

Abstract
Despite considerable excitement over the potential functional significance of copy-number variants (CNVs), we still lack knowledge of the fine-scale architecture of the large majority of CNV regions in the human genome. In this study, we used a high-resolution array-based comparative genomic hybridization (aCGH) platform that targeted known CNV regions of the human genome at approximately 1 kb resolution to interrogate the genomic DNAs of 30 individuals from four HapMap populations. Our results revealed that 1020 of 1153 CNV loci (88%) were actually smaller in size than what is recorded in the Database of Genomic Variants based on previously published studies. A reduction in size of more than 50% was observed for 876 CNV regions (76%). We conclude that the total genomic content of currently known common human CNVs is likely smaller than previously thought. In addition, approximately 8% of the CNV regions observed in multiple individuals exhibited genomic architectural complexity in the form of smaller CNVs within larger ones and CNVs with interindividual variation in breakpoints. Future association studies that aim to capture the potential influences of CNVs on disease phenotypes will need to consider how to best ascertain this previously uncharacterized complexity.
Introduction
Genomic DNA copy-number gains and losses have been studied for more than 30 years (e.g., at the α- and β-globin [MIM 141800 and 149100],1–3 opsin [MIM 303800],4 and a handful of other gene loci5–9). However, it was generally assumed that such genomic imbalances were few in number and had relatively limited impact on the total content of human genetic variation. Now, recent developments and applications of genome-wide structural-variation technologies have led to the identification of thousands of heritable copy-number variants (CNVs) and sparked considerable interest.10–19 In part, this interest has been motivated by observations that CNVs can influence transcriptional or translational levels of overlapping or nearby genes15,20–25 and by initial reports that certain CNVs are associated with differential susceptibility to complex diseases.22,26–31 However, our ability to expand on these observations and understand better the functional significance of human CNVs is hindered considerably by our limited knowledge of their fine-scale architecture. To simultaneously characterize the fine-scale architecture of thousands of CNV regions across multiple individuals, we have constructed a high-density comparative genomic hybridization microarray with 470,163 oligonucleotide probes covering 2191 putative CNV regions with approximately 1 kb spacing and used this array to interrogate the genomic DNAs of 30 HapMap individuals.32
Material and Methods
Microarray Design
We designed a two-chip array-based comparative genomic hybridization (aCGH) set containing 470,163 60-mer oligonucleotide probes (Agilent Technologies, Santa Clara, CA),33 including 444,891 probes with approximately 1 kb spacing through 2191 putative CNV regions that were annotated in the Database of Genomic Variants as of 30 November 2006, and their flanking regions (approximately 1 kb spacing for 5 kb upstream and downstream, with progressively reduced probe density for an additional 15 kb). Probe sequences were based on the human genome reference sequence (hg17). In order to sufficiently cover segmental duplications (SDs),34 which are commonly associated with CNVs (e.g.,35), we allowed probes to have multiple perfect matches within the human genome reference assembly (hg17) when unique probes were not available at the desired density. The probes for chromosomes 1, 4, 5, 7, 11, 13, 15, 16, 17, 18, 19, and 21 were assigned to array A, and probes for the remaining chromosomes were assigned to array B. We also selected 23,804 autosomal and 1198 X chromosome probes from non-CNV regions throughout the genome from Agilent's High-Definition database of 8.4 million aCGH probes that cover exonic, intronic, and intergenic regions and have unique representation in the human genome reference sequence (hg17). Of these autosomal probes, 19,008 were distributed to arrays A and B according to chromosome (as described above). A subset of the non-CNV probes (4796 autosomal probes and the 1198 X chromosome probes) was included on both arrays.
DNA-Sample Labeling and Hybridization
Human DNA samples were selected from the four populations of the International HapMap project.32 Our sample consisted of ten unrelated Yoruba individuals from Ibadan, Nigeria (YRI), ten unrelated European-American individuals from Utah (CEPH), five unrelated Japanese individuals from Tokyo, and five unrelated Chinese individuals from Beijing. For analyses, we considered the Japanese and Chinese samples as one Asian population (ASN). Samples were selected from those thought to be absent of detectable cell-line artifacts, on the basis of karyotype and computational analyses.16 A single reference sample (NA10851, a CEPH male) was used for all aCGH experiments. This individual was also used as the common reference sample in a previous genome-wide study of copy number variation in the HapMap population samples.16 This facilitated direct comparisons between the two datasets. Genomic DNAs were isolated from B lymphoblastoid cell lines obtained from the Coriell Institute for Medical Research (Camden, NJ) with the Puregene DNA Purification Kit (Gentra Systems, Minneapolis, MN).
aCGH experiments were performed according to the manufacturer's instructions. In brief, test and reference genomic DNAs (500 ng) were digested with restriction enzymes AluI and RsaI and fluorescently labeled with Cy5 (test) and Cy3 (reference) with the Agilent DNA Labeling Kit. For each sample, duplicate labeling reactions were mixed and then separated prior to hybridizing to each of the two arrays. Labeled test and reference DNAs were combined, denatured, pre-annealed with Cot-1 DNA (Invitrogen, Carlsbad, CA) and blocking reagent (Agilent), and then hybridized to the arrays for 40 hr in a rotating oven (Agilent Technologies) at 65°C and 20 rpm. Dye-swap experiments (test in Cy3 and reference in Cy5) were performed for each sample. After hybridization and recommended washes, the arrays were scanned at 5 μm resolution with an Agilent G2505A scanner. Images were analyzed with Feature Extraction Software 9.1.1.1 (Agilent Technologies), with the CGH-v4_91 protocol for background subtraction and normalization. All array data passed Agilent recommended quality metrics. The array data have been submitted to the Gene Expression Omnibus under accession number GSE9831.
Algorithm for Calling CNVs
We performed a BLAST analysis36 of all probe sequences against the human genome reference sequence (hg17) to identify all genomic locations with perfect (identical 60 bp) and imperfect (20–59 bp) matches. A total of 512,945 perfect genomic hits were identified. For CNV calling, all perfect genomic matches were included for each probe in the analyses, with the following exceptions. First, to avoid potential sex-linked artifacts, we ignored 5121 probes with a perfect match to an autosome and a perfect or imperfect match to either the X or Y chromosome, a perfect match to either the X or Y chromosome and an imperfect match to an autosome, or matches to both the X and Y chromosomes. Second, we ignored probes mapped to the immunoglobulin loci that might undergo somatic deletion in B lymphoblast cells (hg17: chr2:88,960,288–89,990,012, chr14:105,030,829–106,300,130, and chr22:20,778,738–21,600,000). Finally, for some analyses, we have further restricted the set of probes to those with perfect hits that are either unique to one location or occur only within 2 Mb of each other (the “proximal probe set”).
Log2 intensity ratio measurements for array A and array B were merged and analyzed as a single dataset for each experiment. Features corresponding to the same probe sequences were averaged with the weighted averaging method used in CGH Analytics (Agilent Technologies, Santa Clara, CA). For each probe, a single combined log2 ratio was computed as the mean of the values from the original array and its dye swap. We estimated the sample-specific dye bias for each probe as half of the difference between the two log2 ratios (both computed as test: reference). We also calculated for each probe the median dye bias across all 30 HapMap experiments and the corresponding interquartile range (IQR). For each experiment, we flagged and removed any probe with sample-specific dye bias that (1) was greater than the absolute value of its combined log2 ratio and (2) was greater than 2.5 IQR from the median dye bias. On average, 864 probes were removed per experiment.
We used the ADM2 statistical algorithm37,38 to identify CNVs on the basis of the combined log2 ratios. In brief, ADM2 uses an iterative procedure to identify all genomic regions for which the weighted average of the measured probe signals is different from the expected value of 0 by more than a given threshold. This deviation is measured by a statistical score. Loci with nearby gain or loss intervals and an intervening region of more than 4 probes with log2 ratios not different than 0 were considered two separate CNVs. To select parameters for calling CNVs (i.e., the statistical threshold of the ADM2 algorithm, the minimum ± log2 ratio, and the minimum number of probes in a CNV interval), we iteratively called CNVs across all 30 HapMap samples and in three self-self experiments (NA10851 versus NA10851) for different combinations of these parameters. We estimated the false-positive error rate for each combination based on the average number of CNV calls in the self-self experiments divided by the average number of CNV calls in the HapMap sample experiments. We targeted a false-positive rate of less than 5%, but without dramatic reductions in the number of calls in the HapMap sample experiments (i.e., reducing the false-positive rate to 0 might result in an unacceptably high false-negative rate). By using this approach, we selected the following parameters: statistical threshold = 5.0, minimum ± log2 ratio = 0.25 (theoretically sufficient to distinguish six copies versus five copies; i.e., log2 (6/5) > 0.25), and minimum number of probes = 2, resulting in averages of 34.3 calls for self-self experiments and 710 calls for the HapMap sample experiments (estimated false-positive rate = 4.8%). We do note, however, that this comparison might underestimate the true false-positive rate in our test experiments because the self-self experiments were performed with genomic DNA from a single extraction and thus cannot account for minor differences in DNA quality among our samples. The identified CNV intervals are reported in Table S1 (using genome-wide perfect match probes) and Table S2 (using the proximal probe set) available online. CNVs on the X and Y chromosomes are reported for males only. CNV regions were defined on the basis of the union of all overlapping CNVs across all 30 HapMap individuals (Table S3).
Results
Evaluation of Concordance of Sample-Specific CNV Calls with a Previous Study
We used a high-resolution aCGH platform to compare the genomic DNAs of 30 HapMap individuals to the genomic DNA of a single reference individual, a European-American male (NA10851) also from the HapMap study. Approximately 470,000 oligonucleotide probes were chosen from 2191 previously reported CNV regions throughout the human genome, for in-depth interrogation of these CNVs. Among the 30 HapMap individuals, we identified CNVs in 1153 (53%) of the 2191 regions (Table S4). The remaining CNV regions might contain relatively low-frequency CNVs not present in the 30 individuals sampled in this study. Alternatively, these could be false positives in the previous studies or false negatives in our study.
To explore these possibilities, we compared our CNV calls to those from the Redon et al.16 study that used two genome-wide platforms (a whole-genome tiling-path aCGH platform with approximately 27,000 large-insert clones [WGTP] and an Affymetrix GeneChip array with approximately 500,000 single-nucleotide polymorphism probes [500K EA]) to identify CNVs in the same individuals that we sampled (and for the WGTP platform, using the same reference individual as in our study). We defined “high-confidence” CNVs from the Redon et al.16 study as CNV calls made by both the WGTP and 500K EA platforms in the same direction (i.e., gain or loss) for the same individual. There were 269 such high-confidence CNV calls recorded among the 30 HapMap individuals. In the present study, we identified gains or losses (in the same direction and individual) for 260 of the 269 high-confidence CNV calls (97%; based on WGTP breakpoints; Tables S5 and S6), demonstrating that our measurements have a low false-negative rate for CNVs that were consistently identified across multiple platforms. Next, we examined the CNV calls from Redon et al.16 for the 30 HapMap individuals that were made by only one of the two platforms (i.e., excluding high-confidence CNV calls). As expected, we observed a reduced level of concordance: 1564 of 2237 CNV calls made with the WGTP platform (70%) and 258 of 480 CNV calls made with the 500K EA platform (54%; Tables S5 and S6) were also considered CNVs in our study in the same individual and direction. We note that although the WGTP experiments in the Redon et al.16 study used the same reference individual as our study to make relative gain or loss CNV calls, the calls based on the 500K EA platform were based on average population intensities, which might in part account for the relatively lower level of observed concordance with our calls. Finally, on the basis of CNV call concordance, we were able to identify, with high accuracy, the samples in our study from all 270 HapMap individuals studied by Redon et al.16 (Figure S1 and Table S7).
The Total Genomic Content of Common Human CNVs Might Be Smaller than Previously Thought
We compared the estimated sizes of CNV regions in our dataset to estimates from previous studies for the corresponding regions, on the basis of information in the Database of Genomic Variants (DGV). We found that our estimate of the total amount of copy-number-variable sequence was smaller than the corresponding DGV region for 1020 of the 1153 loci (88%) in which we called CNVs. Strikingly, the total amount of copy-number-variable sequence was reduced by more than 50% for 876 regions (76%; of 1153; Figure 1; Tables S3 and S4).
Figure 1.
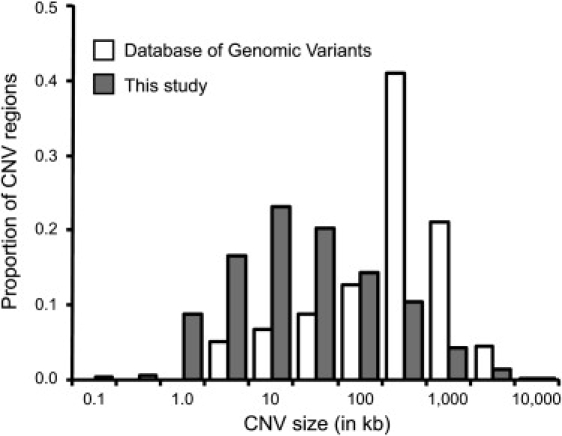
Size Distribution of CNVs from the Database of Genomic Variants, with Corresponding CNVs from This Study
We identified CNVs in at least one individual for 1153 of 2191 putative CNV regions annotated in the Database of Genomic Variants (DGV) as of 30 November 2006. Size distributions for these regions are shown in log scale, with 10-fold multiples of 1 and √10, based on the size of each region from DGV and the estimates from our study of the total amount of copy-number-variable sequence within and overlapping the DGV-defined region. Our estimates were smaller than the corresponding DGV region for 1020 of the 1153 loci (88%) and smaller by more than 50% for 876 regions (76%).
Because the sizes of CNV regions in the DGV represent the combination of calls from previous studies, we repeated the analysis with CNV size estimates from the data of individual studies (Table 1). Although we obtained similar results for studies employing BAC-based aCGH and lower-resolution platforms, better size concordance was observed for studies with potentially increased resolution (such as Conrad et al.14 and McCarroll et al.15, which were based on analyses of HapMap SNP genotypes; see Table 1 for a summary of all comparisons).
Table 1.
Summary of CNV-Size-Estimate Comparisons with Previous Studies
| Previous CNV Study | Platform or Method | Number of Reported CNVsa | Number of CNVs Overlapping with CNVs from This Study (Proportion)b | Number of CNVs Observed in Both Studies with Smaller Estimated Size in This Study (Proportion) | Number of CNVs Observed in Both Studies with Estimated Size in This Study Less Than 50% of the Estimated Size in Previous Study (Proportion) |
|---|---|---|---|---|---|
| Conrad et al.14 | HapMap SNP patterns | 544 | 199 (0.37) | 29 (0.15) | 13 (0.07) |
| de Smith et al.17 | Agilent oligonucleotide arrays | 572 | 322 (0.56) | 158 (0.49) | 75 (0.23) |
| Iafrate et al.10 | BAC-based aCGH | 190 | 99 (0.52) | 76 (0.77) | 70 (0.71) |
| Locke et al.54 | BAC-based aCGH | 253 | 161 (0.64) | 99 (0.61) | 81 (0.50) |
| McCarroll et al.15 | HapMap SNP patterns | 495 | 211 (0.43) | 43 (0.20) | 24 (0.11) |
| Pinto et al.39 | Affymetrix SNP arrays | 774 | 392 (0.51) | 335 (0.85) | 269 (0.69) |
| Redon et al.16—all HapMapc | Affymetrix SNP arrays | 980 | 530 (0.54) | 484 (0.91) | 342 (0.65) |
| Redon et al.16—30 individualsd | Affymetrix SNP arrays | 286 | 259 (0.91) | 194 (0.75) | 77 (0.30) |
| Redon et al.16—all HapMapc | BAC-based aCGH | 913 | 654 (0.72) | 636 (0.97) | 568 (0.87) |
| Redon et al.16—30 individualsd | BAC-based aCGH | 479 | 375 (0.78) | 320 (0.85) | 242 (0.65) |
| Sebat et al.11 | ROMA aCGH | 80 | 58 (0.73) | 28 (0.48) | 24 (0.41) |
| Sharp et al.12 | BAC-based aCGH | 159 | 101 (0.64) | 51 (0.50) | 39 (0.39) |
| Simon-Sanchez et al.61 | Illumina BeadChips | 154 | 70 (0.45) | 58 (0.83) | 49 (0.70) |
| Tuzun et al.13 | Fosmid end mapping | 253 | 124 (0.49) | 73 (0.59) | 48 (0.39) |
| Wang et al.62 | Illumina BeadChips | 749 | 325 (0.43) | 137 (0.42) | 94 (0.29) |
| Wong et al.19 | BAC-based aCGH | 465 | 247 (0.53) | 177 (0.72) | 141 (0.57) |
| Zogopoulos et al.63 | Affymetrix SNP arrays | 273 | 182 (0.67) | 139 (0.76) | 114 (0.63) |
From CNV data in the Database of Genomic Variants as of September 2007. For studies published after our selection of CNV regions for high-density probe coverage in our array design (based on the Database of Genomic Variants, November 2006), we have only included and analyzed their reported CNV regions that are within the originally selected regions.
It is important to note that different samples were used for many of these studies. Therefore, the concordance rate is expected to depend not only on the properties and performance of these platforms and of our CNV-enriched array, but also on the sample composition and the number of samples studied. For example, we have analyzed the data from Redon et al.16 considering (1) all 270 HapMap individuals study and (2) only the 30 individuals that were also included in our study, and observed notably higher concordance in the latter analysis.
Based on data from all 270 HapMap individuals.
Based on data from only the 30 HapMap individuals included in this study. For regions in which CNVs were called in one or more of the 30 HapMap individuals by both the Redon et al.16 500K EA platform (Affymetrix SNP arrays) and in this study, the total estimated size of all CNV regions was 50.3 Mb, based on the 500K EA results and 43.0 Mb in this study, representing a 15% reduction in size. With the same criteria, the total estimated size of all CNV regions was 102.3 Mb, based on the Redon et al.16 WGTP platform and 68.9 Mb in this study (33% reduction).
We also considered the possibility that in some regions, we might have actually identified different and smaller CNVs than those that were detected by previous lower-resolution studies. However, even when we excluded all regions with less than 20 kb of copy-number-variable sequence from our dataset and repeated our comparison with CNVs called by the Redon et al.16 WGTP platform in the same samples, 213 of 264 overlapping CNVs (80%) were smaller in our dataset, with 154 of the 264 CNVs (58%) smaller by more than 50% (Figure S2). Therefore, we conclude that the total genomic content of currently identified common human CNVs is likely lower than previous estimates that were obtained with lower-resolution platforms (e.g., 12% of the genome16) or based on all DGV regions (currently, 18.8% of the genome39).
Refining the Breakpoints of Human CNVs and Mechanisms of CNV Formation
Delineation of CNV breakpoints provides precise identification of the copy-number-variable functional elements in the human genome, which will be important for the generation and testing of hypotheses concerning the roles of CNVs in complex diseases, as well as for global analyses of the properties of human CNVs (e.g., Gene Ontology analyses). Moreover, precise definition of CNV breakpoints will lead to a better understanding of the mechanisms of CNV formation. For example, previous studies have observed that segmental duplications (SDs; low-copy repeats at least 1 kb in size with at least 90% homology34) are enriched within and near CNVs, suggesting nonallelic homologous recombination (NAHR) as a likely mechanism for the genesis of these CNVs (for review, see35). However, only a minority of CNVs overlap SDs—for example, just 25% of the CNVs from the Redon et al. study16 are associated with SDs—and this proportion is likely to decrease as smaller CNVs are identified by platforms with improved resolution.40 In addition, precise breakpoint data are currently available for only a fraction of the known non-SD associated CNVs (e.g.,18,41–44). Therefore, the mechanisms underlying the formation of the majority of human CNVs remain unknown.
With our CNV-enriched array, we were able to estimate breakpoints to approximately 1 kb resolution (Table S1). To evaluate the accuracy of these predictions and advance our understanding of the mechanisms of CNV formation, we developed a strategy for polymerase chain reaction (PCR) amplification and sequencing over the breakpoints of CNVs identified in our study (excluding complex CNVs with interindividual variation in estimated breakpoints and CNVs that are associated with SDs). This strategy was designed to amplify over the breakpoint regardless of whether the CNV was actually a deletion or a tandem duplication (because we had little a priori knowledge of the absolute-copy-number state for each of the CNVs in our reference individual; Figure S3). By using this approach, we successfully sequenced over the breakpoints of 23 of 51 attempted CNVs (Figure 2; Table S8). Twenty of 23 CNVs were sequenced in multiple individuals, with identical breakpoints observed across all samples. Interestingly, all 23 of the successfully sequenced CNVs were deletions rather than duplications (i.e., unique DNA segments from the human genome reference sequence were missing from our sequenced fragments). It is not immediately clear what accounts for this bias. Possible explanations include one or more of the following: (1) that deletions might be more common than duplications in the human genome, at least for non-SD-associated CNVs, (2) that our breakpoint predictions might in general have been more accurate for deletion than duplication CNVs, and (3) that many non-SD-associated duplication CNVs in the human genome might be non-tandemly arranged (and thus not detectable by our strategy).
Figure 2.
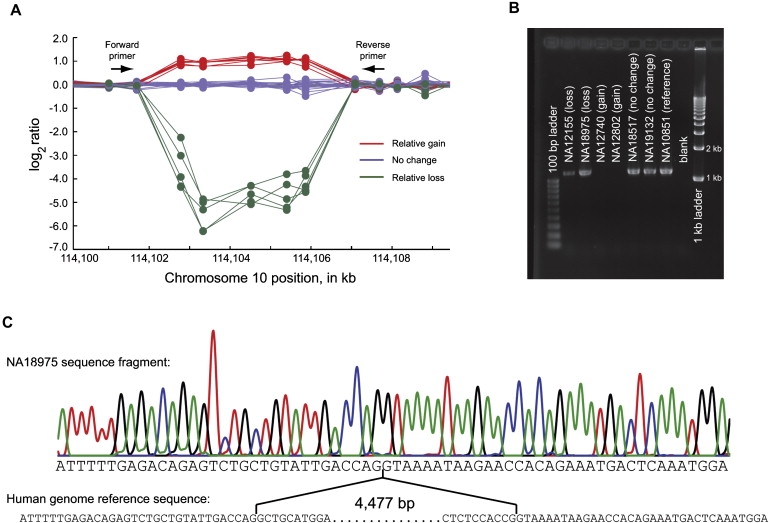
CNV Breakpoint Sequencing
We developed a PCR amplification and sequencing strategy (see Figure S3) for nucleotide-level resolution of CNV breakpoints.
(A) Log2 ratios for 30 HapMap samples for a CNV region on human chromosome 10 (hg17). Probes are depicted as solid circles. The log2 ratios form three distinct clusters (gain, no change, and loss relative to the reference individual NA10851). PCR primer locations are depicted as arrows.
(B) Results of PCR amplification, with a 1.2% agarose gel with ethidium bromide staining. Amplification was successful for individuals with no change and losses relative to the reference individual, as well as for the reference individual. Amplification was unsuccessful for individuals with a relative gain, suggesting that the reference individual is heterozygous for a deletion in this genomic region.
(C) Chromatogram from NA18975 and comparison to the human reference genome sequence (hg17) to precisely identify the CNV breakpoint. All sequenced individuals were observed to have identical breakpoints.
Of the 23 deletions, we observed homologous nucleotide sequences across the two breakpoints of the same CNV in only two cases (9%; one each with flanking LINE and Alu/SINE elements). The lack of crossbreakpoint homology for the other 21 deletions suggests that nonhomologous end joining (NHEJ;45,46) might have been involved in the formation of a large proportion of common human CNVs, consistent with the observations made by a recent paired-end-mapping CNV study.18 For nine of the 21 CNVs (43%) without breakpoint homology, we found inserted segments of between 1 and 76 bp at the breakpoints (Table S8), which likely occurred as part of the NHEJ process.18,47 In the cases with the two largest insertions (one of 50 bp and one of 76 bp), the inserted sequences are homologous to a segment within the deletion but in inverted orientation. Another deletion was found to co-occur with a larger inversion near its 5′ breakpoint (Table S8). Interestingly, we observed that CNVs located on chromosome 2 at 130.3 Mb and chromosome 5 at 151.4 Mb in fact each consisted of two distinct deletions, separated by relatively small nondeleted segments (of 601 bp and 101 bp, respectively). It is unclear whether each of these examples reflects a single deletion event with an associated recovery of some intervening sequence or two independent, nearby deletion events. However, the latter scenario would be consistent with our general observation that many previously described CNV regions are in fact comprised of multiple, smaller CNVs. For example, within the 1153 DGV regions for which we observed at least one CNV, we recorded a total of 2664 distinct and nonoverlapping regions of copy-number variation. Certain genomic regions might be particularly prone to structural rearrangements.
To gain additional insight into the mechanisms of CNV genesis in the human genome, we next interrogated the sequence composition of all the estimated breakpoint regions of our study (approximately 1 kb of sequence for each estimated breakpoint region, between the copy-number-variable probe that defines the CNV boundary and the adjacent non-copy-number-variable probe). We compared these breakpoint-region sequences to a random set of genomic sequences and to sequences constructed from random pairs of adjacent non-CNV probes on the array (in both cases, approximating the original size distribution of the breakpoint-region sequences). We unexpectedly observed a significant enrichment for simple tandem repeats within the individual CNV breakpoint-region sequences (Figure 3). For example, 174 of our breakpoint-region sequences contain two or more perfect repeats of at least 30 bp, compared to 52 of the random genomic sequences [p < 10−16; the hypergeometric tail HGT(N,B,n,b)48 was computed for a universal set of N = 20,195 observed breakpoint-region and random sequences, for B = 10,115 observed breakpoint-region sequences, for n = 226 total sequences containing repeats of at least 30 bp, and for an intersection of b = 174 observed breakpoint-region sequences containing at least 30 bp repeats] and 77 sequences between random sets of probes on the array (p < 10−9). These sequences might lead to non-B DNA conformations,49 and possibly general genomic instability. Although other features thought to be involved in the formation of non-B DNA, such as (R)n, (Y)n, (RY)n, and inverted repeats,49,50 were not found to be significantly enriched within our breakpoint-region sequences (p > 0.05), we did identify a significant enrichment of inverted repeats between the two breakpoint-region sequences of our CNVs (Figure S4). These include many inverted Alu repeats, which are generally depleted in the human genome.51,52 This depletion possibly reflects purifying selection on inverted Alu insertions or the long-term tendency for these regions to be lost through the fixation of deletions, or both.
Figure 3.
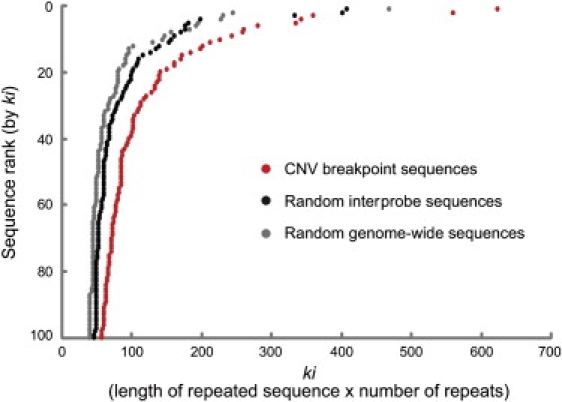
Enrichment for Tandem Repeats within Individual CNV Breakpoint-Region Sequences
This figure depicts the empirical cumulative distribution of the observed longest repeated subsequence ki (k × i), where k = the length of the repeated subsequence and i = the number of recurrences within the sequence, for the sequences between the copy-number-variable probes at CNV boundaries and the adjacent non-copy-number-variable probes estimated to harbor breakpoints in our study (CNV breakpoint sequences; approximately 1 kb each), sequences from between random pairs of adjacent non-CNV probes on the array (random interprobe sequences), and a random set of genome-wide sequences. The random sequences were selected such as to not alter the characteristics of the observed set of CNV calls, in terms of lengths and proximity of the end sequences. The graph reflects only the significant end of the distribution—the top 100 sequences as ranked by ki. A larger proportion of CNV breakpoint-region sequences contain long tandem repeats than the random sequences.
Discussion
There is currently little consensus regarding the true prevalence of CNV architectural complexity and the extent to which this should influence the design of future disease association studies. A subset of previously identified CNVs has been found to be in strong linkage disequilibrium with flanking single-nucleotide polymorphisms (SNPs),15,16,42,53,54 implying a single origin and identical breakpoints among individuals. Many of these simple CNVs could be tagged by adjacent SNPs and thereby be effectively captured by high-throughput SNP genotyping platforms.55 In contrast, CNV loci that were formed by multiple structural-rearrangement events (complex CNVs) might require more direct approaches for accurate measurement and inclusion in genome-wide disease association studies. Although certain previously identified CNVs do appear to harbor some degree of complexity—as evidenced by breakpoint variation and spatial complexity,14,16,56 susceptibility to recurrent origin,57–60 and observations of relatively low linkage disequilibrium with flanking SNPs16,54—the relative contribution of such regions to the total content of human genomic variation remains unclear.
In our dataset, there were 1326 distinct genomic regions in which CNVs were called in two or more of the 30 HapMap individuals. On the basis of our high-resolution aCGH data, 705 of these CNV regions had consistent breakpoints (to within one probe resolution) across all variant samples (Table S3); many of these CNVs are likely to be simple in nature. For these 705 loci, we developed a method for scoring the modality of CNVs that was based on a t test, to identify CNVs for which the mean log2 ratios form discrete clusters (i.e., likely reflecting distinct copy-number states; Figure 4). By using stringent thresholds, we identified 49 CNVs with two mean log2-ratio clusters, 186 CNVs with three clusters, and one CNV with four distinct clusters (Table S3; depictions of mean log2 ratios for all 236 discretely clustering CNVs are available at the Lee Lab Website). The remaining 469 CNVs were not robustly separable into distinct clusters. In future studies, modality analyses for such CNVs might benefit from larger sample sizes and the inclusion of additional probes within the CNV regions.
Figure 4.
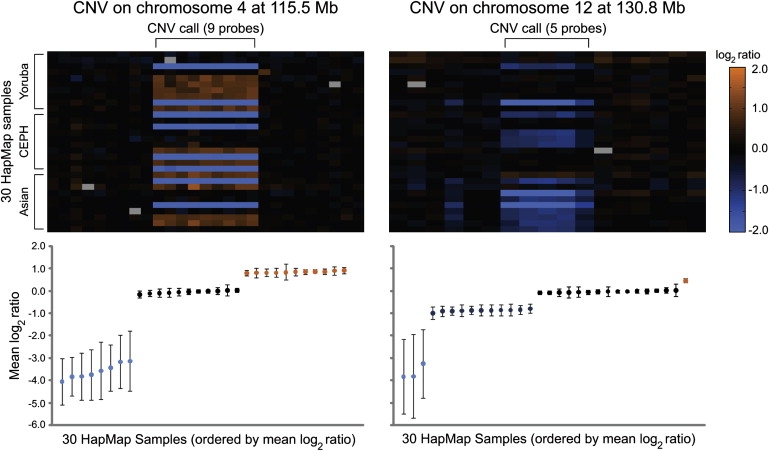
Simple CNVs and Inference of Genotypes, Based on Discrete Log2-Ratio Clustering
For two CNV-containing genomic regions that have similar estimated breakpoints across all individuals, probe-by-probe log2 ratios are depicted in heatmaps (see scale bar) in the upper panel (with rows representing individuals and columns representing probes ordered by genomic position). Mean log2 ratios of the probes within the CNV are provided in the lower panel. The mean log2 ratios form discrete clusters, letting us infer CNV genotypes. For both loci, there is one cluster with strongly negative log2 ratios, suggesting that these individuals have homozygous deletions for this DNA segment. For the CNV on chromosome 4 at 155.5 Mb (hg17), there are three mean log2-ratio clusters, likely reflecting zero, one, and two copies of this DNA segment. For the CNV on chromosome 12 at 130.8 Mb there are four mean log2-ratio clusters, likely reflecting states of zero, one, two, and three copies; therefore, this CNV would be considered to be multiallelic. Error bars represent the standard deviation (SD).
To identify and describe architecturally complex genomic regions, we searched for evidence of smaller CNVs contained within larger ones, CNVs with interindividual breakpoint variation, or CNVs with juxtaposed gains and losses within the same individual. Before conducting this analysis, we eliminated the probes that had perfect matches to multiple chromosomes or to sites more than 2 Mb away on the same chromosome. The inclusion of such probes could result in CNV shadowing effects, or artifactual calling of CNVs in a particular region due to true CNVs in homologous regions of the genome (see Table S9). These shadowing effects could lead to false appearances of complexity. By using the remaining probes (the proximal probe set; see Material and Methods) and a combination of computational filtering and manual curation, we identified 101 CNV regions with evidence for architectural complexity (Figure 5 and Figure S5; Table S10; depictions of all 101 complex CNV regions are available at the Lee Lab Website). This could be considered an underestimate of CNV complexity in the human genome, given our conservative calling approach and a sample size of 30 individuals.
Figure 5.
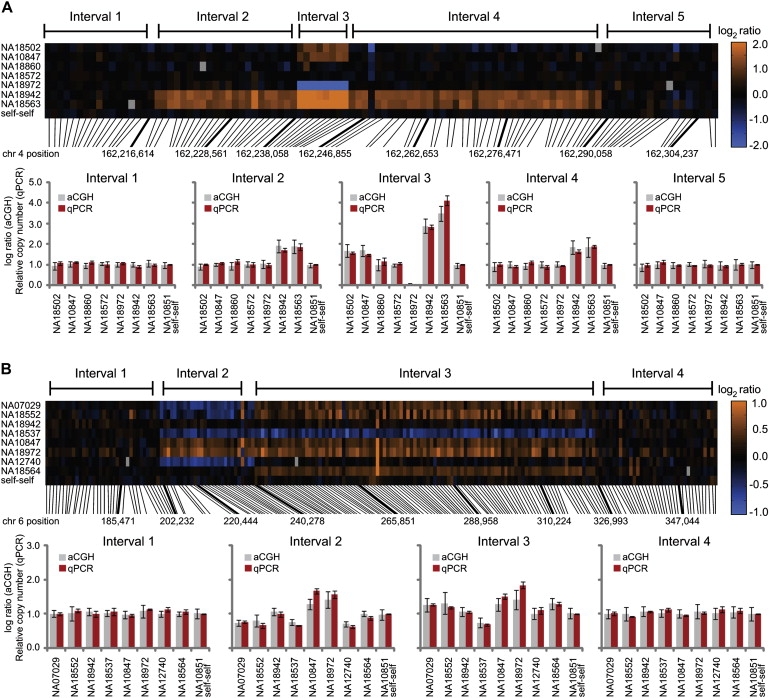
Validation of Architecturally Complex CNV Regions by qPCR
We used a series of quantitative PCR (qPCR) probes positioned across CNV regions to validate the patterns of architectural complexity observed with our CNV-enriched array. The probe-by-probe log2 ratios depicted in the heatmaps (see scale bars) illustrate examples of a smaller CNV inside a larger one on chromosome 4 at 162.2 Mb (A) and a CNV with immediately adjacent and variably present CNVs (i.e., juxtaposed gain and loss CNV calls in the same individual) on chromosome 6 at 0.2 Mb (B). The relative genomic positions of the probes are depicted with black lines, with midpoint positions (hg17) provided for selected probes (thicker lines). For each CNV, qPCR primers were designed at intervals throughout and flanking the CNV region and tested on all individuals depicted in the heatmaps. The qPCR results (i.e., relative copy number to the reference individual NA10851) are consistent with the aCGH results provided as log ratio (i.e., to be on a consistent scale with the qPCR results) for each interval. Error bars represent the SD. See Table S11 for qPCR primers and results.
It should be noted that for this analysis, we did not remove probes with imperfect sequence similarities to elsewhere in the genome, or with perfect sequence similarities that occurred on the same chromosome at distance of less than 2 Mb, because this would have limited our ability to examine tandemly arranged SDs. Therefore, shadowing effects could still explain a subset of the 101 complex regions. However, we believe that many of these regions are truly architecturally complex. For example, SDs are completely absent from 20 of these regions (including both validated regions depicted in Figure 5 and two of the three validated regions in Figure S5), and for many of the remaining regions, segmental duplications cannot fully explain the patterns of complexity. Strategies for elucidating the true underlying structure of these regions will need to be considered for future studies.
In summary, our results suggest that while the majority of human CNVs might be simple in nature, a substantial proportion of previously identified human CNV regions might in fact harbor some degree of architectural complexity. Specifically, approximately 8% of regions containing CNVs in at least 2 individuals were classified as complex on the basis of our conservative criteria. This observation further highlights the structural instability and variation of the human genome and has important implications for future human genetics studies. For example, the functional effects of architecturally complex CNVs might be intricate and unexpected. Moreover, these complex CNV regions will be difficult to incorporate into future genome-wide disease association studies without direct ascertainment and detailed characterization of their fine-scale architecture.
Supplemental Data
Five figures, simple CNVs, complex CNVs, and 11 tables are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
The Database of Genomic Variants, http://projects.tcag.ca/variation/
Lee Lab Website, http://www.chromosome.bwh.harvard.edu/data.htm (for simple and complex CNV supplements)
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Accession Numbers
The aCGH data reported in this paper have been deposited in Gene Expression Omnibus with the accession number GSE9831.
Acknowledgments
G.H.P., A.B.-D., A.T., and N.S. are co-first authors and contributed equally to this work. The authors would like to acknowledge the technical assistance of Stephanie Dallaire and Joëlle Tchinda in the early phases of this study and Arthur Lee for comments on an earlier version of the manuscript. This work was supported in part by the Department of Pathology at Brigham and Women's Hospital and a National Institutes of Health (NIH) grant to C.L. (HG004221). A.B.-D., A.T., N.S., A.S., I.S., P.T., N.A.Y., Z.Y., S.L., and L.B. are employees of Agilent Technologies.
References
- 1.Ottolenghi S., Lanyon W.G., Paul J., Williamson R., Weatherall D.J., Clegg J.B., Pritchard J., Pootrakul S., Boon W.H. The severe form of alpha thalassaemia is caused by a haemoglobin gene deletion. Nature. 1974;251:389–392. doi: 10.1038/251389a0. [DOI] [PubMed] [Google Scholar]
- 2.Taylor J.M., Dozy A., Kan Y.W., Varmus H.E., Lie-Injo L.E., Ganesan J., Todd D. Genetic lesion in homozygous alpha thalassaemia (hydrops fetalis) Nature. 1974;251:392–393. doi: 10.1038/251392a0. [DOI] [PubMed] [Google Scholar]
- 3.Ottolenghi S., Comi P., Giglioni B., Tolstoshev P., Lanyon W.G., Mitchell G.J., Williamson R., Russo G., Musumeci S., Schillro G. Delta-beta-thalassemia is due to a gene deletion. Cell. 1976;9:71–80. doi: 10.1016/0092-8674(76)90053-2. [DOI] [PubMed] [Google Scholar]
- 4.Nathans J., Thomas D., Hogness D.S. Molecular genetics of human color vision: The genes encoding blue, green, and red pigments. Science. 1986;232:193–202. doi: 10.1126/science.2937147. [DOI] [PubMed] [Google Scholar]
- 5.Awdeh Z.L., Alper C.A. Inherited structural polymorphism of the fourth component of human complement. Proc. Natl. Acad. Sci. USA. 1980;77:3576–3580. doi: 10.1073/pnas.77.6.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groot P.C., Bleeker M.J., Pronk J.C., Arwert F., Mager W.H., Planta R.J., Eriksson A.W., Frants R.R. The human alpha-amylase multigene family consists of haplotypes with variable numbers of genes. Genomics. 1989;5:29–42. doi: 10.1016/0888-7543(89)90083-9. [DOI] [PubMed] [Google Scholar]
- 7.Colin Y., Cherif-Zahar B., Le Van Kim C., Raynal V., Van Huffel V., Cartron J.P. Genetic basis of the RhD-positive and RhD-negative blood group polymorphism as determined by Southern analysis. Blood. 1991;78:2747–2752. [PubMed] [Google Scholar]
- 8.Trask B.J., Friedman C., Martin-Gallardo A., Rowen L., Akinbami C., Blankenship J., Collins C., Giorgi D., Iadonato S., Johnson F. Members of the olfactory receptor gene family are contained in large blocks of DNA duplicated polymorphically near the ends of human chromosomes. Hum. Mol. Genet. 1998;7:13–26. doi: 10.1093/hmg/7.1.13. [DOI] [PubMed] [Google Scholar]
- 9.Buckland P.R. Polymorphically duplicated genes: Their relevance to phenotypic variation in humans. Ann. Med. 2003;35:308–315. doi: 10.1080/07853890310001276. [DOI] [PubMed] [Google Scholar]
- 10.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 11.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Maner S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 12.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 14.Conrad D.F., Andrews T.D., Carter N.P., Hurles M.E., Pritchard J.K. High-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 15.McCarroll S.A., Hadnott T.N., Perry G.H., Sabeti P.C., Zody M.C., Barrett J.C., Dallaire S., Gabriel S.B., Lee C., Daly M.J. Common deletion polymorphisms in the human genome. Nat. Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 16.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Smith A.J., Tsalenko A., Sampas N., Scheffer A., Yamada N.A., Tsang P., Ben-Dor A., Yakhini Z., Ellis R.J., Bruhn L. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: Implications for association studies of complex diseases. Hum. Mol. Genet. 2007;16:2783–2794. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 18.Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong K.K., deLeeuw R.J., Dosanjh N.S., Kimm L.R., Cheng Z., Horsman D.E., MacAulay C., Ng R.T., Brown C.J., Eichler E.E. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollox E.J., Armour J.A., Barber J.C. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am. J. Hum. Genet. 2003;73:591–600. doi: 10.1086/378157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldred P.M., Hollox E.J., Armour J.A. Copy number polymorphism and expression level variation of the human alpha-defensin genes DEFA1 and DEFA3. Hum. Mol. Genet. 2005;14:2045–2052. doi: 10.1093/hmg/ddi209. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez E., Kulkarni H., Bolivar H., Mangano A., Sanchez R., Catano G., Nibbs R.J., Freedman B.I., Quinones M.P., Bamshad M.J. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 23.Linzmeier R.M., Ganz T. Human defensin gene copy number polymorphisms: comprehensive analysis of independent variation in alpha- and beta-defensin regions at 8p22-p23. Genomics. 2005;86:423–430. doi: 10.1016/j.ygeno.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Perry G.H., Dominy N.J., Claw K.G., Lee A.S., Fiegler H., Redon R., Werner J., Villanea F.A., Mountain J.L., Misra R. Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aitman T.J., Dong R., Vyse T.J., Norsworthy P.J., Johnson M.D., Smith J., Mangion J., Roberton-Lowe C., Marshall A.J., Petretto E. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 27.Fellermann K., Stange D.E., Schaeffeler E., Schmalzl H., Wehkamp J., Bevins C.L., Reinisch W., Teml A., Schwab M., Lichter P. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am. J. Hum. Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J., Chen L., Ratnashinge L., Sellers T.A., Tanner J.P., Lee J.H., Dossett N., Lang N., Kadlubar F.F., Ambrosone C.B. Deletion polymorphism of UDP-glucuronosyltransferase 2B17 and risk of prostate cancer in African American and Caucasian men. Cancer Epidemiol. Biomarkers Prev. 2006;15:1473–1478. doi: 10.1158/1055-9965.EPI-06-0141. [DOI] [PubMed] [Google Scholar]
- 29.Fanciulli M., Norsworthy P.J., Petretto E., Dong R., Harper L., Kamesh L., Heward J.M., Gough S.C., de Smith A., Blakemore A.I. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat. Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y., Chung E.K., Wu Y.L., Savelli S.L., Nagaraja H.N., Zhou B., Hebert M., Jones K.N., Shu Y., Kitzmiller K. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 2007;80:1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollox, E.J., Huffmeier, U., Zeeuwen, P.L.J.M., Palla, R., Lascorz, J., Rodijk-Olthuis, D., van de Kerkhof, P.C.M., Traupe, H., de Jongh, G., den Heijer, M., et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 40, 23–25. [DOI] [PMC free article] [PubMed]
- 32.HapMap A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrett M.T., Scheffer A., Ben-Dor A., Sampas N., Lipson D., Kincaid R., Tsang P., Curry B., Baird K., Meltzer P.S. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc. Natl. Acad. Sci. USA. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 35.Cooper G.M., Nickerson D.A., Eichler E.E. Mutational and selective effects on copy-number variants in the human genome. Nat. Genet. 2007;39:S22–S29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- 36.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 37.Lipson, D., Tsalenko, A., Yakhini, Z., and Ben-Dor, A. (2005). Interval scores for quality annotated CGH data. In Workshop on Genomic Signal Processing and Statistics (GENSIPS) (Newport, Rhode Island).
- 38.Lipson D., Aumann Y., Ben-Dor A., Linial N., Yakhini Z. Efficient calculation of interval scores for DNA copy number data analysis. J. Comput. Biol. 2006;13:215–228. doi: 10.1089/cmb.2006.13.215. [DOI] [PubMed] [Google Scholar]
- 39.Pinto D., Marshall C., Feuk L., Scherer S.W. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- 40.Conrad D.F., Hurles M.E. The population genetics of structural variation. Nat. Genet. 2007;39:S30–S36. doi: 10.1038/ng2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khaja R., Zhang J., MacDonald J.R., He Y., Joseph-George A.M., Wei J., Rafiq M.A., Qian C., Shago M., Pantano L. Genome assembly comparison identifies structural variants in the human genome. Nat. Genet. 2006;38:1413–1418. doi: 10.1038/ng1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newman T.L., Rieder M.J., Morrison V.A., Sharp A.J., Smith J.D., Sprague L.J., Kaul R., Carlson C.S., Olson M.V., Nickerson D.A. High-throughput genotyping of intermediate-size structural variation. Hum. Mol. Genet. 2006;15:1159–1167. doi: 10.1093/hmg/ddl031. [DOI] [PubMed] [Google Scholar]
- 43.Urban A.E., Korbel J.O., Selzer R., Richmond T., Hacker A., Popescu G.V., Cubells J.F., Green R., Emanuel B.S., Gerstein M.B. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc. Natl. Acad. Sci. USA. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korbel J.O., Urban A.E., Grubert F., Du J., Royce T.E., Starr P., Zhong G., Emanuel B.S., Weissman S.M., Snyder M. Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome. Proc. Natl. Acad. Sci. USA. 2007;104:10110–10115. doi: 10.1073/pnas.0703834104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfeiffer P., Goedecke W., Obe G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis. 2000;15:289–302. doi: 10.1093/mutage/15.4.289. [DOI] [PubMed] [Google Scholar]
- 46.Rothkamm K., Kruger I., Thompson L.H., Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Linardopoulou E.V., Williams E.M., Fan Y., Friedman C., Young J.M., Trask B.J. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005;437:94–100. doi: 10.1038/nature04029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eden E., Lipson D., Yogev S., Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bacolla A., Wells R.D. Non-B DNA conformations, genomic rearrangements, and human disease. J. Biol. Chem. 2004;279:47411–47414. doi: 10.1074/jbc.R400028200. [DOI] [PubMed] [Google Scholar]
- 50.Bacolla A., Jaworski A., Larson J.E., Jakupciak J.P., Chuzhanova N., Abeysinghe S.S., O'Connell C.D., Cooper D.N., Wells R.D. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc. Natl. Acad. Sci. USA. 2004;101:14162–14167. doi: 10.1073/pnas.0405974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lobachev K.S., Stenger J.E., Kozyreva O.G., Jurka J., Gordenin D.A., Resnick M.A. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J. 2000;19:3822–3830. doi: 10.1093/emboj/19.14.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stenger J.E., Lobachev K.S., Gordenin D., Darden T.A., Jurka J., Resnick M.A. Biased distribution of inverted and direct Alus in the human genome: Implications for insertion, exclusion, and genome stability. Genome Res. 2001;11:12–27. doi: 10.1101/gr.158801. [DOI] [PubMed] [Google Scholar]
- 53.Hinds D.A., Kloek A.P., Jen M., Chen X., Frazer K.A. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 54.Locke D.P., Sharp A.J., McCarroll S.A., McGrath S.D., Newman T.L., Cheng Z., Schwartz S., Albertson D.G., Pinkel D., Altshuler D.M. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCarroll S.A., Altshuler D. Copy number variation and association studies of human disease. Nat. Genet. 2007;39:S37–S42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 56.Goidts V., Cooper D.N., Armengol L., Schempp W., Conroy J., Estivill X., Nowak N., Hameister H., Kehrer-Sawatzki H. Complex patterns of copy number variation at sites of segmental duplications: An important category of structural variation in the human genome. Hum. Genet. 2006;120:270–284. doi: 10.1007/s00439-006-0217-y. [DOI] [PubMed] [Google Scholar]
- 57.Perry G.H., Tchinda J., McGrath S.D., Zhang J., Picker S.R., Caceres A.M., Iafrate A.J., Tyler-Smith C., Scherer S.W., Eichler E.E. Hotspots for copy number variation in chimpanzees and humans. Proc. Natl. Acad. Sci. USA. 2006;103:8006–8011. doi: 10.1073/pnas.0602318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Repping S., van Daalen S.K., Brown L.G., Korver C.M., Lange J., Marszalek J.D., Pyntikova T., van der Veen F., Skaletsky H., Page D.C. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat. Genet. 2006;38:463–467. doi: 10.1038/ng1754. [DOI] [PubMed] [Google Scholar]
- 59.Egan C.M., Sridhar S., Wigler M., Hall I.M. Recurrent DNA copy number variation in the laboratory mouse. Nat. Genet. 2007;39:1384–1389. doi: 10.1038/ng.2007.19. [DOI] [PubMed] [Google Scholar]
- 60.Jobling M.A., Lo I.C., Turner D.J., Bowden G.R., Lee A.C., Xue Y., Carvalho-Silva D., Hurles M.E., Adams S.M., Chang Y.M. Structural variation on the short arm of the human Y chromosome: recurrent multigene deletions encompassing Amelogenin Y. Hum. Mol. Genet. 2007;16:307–316. doi: 10.1093/hmg/ddl465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simon-Sanchez J., Scholz S., Del Mar Matarin M., Fung H.C., Hernandez D., Gibbs J.R., Britton A., Hardy J., Singleton A. Genomewide SNP assay reveals mutations underlying Parkinson disease. Hum. Mutat. 2007 doi: 10.1002/humu.20626. Published online November 9, 2007. [DOI] [PubMed] [Google Scholar]
- 62.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zogopoulos G., Ha K.C., Naqib F., Moore S., Kim H., Montpetit A., Robidoux F., Laflamme P., Cotterchio M., Greenwood C. Germ-line DNA copy number variation frequencies in a large North American population. Hum. Genet. 2007;122:345–353. doi: 10.1007/s00439-007-0404-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.