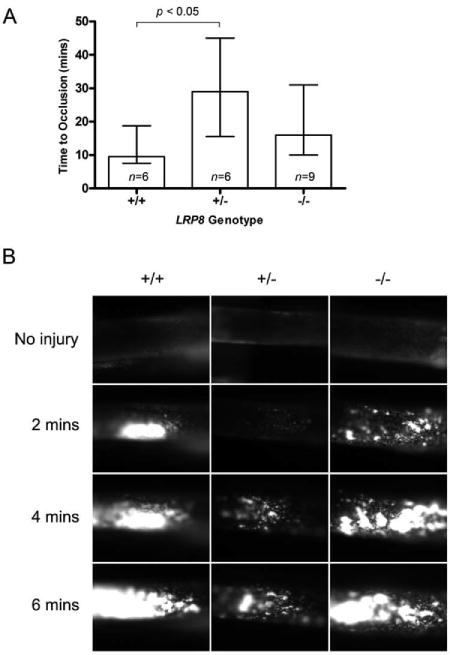
Platelets were labeled with Rhodamine 6G, and thrombosis was initiated with 12.5% FeCl3 applied to the carotid artery. Time to cessation of flow is plotted as median ± Q1-Q3 (A). Representative images of thrombus formation are shown (B).
Despite the differences observed for the occlusion time, the median bleeding times were 80, 103 and 101 seconds for the LRP8+/+ (n=29), LRP8+/- (n=53) and LRP8-/- (n=34) mice, respectively, and these differences were not statistically significant by a non-parametric ANOVA (p=0.35, Figure 4).
Figure 4. Effects of genotype on in vivo tail bleeding assays.
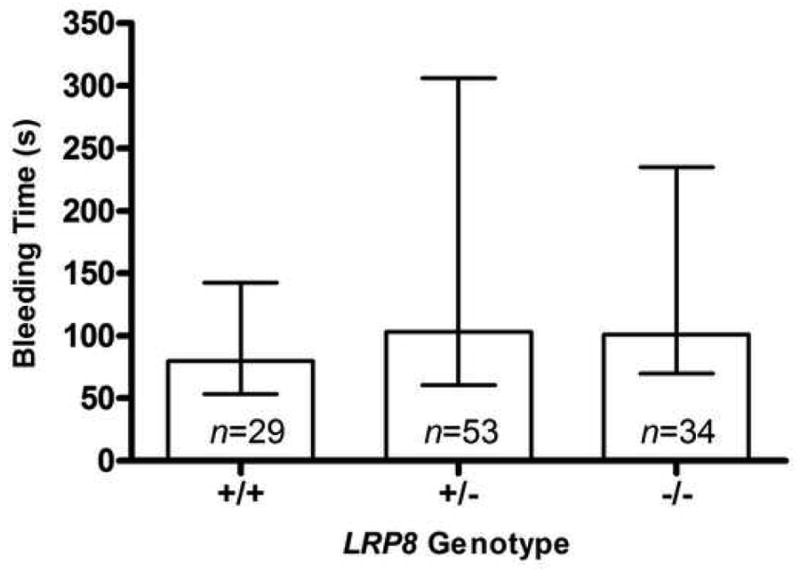
The bleeding time was defined as time to cessation of initial bleeding. Plots show median ± Q1-Q3.
apoE3 inhibits platelet activation through LRP8
In order to confirm that apoE signals through LRP8 to inhibit platelet activation, we used flow cytometry to measure activated αIIbβ3 integrin in response to thrombin activation, following pretreatment with lipidated human apoE3. We observed a dose-dependent reduction of activation when gel-filtered platelets were pre-treated with apoE3/DMPC complexes. LRP8-/- mice demonstrated 50% less inhibition than the wild-type controls in the presence of the highest dose of apoE3/DMPC (Figure 5A). A two-way ANOVA demonstrated interaction between apoE3/DMPC dose and genotype at a significance level of p=0.0118. Bonferroni post-tests confirmed a difference between LRP8+/+ and LRP8-/- platelets at p<0.01 for doses of apoE3 ranging from 10-20 μg/mL and at p<0.001 for 30 μg/mL of apoE3. Inhibition of LRP8+/- platelet activation by apoE3/DMPC was comparable to that of LRP8+/+ platelets and significantly different from LRP8-/- platelets (p <0.05 for doses of apoE3 between 10-20 μg/mL and p <0.001 for 30 μg/mL of apoE3). DMPC liposomes in the absence of apoE only mildly inhibited activation of LRP8+/+ platelets (Figure 5A).
Figure 5. LRP8-deficiency attenuates apoE-mediated inhibition of activation of platelets.
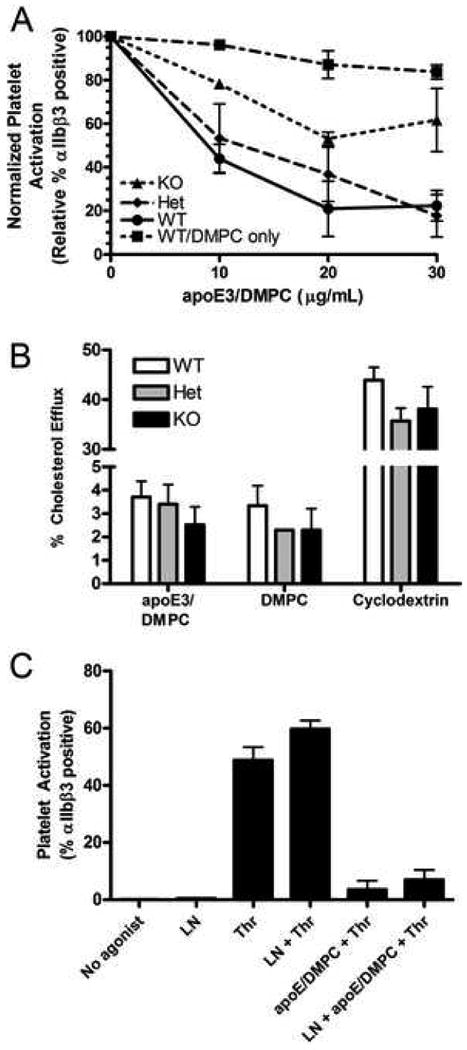
Gel filtered platelets were pre-incubated with lipidated apoE3 or equivalent DMPC before activation with thrombin and flow cytometric analysis (n=3, A). Genotype had no effect on cholesterol efflux from platelets, and efflux to lipid complexes with and without apoE3 was not significantly different (n=2, B). ApoE-mediated inhibition of platelet activation is not NO-dependent (n=4, C). LN, L-NAME.
To ensure that the inhibitory effect of apoE was not due extraction of cholesterol from the platelet membrane, we measured cholesterol efflux in response to apoE3/DMPC complexes, DMPC liposomes, and methyl-β-cyclodextrin (Figure 5B). Cholesterol efflux to apoE3/DMPC complexes was approximately 3% over background levels, and was similar to efflux to DMPC liposomes. Thus, the degree of cholesterol efflux did not correlate with the inhibition of platelet activation. There was no LRP8 genotype effect on cholesterol efflux to any of these acceptors (Figure 5B).
Nitric oxide does not mediate effect of apoE3
The nitric oxide inhibitor L-NAME was used to determine if NO played an important role in apoE3 mediated inhibition of murine platelet activation by thrombin (Figure 5C). The addition of L-NAME did not significantly affect thrombin mediated activation of LRP8+/+ platelets. Furthermore, L-NAME did not reverse the inhibition of platelet activation caused by lipidated apoE3.
Discussion
Our group has previously associated polymorphisms in the LRP8 gene with increased risk for MI [2, 3]. Since LRP8 has been identified on platelets [5], we sought to determine if it has a modulatory role in platelet activation and thrombus formation by using LRP8-deficient mice, for which we could find no prior reports regarding platelet activation or thrombosis.
Although LRP8 has not been extensively studied in human platelets, it has been proposed that both apoE and LDL are ligands for platelet LRP8 [5, 10, 14]. Data suggest that apoE attenuates the response of platelets to agonists [5, 14], while LDL activates p38 mitogen-activated protein kinase (MAPK) phosphorylation in platelets [10], both presumably through LRP8. In our activation studies, platelet preparations were washed, filtered and diluted in order to separate them from any lipoproteins present in the plasma; although we observed no effect of human LDL on platelet activation as measured by flow cytometry (data not shown). In the absence of lipoproteins, we observed a reduction in the rate and extent of activation for both LRP8+/- and LRP8-/- platelets in response to thrombin and ADP compared to wild-type platelets (Figures 1-2 and Table 1). One potential explanation for this finding is that platelets secrete, or expose on their surfaces, a ligand for LRP8, resulting in autocrine regulation that augments platelet activation. Of the many adhesion proteins, mitogens and protease inhibitors released from the α-granules [17], several are known to interact with other members of the LRP family [18-21], but further investigation would be required to determine if any of these molecules are ligands for LRP8. Alternatively, reduced presence or absence of LRP8 on the platelet surface may alter platelet activation in a ligand independent fashion. Attempts at quantifying levels of LRP8 on platelet membranes (and in brain extracts as a positive control) were unsuccessful, as several commercially available antibodies were unable to detect LRP8 by Western blot or flow cytometry.
Examination of thrombosis in vivo in LRP8-deficient mice confirmed the importance of LRP8 in modulating thrombus formation. This data clearly demonstrates deviations from the wild-type thrombosis phenotype in LRP8+/- mice (Figure 3A) and shows qualitative differences early in thrombosis in both LRP8+/- and LRP8-/- mice (Figure 3B), affirming the functional importance of the observed in vitro reduction in platelet activation.
The inhibition of activation of gel-filtered platelets that we observed in response to apoE (Figure 5A) was comparable to the effect previously observed with human platelets [14]. We used apoE phospholipid complexes, which mimic the form of apoE secreted by macrophages [22], as lipidation of apoE has previously been demonstrated to be required for binding to LRP8 [5, 14, 16]. As expected, this effect was significantly attenuated in LRP8-/- platelets, confirming that apoE does indeed signal through LRP8 to inhibit platelet activation by agonists. Yet, some level of inhibition was still evident even in LRP8-/- platelets, suggesting that apoE may also signal platelets through non-LRP8 pathways. This is consistent with previous human platelet studies in which receptor-associated protein (RAP), a specific inhibitor of LRP family members, only reversed about 60% of the inhibition of activation caused by apoE [5]. The remainder of the apoE effect cannot be accounted for by cholesterol sequestration from the platelet membrane, which is known to diminish response to agonists [23-25], as the highest dose of DMPC liposomes caused only minor inhibition of platelet activation, but led to the same amount of cholesterol efflux as observed for lipidated apoE (Figure 5B).
Nevertheless, a few differences were noted between human and murine platelets with regard to apoE mediated inhibition. Riddell, et al have previously reported that thrombin mediated activation of human platelets was only inhibited using very high doses of apoE/DMPC (500 μg/mL) or incubation times reaching 30 minutes [14]. In contrast to that study, we found that physiological doses of apoE3/DMPC complexes and a relatively short pre-incubation period were sufficient to inhibit thrombin induced aggregation in murine platelets (Figure 5A). This difference could be accounted for by differences in the concentration of thrombin used or by innate differences between human and murine platelets. Thrombin activates human platelets through activation of the PAR-4 and PAR-1 receptors, with signaling through the PAR-1 receptor predominating at low doses of thrombin, but in murine platelets thrombin signals entirely through PAR-4, with PAR-3 facilitating cleavage of PAR-4 at low doses [26]. Moreover, our results suggest that NO does not play a role in mediating inhibition of platelet activation by lipidated apoE in murine platelets (Figure 5C). This differed from the data reported using human platelets, in which an L-arginine:NO signal transduction pathway for apoE signaling was found [14].
The smaller increase in occlusion time seen in the LRP8-/- mice (compared to the large increase seen in the LRP8+/- mice, Figure 3A) can potentially be explained by the effect of apoE on platelet activation. Both LRP8+/- and LRP8-/- platelets exhibit similarly reduced activation in response to agonists (Figures 1 and 2), but LRP8-/- platelets alone show evidence of an attenuated response to regulation by apoE (Figure 5A). It is only the hemizygous LRP8+/- mice that have two factors inhibiting platelet aggregation: 1) the basal reduction to agonist activation and 2) intact inhibition by apoE, leading to longer occlusion time than in either the wild-type or knockout mice. Therefore, we hypothesize that LRP8-/- mice undergo carotid artery thrombosis faster than LRP8+/- mice because platelet activation is no longer under negative regulation by apoE, thereby counterbalancing the reduced aggregation observed in response to platelet agonists. Interestingly, we observed no significant effect on bleeding time between genotypes (Figure 4), suggesting that alteration of LRP8 is able to modulate thrombosis without altering this response to wounding.
It is apparent from this work that polymorphisms in LRP8 have the potential to modify platelet function and thrombus formation, and thereby contribute to the development of early-onset MI. It is currently unknown whether several implicated polymorphisms in LRP8 are associated with altered expression levels or protein coding changes that could alter LRP8 function, but either would theoretically have the potential to contribute pathologically to an MI. Increased expression or a gain of function of this receptor could augment agonist-induced platelet activation (as we observed for LRP8+/+ vs. LRP8+/- and LRP8-/- mice). One important human variant about which slightly more is known, the R952Q variant of LRP8, has recently been demonstrated to increase p38 MAPK phosphorylation in vitro and increase platelet aggregation by 14% in patients not on anti-platelet therapy when compared to the common variant not associated with disease [3]. This has clear potential to contribute to myocardial infarction. ApoE signaling through LRP8 may be a key regulator of platelet activation, and since apoE is present in blood and secreted locally in lesions by macrophages [27], it has the potential to be an important regulator of thrombosis during a plaque rupture.
In conclusion, our use of a mouse knockout model demonstrated that LRP8 can modulate platelet activation in vitro and thrombosis in vivo. These findings lend support to the hypothesis that polymorphisms in LRP8 may play a role in susceptibility to MI, and they provide a rational mechanism for this association. At the same time, our data show that partial LRP8 deficiency is capable of altering thrombosis in the context of intravascular injury without altering bleeding time, indicating that a partial inhibitor of LRP8 could have potential clinical applications.
Acknowledgments
We thank Drs. Eugene Podrez and Edward Plow for their advice and discussion. This work was supported by grant P50-HL077107 from the National Heart Lung and Blood Institute of the National Institutes of Health. Jason Robertson was supported in his work by a medical student fellowship from the Howard Hughes Medical Institute.
Abbreviations
- LRP8
Lipoprotein receptor related protein 8
- ApoER2
Apolipoprotein E receptor 2
- CAD
coronary artery disease
- MI
myocardial infarction
- SNPs
single nucleotide polymorphisms
- LDL
low density lipoprotein
- Q1-Q3
interquartile range
- PE
phycoerythrin
- DMPC
1,2-Dimyristoyl-sn-glycero-3-phosphocholine
- L-NAME
N-nitro-L-arginine methyl ester
Footnotes
Conflict of Interest: None of the authors have any personal or financial relationships to disclose that would have biased our work.
This manuscript was presented at the April 2007 American Heart Association Atherosclerosis, Thrombosis and Vascular Biology Conference in Chicago, IL.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–6. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Rao S, Shen GQ, Li L, Moliterno DJ, Newby LK, Rogers WJ, Cannata R, Zirzow E, Elston RC, Topol EJ. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. Am J Hum Genet. 2004;74:262–71. doi: 10.1086/381560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen GQ, Li L, Girelli D, Seidelmann SB, Rao S, Fan C, Park JE, Xi Q, Li J, Hu Y, Olivieri O, Marchant K, Barnard J, Corrocher R, Elston R, Cassano J, Henderson S, Hazen SL, Plow EF, Topol EJ, Wang QK. An LRP8 variant is associated with familial and premature coronary artery disease and myocardial infarction. Am J Hum Genet. 2007;81:780–91. doi: 10.1086/521581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 5.Riddell DR, Vinogradov DV, Stannard AK, Chadwick N, Owen JS. Identification and characterization of LRP8 (apoER2) in human blood platelets. J Lipid Res. 1999;40:1925–30. [PubMed] [Google Scholar]
- 6.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 7.Riddell DR, Sun XM, Stannard AK, Soutar AK, Owen JS. Localization of apolipoprotein E receptor 2 to caveolae in the plasma membrane. J Lipid Res. 2001;42:998–1002. [PubMed] [Google Scholar]
- 8.Andersen OM, Yeung CH, Vorum H, Wellner M, Andreassen TK, Erdmann B, Mueller EC, Herz J, Otto A, Cooper TG, Willnow TE. Essential role of the apolipoprotein E receptor-2 in sperm development. J Biol Chem. 2003;278:23989–95. doi: 10.1074/jbc.M302157200. [DOI] [PubMed] [Google Scholar]
- 9.Andrade N, Komnenovic V, Blake SM, Jossin Y, Howell B, Goffinet A, Schneider WJ, Nimpf J. ApoER2/VLDL receptor and Dab1 in the rostral migratory stream function in postnatal neuronal migration independently of Reelin. Proc Natl Acad Sci U S A. 2007;104:8508–13. doi: 10.1073/pnas.0611391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korporaal SJ, Relou IA, van Eck M, Strasser V, Bezemer M, Gorter G, van Berkel TJ, Nimpf J, Akkerman JW, Lenting PJ. Binding of low density lipoprotein to platelet apolipoprotein E receptor 2′ results in phosphorylation of p38MAPK. J Biol Chem. 2004;279:52526–34. doi: 10.1074/jbc.M407407200. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Lu W, Marzolo MP, Bu G. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J Biol Chem. 2001;276:18000–6. doi: 10.1074/jbc.M101589200. [DOI] [PubMed] [Google Scholar]
- 12.Sun XM, Soutar AK. Expression in vitro of alternatively spliced variants of the messenger RNA for human apolipoprotein E receptor-2 identified in human tissues by ribonuclease protection assays. Eur J Biochem. 1999;262:230–9. doi: 10.1046/j.1432-1327.1999.00394.x. [DOI] [PubMed] [Google Scholar]
- 13.Pennings MT, Derksen RH, Urbanus RT, Tekelenburg WL, Hemrika W, de Groot PG. Platelets express three different splice variants of ApoER2 that are all involved in signaling. J Thromb Haemost. 2007 doi: 10.1111/j.1538-7836.2007.02605.x. [DOI] [PubMed] [Google Scholar]
- 14.Riddell DR, Graham A, Owen JS. Apolipoprotein E inhibits platelet aggregation through the L-arginine:nitric oxide pathway. Implications for vascular disease. J Biol Chem. 1997;272:89–95. doi: 10.1074/jbc.272.1.89. [DOI] [PubMed] [Google Scholar]
- 15.Zhu K, Brubaker G, Smith JD. Large disk intermediate precedes formation of apolipoprotein A-I-dimyristoylphosphatidylcholine small disks. Biochemistry. 2007;46:6299–307. doi: 10.1021/bi700079w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Innerarity TL, Pitas RE, Mahley RW. Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. J Biol Chem. 1979;254:4186–90. [PubMed] [Google Scholar]
- 17.Jurk K, Kehrel BE. Platelets: physiology and biochemistry. Semin Thromb Hemost. 2005;31:381–92. doi: 10.1055/s-2005-916671. [DOI] [PubMed] [Google Scholar]
- 18.Bu G, Williams S, Strickland DK, Schwartz AL. Low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor is an hepatic receptor for tissue-type plasminogen activator. Proc Natl Acad Sci U S A. 1992;89:7427–31. doi: 10.1073/pnas.89.16.7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabello-Verrugio C, Brandan E. A Novel Modulatory Mechanism of Transforming Growth Factor-beta Signaling through Decorin and LRP-1. J Biol Chem. 2007;282:18842–50. doi: 10.1074/jbc.M700243200. [DOI] [PubMed] [Google Scholar]
- 20.Loukinova E, Ranganathan S, Kuznetsov S, Gorlatova N, Migliorini MM, Loukinov D, Ulery PG, Mikhailenko I, Lawrence DA, Strickland DK. Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP). Evidence for integrated co-receptor function betwenn LRP and the PDGF. J Biol Chem. 2002;277:15499–506. doi: 10.1074/jbc.M200427200. [DOI] [PubMed] [Google Scholar]
- 21.Mikhailenko I, Kounnas MZ, Strickland DK. Low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor mediates the cellular internalization and degradation of thrombospondin. A process facilitated by cell-surface proteoglycans. J Biol Chem. 1995;270:9543–9. doi: 10.1074/jbc.270.16.9543. [DOI] [PubMed] [Google Scholar]
- 22.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 23.Higashihara M, Kinoshita M, Teramoto T, Kume S, Kurokawa K. The role of apoE in inhibitory effects of apoE-rich HDL on platelet function. FEBS Lett. 1991;282:82–6. doi: 10.1016/0014-5793(91)80449-d. [DOI] [PubMed] [Google Scholar]
- 24.Stuart MJ, Gerrard JM, White JG. Effect of cholesterol on production of thromboxane b2 by platelets in vitro. N Engl J Med. 1980;302:6–10. doi: 10.1056/NEJM198001033020102. [DOI] [PubMed] [Google Scholar]
- 25.Tandon N, Harmon JT, Rodbard D, Jamieson GA. Thrombin receptors define responsiveness of cholesterol-modified platelets. J Biol Chem. 1983;258:11840–5. [PubMed] [Google Scholar]
- 26.Brass LF. Thrombin and platelet activation. Chest. 2003;124:18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- 27.Rosenfeld ME, Butler S, Ord VA, Lipton BA, Dyer CA, Curtiss LK, Palinski W, Witztum JL. Abundant expression of apoprotein E by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb. 1993;13:1382–9. doi: 10.1161/01.atv.13.9.1382. [DOI] [PubMed] [Google Scholar]